
Abstract
Primary breast lymphoma (PBL) is a rare malignant lymphoid neoplasm limited to the breast, accounting for about 0.15% of all malignant breast tumors and 1.7% to 2.2% of extra-nodal lymphomas. PBL must be distinguished from conventional breast carcinomas due to different therapeutic approaches. A 25-year-old female presented with a left breast mass. Histopathology and immunohistochemical tests confirmed the diagnosis of diffuse large B-cell lymphoma (DLBCL). She had no similar lesions elsewhere in the body. She received 1 cycle of R-CHOP chemotherapy but absconded from the treatment and succumbed afterward while at home. Recent developments in DLBCL treatment have greatly improved patient outcomes by incorporating targeted medicines like rituximab, increased chemotherapy regimens, new drugs, and individualized treatment techniques. PBL appears to have a worse prognosis; thus, delay or abscondment from treatment is of serious concern when it comes to improving the prognosis of patients with PBL.
Introduction
Primary breast lymphoma (PBL) is a rare form of extranodal lymphoma, defined as lymphoma limited to the breast with or without axillary lymph node involvement in a patient without evidence of distant disease at presentation and without prior history of lymphoma. 1 Some scholars accept lymphoma as primary if the initial presentation and the dominant mass or symptoms occur in the breast, even if other involved sites are identified. PBL is a rare clinical entity, accounting for about 0.5% of all breast malignancies.1,2 It is rare because lymphoid tissue is not present in the breast. The etiology of PBL is not clearly understood. Malignant lymphoma of the breast may present as a primary or secondary tumor, both of which are extremely rare. Unfortunately, there is no morphological criterion to differentiate between primary and secondary breast lymphoma. 3 Clinically, PBL patients present with painless, palpable masses similar to breast carcinoma. Potential differential diagnoses for PBL include (i) poorly differentiated invasive ductal carcinoma that morphologically mimics high-grade lymphoma; (ii) invasive lobular carcinoma mimicking low-grade lymphoma; (iii) reactive inflammatory conditions; and (iv) nodal lymphoma extending to the breast. Therefore, a thorough clinico-pathological examination coupled with immunohistochemical testing with markers such as cytokeratin, CD45, CD20, CD3, among others, is essential for a definitive diagnosis. 4 Herein, the authors present a case of PBL in a 25-year-old African patient and a brief review of the literature.
Presentation of Case
A 25-year-old female patient presented to our surgical clinic complaining of a left breast mass. The mass started 4 months earlier and was progressively increasing in size with time. The mass was associated with dull pain radiating to her left arm but was not associated with nipple discharge. She had no history of chest tightness, difficulty breathing, coughs, fevers, or night sweats. Her past medical history was essentially unremarkable. She was nulliparous, with normal menses reported. On examination, a palpable, painless left breast mass measuring 6 × 7 cm (Figure 1) with ipsilateral axillary lymphadenopathy was noted. The contralateral breast and axillar examination were essentially normal. During admission, her resting blood pressure was 131/87 mmHg, her pulse rate was 87 beats per minute, and her axillary temperature was 36.2ºC. She was saturating at 99% on room air. She was not pale or jaundiced. The cardio-respiratory examination was fairly normal with no upper limb edema. Initial laboratory blood test results showed elevated lactate dehydrogenase levels of 974 U/L. The leucocyte count, erythrocyte count, hemoglobin, and platelet count were within normal limits. The chest X-ray showed normal lung fields and a cardiac shadow with a soft tissue dense mass over the left chest (Figure 2). Histopathology evaluation of the incisional biopsy from the breast mass demonstrated diffuse proliferation of atypical lymphoid cells (Figure 3A and B). The cells were large centroblasts infiltrating the breast tissue. The tumor cells were strongly positive for CD20 (Figure 3C), BCL2, CD45, PAX5, and Ki67. However, the tumor cells were negative for TdT, CD10, BCL6, and cytokeratin. The diagnosis of diffuse large B-cell lymphoma (DLBCL) was considered. Metastatic workups that included computed tomography (CT) scans of the chest, abdomen, and pelvis were all negative. However, due to financial constraints, magnetic resonance imaging (MRI) was not conducted. A contrast-enhanced mammography (CEM) was considered inappropriate because of the younger age of the patient. The diagnosis of PBL was established. The patient was transferred to the oncology department for oncological care. A multidisciplinary tumor board discussion recommended chemotherapy and radiotherapy. She was kept on pre-phase prednisolone and allopurinol, and later she received 1 cycle of R-CHOP regimen (ie, cyclophosphamide, doxorubicin hydrochloride, vincristine, and prednisone). She was discharged and instructed to come after 3 weeks for the second cycle. However, she did not show up. An attempt was made to reach out to her via a phone call, and we learned that she was not ready to continue with oncological care. Unfortunately, she succumbed about 60 days post-hospital discharge while at home, denying us the opportunity to ascertain the actual cause of death.
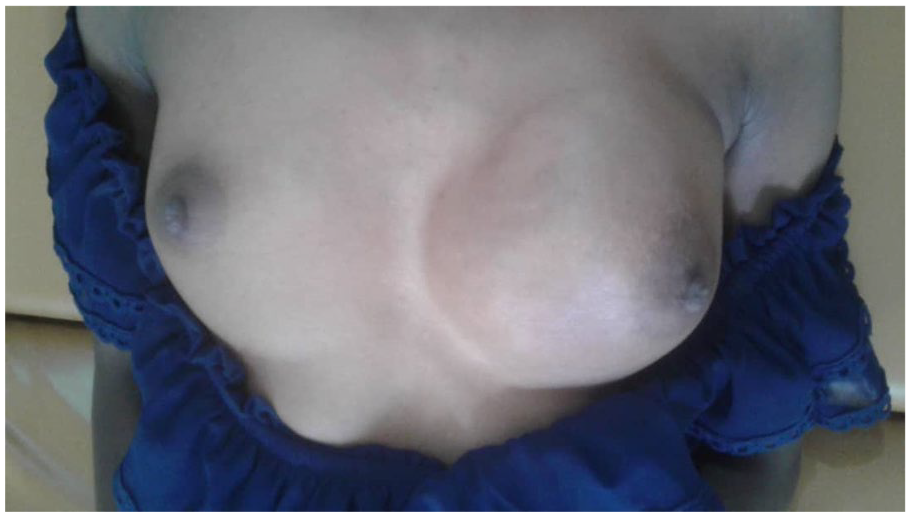
A photograph of the patient highlighting an enlarged left breast.

A plain chest X-ray showing normal lung fields and cardiac shadow with a soft tissue dense mass over the left chest.
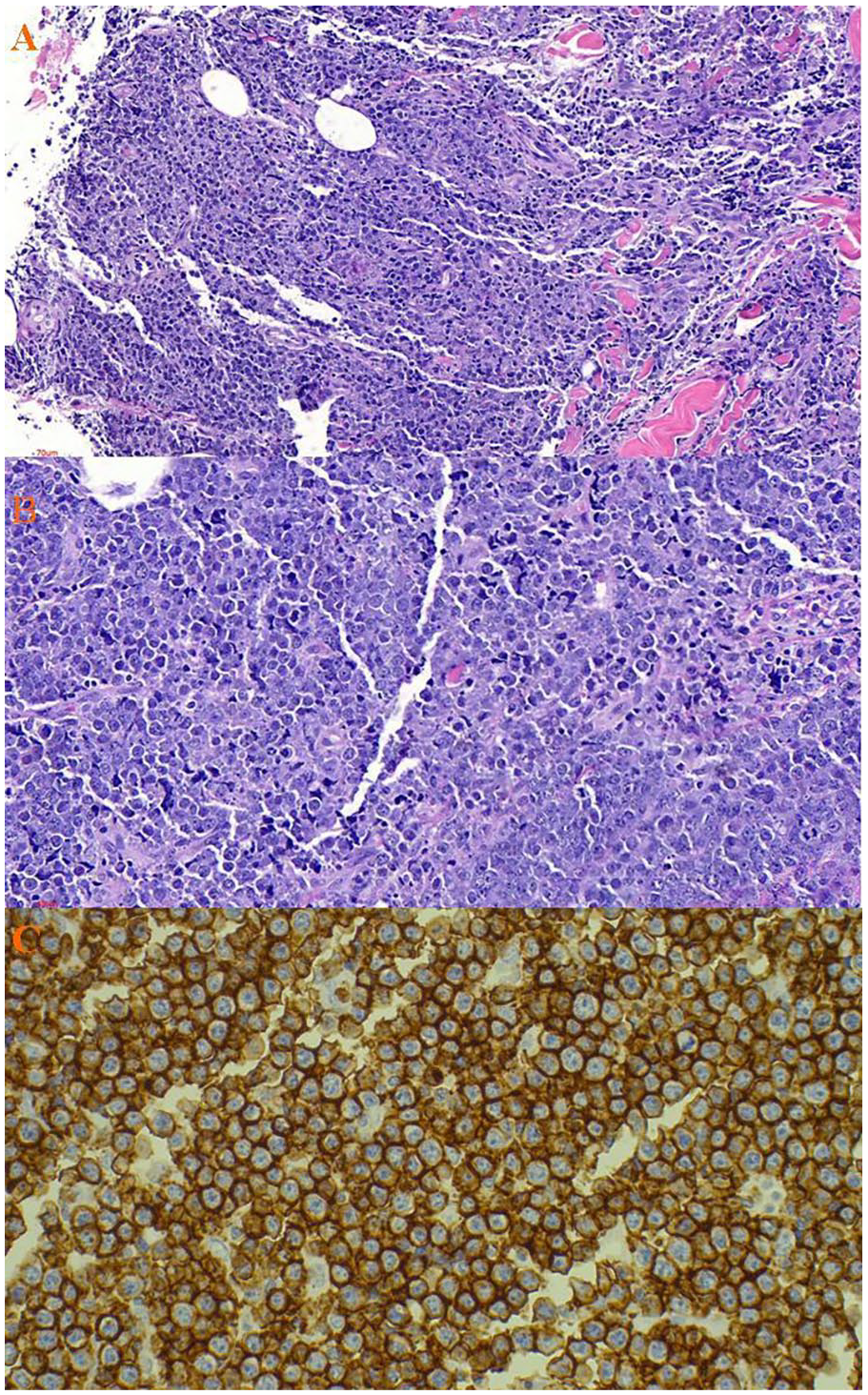
Histopathology of the tumor displaying aggressive high-grade lymphoma characterized by a dense malignant lymphoid infiltrates fibro-adipose tissue and skeletal muscle with only scanty preserved breast tissue (H&E stained 40× original magnification) (A); a high power magnification of the tumor displaying population of diffuse large lymphocytic cells, moderately pleomorphic with scant dense cytoplasm and have enlarged hyperchromatic nuclei, with irregular contours, coarse chromatin and prominent nucleoli (H&E stained ×200 original magnification) (B); immunohistochemical stains with CD20 highlighting strong membrane positivity confirming the diffuse large B-cell lymphoma (IHC 400× original magnification (C).
Discussion
Primary breast lymphoma (PBL) is one of the rarest disorders of the breast, with a prevalence of 0.15% to 0.5% of all breast malignancies.1 -3 To date, only a few cases have been documented in the English literature.1 -15 PBL may appear at any age, but the majority of patients are postmenopausal women. A subset of patients is represented by pregnant or lactating women with massive bilateral breast swelling; most of these cases were reported from Africa, 8 although non-African cases are also on record. PBL mostly affects females; contrary to the index case, it is seen to affect people mostly in their sixth decade of life; our patient presented with the disease in her third decade. The disease is exceedingly rare in men. 5
PBL is usually non-Hodgkin’s B-cell type, with diffuse large B-cell lymphoma (DLBCL) being the most common subtype, as presented in our case. 6 Other rare lymphoma subtypes that may involve the breast include Burkitt lymphoma, extranodular marginal-zone B-cell lymphoma of mucosa-associated lymphoid tissue (MALT) type, follicular lymphoma, lymphoblastic lymphoma of either B or T type, and implant-associated anaplastic large cell lymphoma. 7 Extremely rarely, T-cell lymphomas of variable subtypes according to the current WHO classification have been documented.
A widely accepted criteria for defining PBL includes the presence of breast tissue in, or adjacent to, the lymphoma infiltrates, the absence of concurrent nodal disease except for the involvement of ipsilateral axillary lymph nodes, and the absence of a previous history of lymphoma involving other organs. 4 However, such criteria seem to be too restrictive and thus leave no room for PBL at higher stages. On the other hand, other scholars consider PBL in cases where the breast is the first or major site of presentation, even if, on subsequent staging procedures, involvement of distant nodal sites or bone marrow is discovered.9,10 The clinical presentation of PBL usually does not differ from that of breast carcinoma. It usually presents with a painless lump, sometimes multinodular, which is bilateral in approximately 10% of cases. 4 Imaging usually reveals no feature that helps distinguish primary from secondary lymphoma. Unfortunately, there are no histomorphological criteria to differentiate between primary and secondary breast lymphoma. The value of magnetic resonance imaging (MRI) in breast lymphomas has not been clearly determined. Having an almost similar clinical presentation as well as imaging as that of carcinoma of the breast, histopathology coupled with immunohistochemistry, as was evidenced in the index case, is essential to establishing the definitive diagnosis.10,11 As PBL is exceedingly uncommon, it must be distinguished from secondary breast involvement from another primary site in the setting of relapse or widespread diseases that are more common. Also, PBL must be distinguished from breast carcinomas, as therapeutic approaches are different. Thus, the use of immunohistochemistry is the key to confirming the diagnosis.
The treatment of PBL is still a topic of controversy. The treatment depends on the histopathology diagnosis and is generally the same as lymphoma at any other site. Recent advances in treatment strategies for primary breast DLBCL have focused on improving patient outcomes through targeted therapies, novel drug combinations, and optimized treatment protocols. Some of the most recent advances and their impact on patient outcomes include: (i) Targeted therapies, that is, rituximab. The addition of rituximab, an anti-CD20 monoclonal antibody, to the standard chemotherapy regimen (CHOP) has significantly improved outcomes for PBL patients. Rituximab-CHOP (R-CHOP) has become the standard first-line therapy, leading to better overall survival and progression-free survival rates compared to CHOP alone. Studies have shown that R-CHOP results in a higher complete remission (CR) rate and reduces the risk of relapse, especially in patients with localized disease.12,13 (ii) Intensified chemotherapy regimens. DA-EPOCH-R: Dose-adjusted EPOCH (etoposide, prednisone, vincristine, cyclophosphamide, and doxorubicin) combined with rituximab (DA-EPOCH-R) is used for high-risk DLBCL, including PBL. This regimen allows for dose adjustments based on patient tolerance, potentially leading to better efficacy and reduced toxicity. Early studies suggest improved progression-free survival and overall survival in patients treated with DA-EPOCH-R compared to standard R-CHOP, particularly in high-risk cases. 14 (iii) Novel agents and combinations, such as polatuzumab vedotin. This antibody-drug conjugate targets CD79b, a component of the B-cell receptor. When combined with bendamustine and rituximab (BR), it has shown promising results in relapsed/refractory DLBCL. Polatuzumab vedotin plus BR has been associated with improved response rates and durability of response in relapsed/refractory settings. 15 (iv) Radiotherapy, is often used as an adjunct to systemic therapy in PBL-DLBCL, especially for localized disease. Modern techniques such as intensity-modulated radiotherapy (IMRT) allow for precise targeting of the tumor while sparing surrounding tissues. Combining radiotherapy with systemic therapy can improve local control and reduce the risk of recurrence, leading to better long-term outcomes. 16 (v) Prognostic markers and personalized medicine; advances in molecular profiling have identified specific genetic and molecular markers that predict response to therapy and prognosis. For example, the presence of MYC, BCL2, and BCL6 rearrangements can influence treatment decisions. Tailoring treatment based on these biomarkers can lead to more personalized and effective therapeutic strategies, improving patient outcomes. 17 Some scholars argue for the use of surgical approaches, in which it is seen that they offer no benefit to the overall survival rate and have a high chance of recurrence.10,11,18 Some studies showed that mastectomy with radiation therapy had an increased survival rate compared to when the surgical technique was used alone.18,19 We were able to treat our patient with anthracycline-based chemotherapy (CHOP); however, only 1 cycle was received.
The prognosis of PBL is still a challenge to determine, as there are few cases of this uncommon breast malignancy. 9 The prognosis is dependent on the stage and type of lymphoma. A study done by Hosein et al showed that none of the treatments that involved chemotherapy, radiotherapy, or immunotherapy (rituximab) were associated with overall survival. 20 Caon et al showed that for indolent breast lymphoma, radiation therapy alone had high chances of local control and survival compared to aggressive histology, which received radiation therapy with chemotherapy and had excellent local control but poor overall survival rates, with a 5-year local control rate of 92% in indolent and 96% in aggressive form. 11 Current literature recommends the use of anthracycline-based chemotherapy with rituximab and radiation therapy for aggressive forms of breast lymphoma, although few studies support its advantage. Our patient succumbed before the prescribed treatment was finished. We were unable to establish whether the patient experienced adverse reactions since she died at home.
There are several potential limitations to consider with the present study. Our case study lacks some essential information. For instance, it is difficult to confidently establish the cause of death in the index case since she absconded from her treatment and died at home. Unfortunately, postmortem was out of options. Thus, it is not clear whether her demise was attributed to the PBL disease, whether the chemotherapy was kept on, or other causes. However, based on previous published data, it is plausible to speculate that the most likely cause of death was PBL disease that was left untreated.
Conclusion
Primary breast lymphoma is a rare disease of the breast whose diagnosis and treatment may be challenging. The recent advances in treatment of the primary DLBCL strategies have significantly improved patient outcomes through the integration of targeted therapies like rituximab, intensified chemotherapy regimens, novel agents, and personalized treatment approaches. These advancements have led to higher remission rates, prolonged survival, and a better quality of life for patients. Ongoing research and clinical trials continue to push the boundaries of what is possible in the treatment of this rare lymphoma. This case report highlights the awareness of primary breast lymphoma as a differential diagnosis and the importance of histopathology and immunohistochemistry analysis in establishing the definitive diagnosis in patients presenting with breast masses.
Footnotes
Acknowledgements
The authors would like to thank the patient for permission to share her medical history for educational purposes and publication.
Funding:
The author(s) disclosed receipt of the following financial support for the research, authorship, and/or publication of this article: The project on which this publication is based was in part funded by the German Federal Ministry of Education and Research 01KA2220B to the NORA Programme. This research was funded in part by Science for Africa Foundation to the Programme REACCT-CAN with support from Wellcome Trust and the UK Foreign, Commonwealth & Development Office and is part of the EDCPT2 programme supported by the European Union.
Declaration of conflicting interests:
The author(s) declared no potential conflicts of interest with respect to the research, authorship, and/or publication of this article.
Author Contributions
AM, JL, and HC conceptualized and drafted the manuscript and EU and EM reviewed medical records. AM reviewed and reported the histology films. All authors have read and approved the final manuscript.
Ethical Approval
Ethics Committee approval was not required for this case report. The surgical procedure was conducted in accordance with the principles of the Declaration of Helsinki.
Informed Consent
Written informed consent was obtained from the patient’s legally authorized representative for the publication of this case report. A copy of the written consent is available for review by the Editor-in-Chief of this journal on request.
Guarantor
AM accepts full responsibility for the work and the conduct of the study, he had access to the data, and controlled the decision to publish.