
Abstract
Non-Hodgkin lymphomas are most frequently classified based on the lineage marker expression. However, lymphomas with aberrant marker expression as well as monoclonal IgH/IgΚ and TCR gene rearrangements may co-exist which can be misleading and confusing. Primary CD20 negative diffuse large B-cell lymphomas (DLBCL) represent a rare entity, and they account for 1% to 3% of cases. However, some CD20 negative DLBCLs could not be classified into known variants, creating both diagnostic and therapeutic dilemma’s. Primary CD20 negative DLBCL are more likely to have a non-germinal centre subtype, a higher proliferation index, more frequent extra-nodal involvement, a poorer response, and poorer prognosis to conventional treatment compared to CD20 positive DLBCL. A 66- year-old postmenopausal lady, presented with palpable, bilateral neck lymphadenopathy and difficulty swallowing. She also had left leg lymphoedema, poor appetited, fatigue and weight loss. Her symptoms lasted approximately 1 month. After histological, immunohistochemical and clonality analysis of the lymph node the patient was diagnosed with primary nodal CD20 and PAX-5 negative DLBCL with dual immunoglobulin light-chain kappa (IgK) and T-cell receptor (TCR) gene rearrangement. This unusual and unique case presented a diagnostic challenge because it was CD20 and PAX-5 negative, had dual IgK and TCR gene rearrangement and, it could not be classified within the known and well established CD20 negative DLBCL variants. Describing such cases emphasises the fact that lymphomas unclassifiable within known variants of CD20 negative DLBCL do exist and that range and heterogeneity of CD20 negative DLBCL continues to evolve, and pathologist should be aware of these uncommon, atypical mature B-cell neoplasms.
Introduction
Diffuse large B-cell lymphoma is the most common non-Hodgkin lymphoma (NHL), accounting for 30% to 35% of the cases. Primary CD20 negative DLBCL is a rare entity, and it accounts for 1% to 3% of cases. 1 The well-known variants of CD20 negative DLBCL include plasmablastic lymphoma (PBL), primary effusion lymphoma (PEL), large B-cell lymphoma arising in human herpesvirus-8 (HHV-8) associated multicentric Castleman disease (MCD) and anaplastic lymphoma kinase (ALK)-positive DLBCL. 2 This group of lymphomas usually have aggressive clinical behaviour, extranodal disease presentation and unusual morphology. 3 In addition to the above mentioned CD20 negative lymphomas, CD20 positive lymphoma can relapse as CD20 negative lymphoma after CD20 antibody therapy.4,5 However, some CD20 negative DLBCL could not be classified into known variants, and they pose diagnostic as well as therapeutic dilemma. On the other hand, the proper treatment of patients suffering from lymphoid neoplasms is based on the correct classification of lymphoid malignancies as well as lineage and maturity determination. Lymphomas are broadly classified as B-cell or T-cell lymphomas, based on histology, immunohistochemistry, flow cytometry and molecular analyses. The most common immunohistochemical markers used for determining the B or T cell lineage are CD20, CD79a or PAX-5 and CD3 or CD5, respectively. However, in some lymphoid malignancies expression of lineage specific markers deviates from one expected either due to loss of cell antigens or acquiring lineage markers which are usually absent in their benign counterparts. 6 The so called ‘lineage infidelity’ of surface markers is well documented in lymphomas and quite often leads to confusions and serious diagnostic difficulties.7 -9 In such cases of phenotypical equivocal cases, molecular investigations of immunoglobulin heavy chain (IgH) or light-chain kappa (IgΚ) and T-cell receptor (TCR) gene rearrangement should be used to establish B or T cell histogenesis.9,10 However, cases with concurrent rearrangement of IgH/IgΚ and TCR are reported with a frequency of approximately 10% for both B-cell and T-cell lymphoproliferative neoplasms, respectively.11 -13
Here we describe unusual and unique case of primary nodal CD20 and PAX-5 negative but CD79a and CD38 positive DLBCL with dual IgK and TCR rearrangement which could not be classified within the well established CD20 negative DLBCL variants. The patient was 66-year old, immunocompetent female with no prior history of lymphoproliferative disease. This case represented a diagnostic challenge because DLBCL was negative for CD20 and PAX-5, the 2 most used markers, to diagnose B-cell lineage lymphoma, while at the same time B-cell and T-cell clonality analyses showed dual IgK and TCR rearrangement.
Case Presentation
A 66-year-old postmenopausal lady, presented with palpable, bilateral neck lymphadenopathy and difficulty swallowing. She also had left leg lymphoedema and complained of having poor appetite, fatigue, and lost 4 kg within 4 weeks. Patient denied having fever and night sweats. All these difficulties lasted approximately 1 month. Complete blood count (CBC) showed mild anaemia while all other laboratory findings were within normal limits. Also, HBV, HCV and HIV serology were negative. Colour doppler of lower extremities showed no signs of deep vein thrombosis and cardiac ECHO showed ejection fraction of 65%. Abdominal ultrasound showed para-aortic, bilateral pelvic and inguinal lymphadenopathy.
Pathological findings
An excisional biopsy of enlarged axillary and cervical lymph node was performed. The cervical lymph node measured 1.8 × 1 × 0.7 cm and axillary node measured 3.6 × 2.6 × 2 cm. Nodes had firm consistency with no signs of necrosis or haemorrhage. Histological examination of lymph nodes showed effaced architecture, due to infiltration of polymorphic tumour lymphoid cells. The interfollicular area of lymph node was infiltrated with medium to large sized lymphocytes, having blastoid morphology indicative of centroblasts and immunoblasts and intermingled with plasmablast-like cells. Also, there were scattered large bi-nucleated cells similar to Reed-Sternberg cells. In between there were many eosinophils, histiocytes and plasma cells. Furthermore, within interfollicular areas, many mitoses and apoptosis were found. Four-micrometer sections from paraffin-embedded blocks were stained with each antibody listed below, using the streptavidin-biotin complex method. By immunohistochemistry, large atypical lymphoid cells were CD1a-, CD2-, CD3-, CD4−/+, CD5−, CD8−, CD20−, CD79a+, PAX-5−, CD38+, CD138−, CD30−/+, CD56−, MUM1+, PD-1−/+, Bcl-2−, ALK−, c-myc−, TdT−, CD45+, Bcl-6−, CD10− and HHV-8−. The Epstein-Barr encoding region (EBER) in situ hybridisation was negative (Figure 1). Kappa and lambda light chain in situ hybridisation did not show restriction. CD68 staining showed lot of histiocytes and Ki-67 proliferation index was high, approximately 65% (Figure 1).
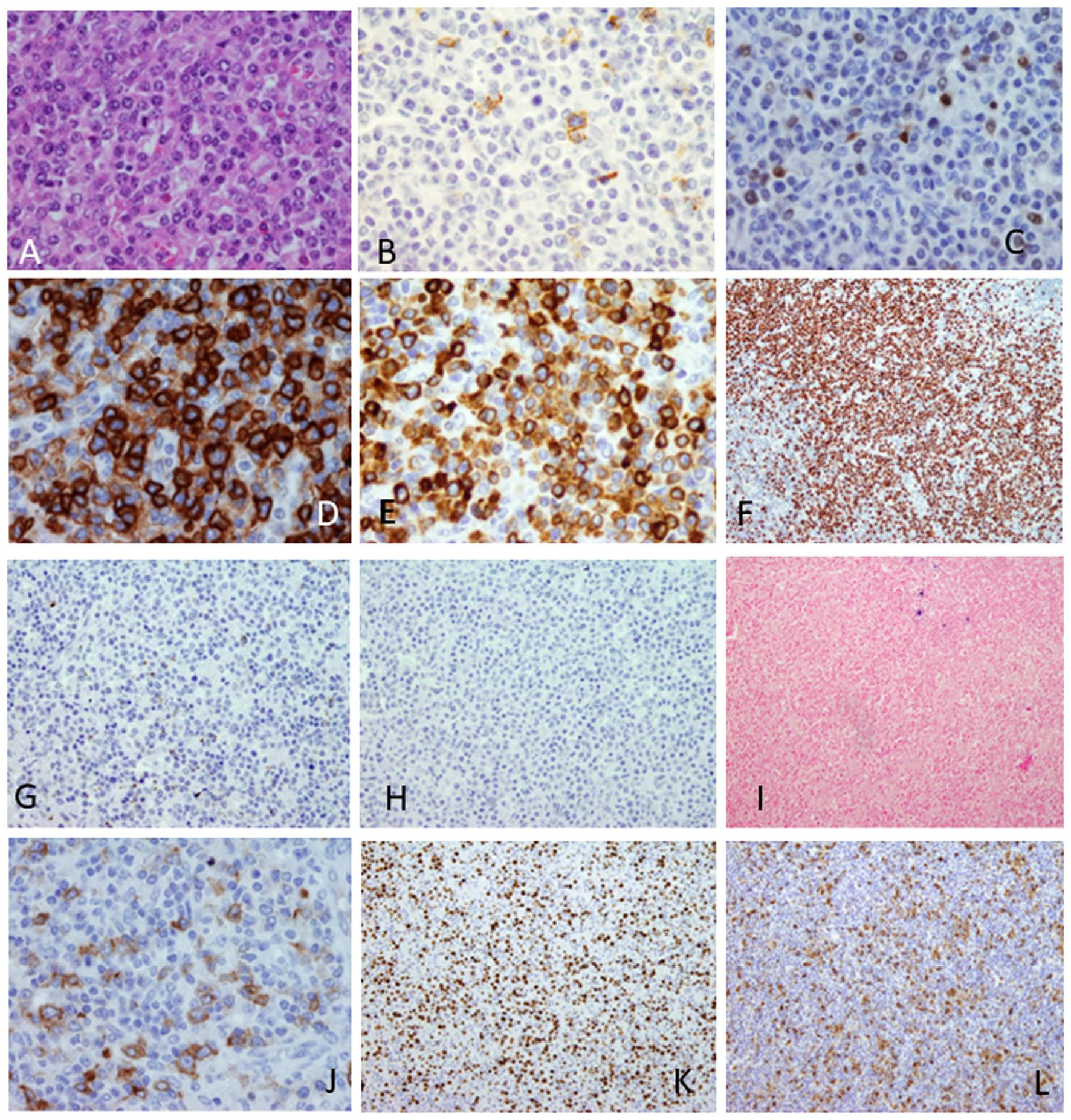
Lymph nodes histological and immunohistologic features. (A) The lymph node architecture was effaced due to infiltrate of medium to large sized atypical lymphatic cells with interfollicular growth pattern and cells having blastoid morphology (HE staining, ×40 magnification). ((B-E), ×40 magnification) Immunohistochemical staining of the interfollicular lymphatic infiltrate for CD20 (image (B)) and PAX-5 (image (C)) was negative while CD38 (image D) and CD79a (image E) staining were positive. Also, tumour cells were diffusely and strongly MUM1 positive (image (F), ×10 magnification). ((G-I), ×20 magnification) The staining for CD138 (image (G)), HHV-8 (image (H)) and EBER in situ hybridisation (image (I)) were negative. The CD30 stained only blastoid cells and in less than 5% of tumour cell population (image (J), ×40 magnification). There was high Ki-67 proliferation index, approximately 65% (image (K), ×10 magnification) and between tumour cells there was lot of histiocytes (image (L), ×10 magnification).
Ig/TCR clonality analysis
Due to the unusual morphology, as well as the immunophenotype of large, polymorphic cells, with blast and plasmablast-like appearance (CD20-/PAX-5-/CD79a+/CD38+/MUM-1+/CD4-/+) a multiplex polymerase chain reaction (PCR) analyses of B-cell and T-cell receptor rearrangement were performed using BIOMED-2 protocol. 14 Briefly, BIOMED-2 multiplex PCR reactions using fluorescently labelled oligonucleotide primers were performed in triplicates with 50 ng and 200 ng of DNA per 50 µL reaction. Following PCR amplification, the PCR products were resolved using capillary electrophoresis on the SeqStudio (Applied Biosystems, USA) genetic analyser. GeneMapper software (Applied Biosystems, USA) was then used to analyse PCR products by peak height and size determination. The clonality analysis of IgH gene in FR2 and FR3 regions showed polyclonal distribution of PCR products (gaussian distribution of multiple peaks representing multiple PCR products) while the IgΚ gene clonality analysis showed only one prominent peak (monoclonal result consisting of one type of PCR product) (Figure 2). However, the T cell receptor analysis of TCR gamma region showed bi-clonal and TCR beta region monoclonal distribution of PCR products (Figure 3).

GeneScaning analysis of multiplex IgH and IgΚ PCR products. This figure shows the B cell clonality analysis which is PCR based high resolution fragment analysis of immunoglobulin heavy chain (IgH) and Ig kappa (Κ) light chain gene rearrangement using multiplex primers from BIOMED2 protocol. The top 2 electropherograms show polyclonal population (characteristic Gaussian size distribution) of lymphocytes with multiple PCR product using IgH FR2 (image (A)) and IgH FR3 (image (B)) primers. However, the immunoglobulin light chain kappa gene clonality analysis showed typical results for clonal population of lymphocytes. Arrows indicate clonality peaks of specific size for IgΚ tube A (149 bp in size) (image (C)) and IgΚ tube B (235 and 287 bp in size) (image (D)). All the gene rearrangement analyses were performed in triplicates. The orange, regularly spaced peaks represent the size standards.
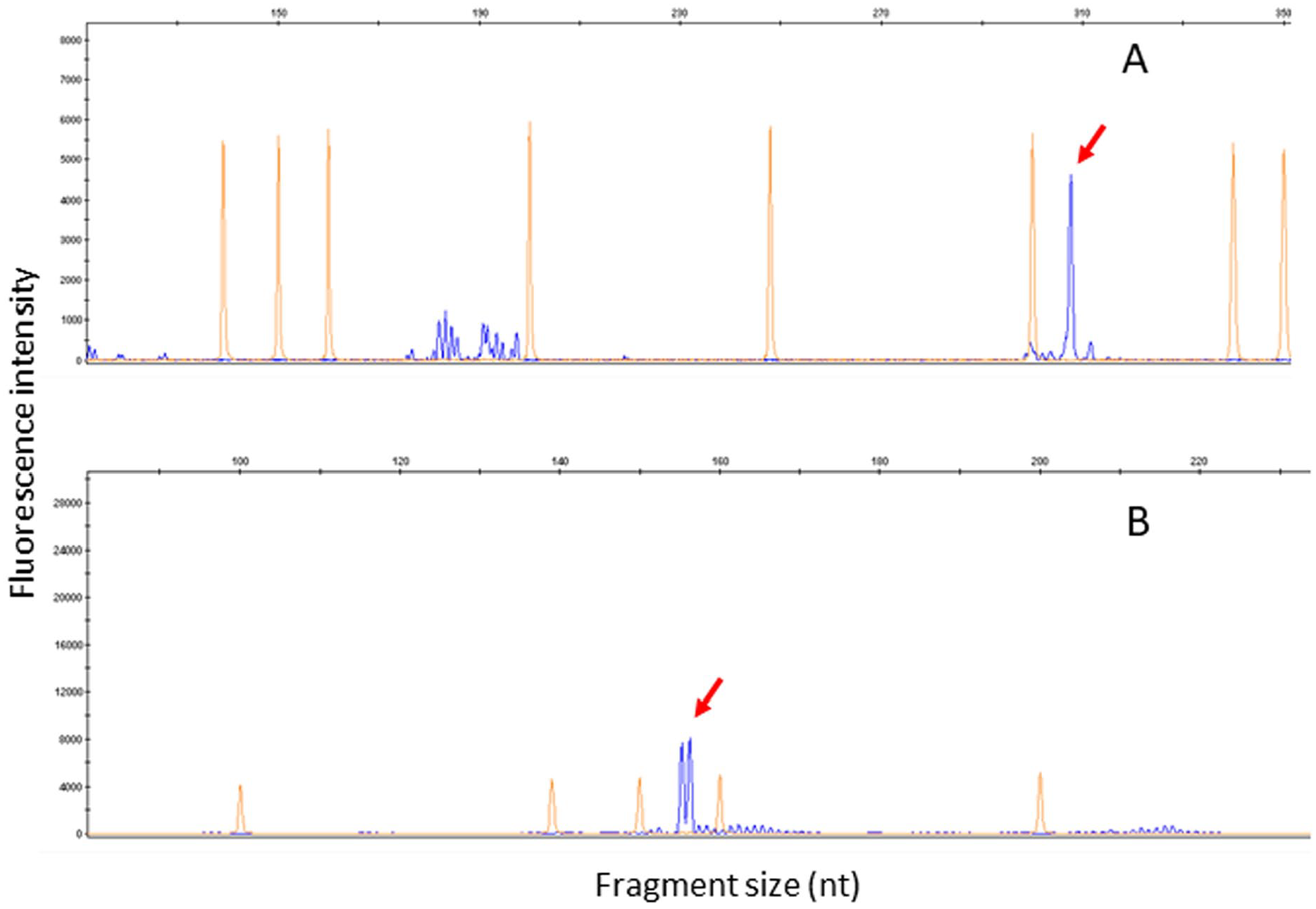
GeneScaning analysis of multiplex TCR PCR products. Figure shows PCR based T cell clonality analysis using multiplex primers for T-cell receptor (TCR) gene rearrangement. In TCRβ tube C (image (A)) high resolution fragment analysis showed one prominent clonal PCR product of 307 bp in size (red arrow). For TCRγ tube A (image (B)) high resolution fragment analysis showed the monoclonal (biallelic) rearrangement (154 bp and 156 bp in size-red arrows). All of the gene rearrangement analyses were performed in triplicates. The orange, regularly spaced peaks represent the size standards.
Patient treatment and follow-up
Considering histological, immunohistochemical and clonality analysis results our final diagnosis was primary nodal CD20 and PAX-5 negative DLBCL with dual IgK and TCR rearrangement. The whole-body computer tomography (CT) scan showed generalised lymphadenopathy and bone marrow biopsy showed no evidence of infiltration. Thus, a stage IIIB of disease was confirmed. Considering, poor prognosis of primary CD20 negative DLBCL, patient was treated with 6 cycles of the R-DA-EPOCH (dose adjusted rituximab, etoposide prednisolone, vincristine, cyclophosphamide, doxorubicin) chemotherapy protocol. The subsequent follow up laboratory tests as well as positron emission tomography and computed tomography scans (PET-CT) showed no evidence of lymphoproliferative disease 4 years after initial diagnosis. The collateral finding at the last control PET-CT scan was diffuse thickening of the large colon at sigmoid region. A colon surgery was performed, and histological examination showed colon adenocarcinoma (pT2, pN0). Other than that, patient did not have any health-related difficulties.
Discussion
NHLs are most frequently classified based on the lineage marker expression, however lymphomas with aberrant expression are reported as well. In such cases, molecular analysis of IgH/IgΚ and TCR gene rearrangements should be performed to determine histogenesis. However concomitant monoclonal IgH/IgΚ and TCR gene rearrangements may co-exist and are reported for B-cell and T-cell lymphomas. 15 In such equivocal cases, final determination of lineage should be based on the multimodal approach considering morphology findings, immunophenotypic and genotypic features [9]. CD20 is expressed from early pre-B development and is lost during terminal B-cell differentiation into plasma cells. 16 Downregulation of CD20 in B-cell neoplasms is often related to plasmablastic features and terminal B-cell differentiation of the tumour cells. 17 It is speculated that genetic mutations of this gene lead to conformational changes of the surface protein resulting in CD20 negative staining. 3 On the other hand, PAX-5 is thought to be the most specific B-cell lineage marker expressed from pro-B to the mature B-cell stage and then silenced in the plasma cells. 9 Besides being the critical B-cell lineage factor, PAX-5 also upregulates the expression of various B-cell differentiation markers. 18 Studies indicate that B cells showing damaged or deregulated PAX-5 activity may occur in bi-phenotypic lymphomas. 19 Moreover, this case was PAX-5 negative which might have caused the downregulation of CD20 expression. CD79a is found in the cytoplasm of pro-B cells before IgH gene rearrangement, and its expression is lost in the terminally differentiated plasma cells. 16 Additionally, it has been suggested, if light chain restriction is detected on the cell surface or cytoplasm the neoplasia can be designated as B cell lineage even though all pan B cell antigens are lost. 6 We then decided to perform molecular investigation of B-cell and T-cell gene rearrangement because the morphology and immunohistochemistry findings were not convincing. This case was complex, not just due to negativity for 2 most important B-cell lineage markers, CD20 and PAX-5, but because clonality analyses showed dual IgK and TCR rearrangement as well. When confronted by such rearrangement data, it is suggested that it is better to interpret these as markers of clonality rather than lineage and that immunophenotypic features are perhaps more robust determinants of the latter. 15
This case was positive for CD79a but at the same time positive for CD38 and MUM-1 as well. The most frequent variant of CD20 negative lymphomas is PBL which is positive for CD138 in almost all cases and frequently express CD38 and MUM-1. 20 MUM-1 is one of the markers of plasma cell differentiation, but previous studies showed that specificity of MUM-1 as plasma cell marker is limited. 20 However, in this case a PBL diagnosis was unlikely because it did not have a specific plasmablastic morphologic features, cells were CD138 and EBV negative while Ki-67 proliferation index was below 90%. One more clinical feature that goes into favour of DLBCL diagnosis, is that majority of patients with PBL do not survive more than 12 months. It is well known that CD38 is expressed at high levels in most cases of DLBCL. Studies using flow cytometry have shown the association of the intensity of CD38 expression with more aggressive DLBCL variants or high CD38 expression to be an independent predictor of poor outcomes in de novo DLBCL.21,22 This case could not be categorised into known variants of CD20 negative DLBCL and in the literature there are few reports describing such new cases of DLBCL,20,23,24 Nakatsuka et al reported an EBV-positive DLBCL case that was negative for various B-cell (CD19, CD20, CD79a, CD38, CD138) and T-cell (CD3, CD4, CD5, CD8) markers. 20 This case was positive only for Oct-2 and BOB-1. 20 Also, their case had clonal IgH and light gene rearrangement while there was no clonal rearrangement of TCR genes. 20 It has been shown that Oct-2, BOB-1 and PAX-5 although considered to be B-cell associated transcription factors have been detected in the mature T-cell lymphomas as well. 9 Also, Starr et al described a case of CD20/CD45 negative EBV positive DLBCL but their case was positive for PAX-5 and CD79a while ours was negative for PAX-5. 25 Furthermore, it is still not clear why B-cell markers are downregulated in EBV positive B-cell lymphomas and association of B-cell marker suppression with EBV infection. 20 However, in our patient the serology was EBV and HIV negative as it was EBER in situ hybridisation (Figure 1). Primary CD20 negative DLBCL represent an aggressive group of lymphomas usually refractory to different chemotherapy protocols with median survival time range from 4 to 20 months.2,5 A retrospective study, of de novo DLBCL patients, demonstrated that patients with CD20 negative DLBCL are more likely to have a non-germinal centre subtype, a higher proliferation index, more frequent extra-nodal involvement, a poorer response, and poorer prognosis to conventional treatment compared to CD20 positive DLBCL. 17 There is still no standard of care for CD20 negative DLBCL lymphomas, thus further studies are warranted to investigate optimal therapy of CD20 negative de novo DLBCL. In this case, considering poor prognosis of primary CD20 negative DLBCL, with unusual morphology, immunohistochemistry and clonality findings, the patient was treated with 6 cycles of the R-DA-EPOCH (dose adjusted rituximab, etoposide prednisolone, vincristine, cyclophosphamide, doxorubicin) chemotherapy protocol. Our patient successfully completed chemotherapy protocol and was disease-free at 4 years of close follow up.
Conclusion
In the case of lymphomas with unusual morphology, immunophenotype and presence of unexpected gene rearrangement the lineage determination can be a rather difficult to confirm. This case is unique because its histology and immunohistochemistry led to a diagnostic dilemma which directed us to perform additional molecular analyses of IgH/IgΚ and TCR gene rearrangement. Based on the histological features, expression of CD79a, CD38 and MUM-1 and IgΚ gene rearrangement, a final diagnosis of DLBCL was favoured despite negative CD20 and PAX-5 staining and TCR gene clonal rearrangement. Therefore, it is evident that lymphomas unclassifiable within known variants of CD20 negative DLBCL do exist. These lymphomas usually display an aberrant immunoprofile with frequent loss of lineage specific markers. Such unusual cases suggest that the range and heterogeneity of CD20 negative DLBCLs continue to evolve. 26
Footnotes
Acknowledgements
The authors thank the technical staff of the immunohistochemical and molecular laboratory of the Department of Pathology for their skilful assistance.
Funding:
The author(s) disclosed receipt of the following financial support for the research, authorship, and/or publication of this article: The publication of this article was funded by University of Rijeka Grants; uniri-biomed-18-104 and uniri-biomed-18-242.
Declaration of conflicting interests:
The author(s) declared no potential conflicts of interest with respect to the research, authorship, and/or publication of this article.
Author Contributions
Material preparation, conception, immunohistochemistry, editing, supervision, molecular analysis, review (I.H.); data collection and material preparation (L.K.); figures, review and editing (E.B.); review, editing, lymph node biopsy (B.M.); review, editing, patient management (T.V.); review, editing, histopathological diagnosis (N.J.). The original draft of the manuscript was written by Lucia Klarica and Ita Hadžisejdić. All authors have read and agreed to the submitted version of the manuscript.
Consent for Publication
Written informed consent has been obtained from the patient to publish this paper.
Data Availability
All data that support the findings of this case are included in this article. Further enquiries can be directed to the corresponding author.
Ethics Approval and Consent to Participate
Ethical review and approval were waived for this case report, due to inclusion of only one patient so according to Faculty of Medicine and Clinical Hospital Centre Rijeka Ethical committees the patient’s informed consent is sufficient. Also, after waiver we are obliged to act according to the guidelines of the Declaration of Helsinki.