
Abstract
Early diets in honeybees have effects on epigenome with consequences on their phenotype. Depending on the early larval diet, either royal jelly (RJ) or royal worker, 2 different female castes are generated from identical genomes, a long-lived queen with fully developed ovaries and a short-lived functionally sterile worker. To generate these prominent physiological and morphological differences between queen and worker, honeybees utilize epigenetic mechanisms which are controlled by nutritional input. These mechanisms include DNA methylation and histone post-translational modifications, mainly histone acetylation. In honeybee larvae, DNA methylation and histone acetylation may be differentially altered by RJ. This diet has biologically active ingredients with inhibitory effects on the de novo methyltransferase DNMT3A or the histone deacetylase 3 HDAC3 to create and maintain the epigenetic state necessary for developing larvae to generate a queen. DNMT and HDAC enzymes work together to induce the formation of a compacted chromatin structure, repressing transcription. Such dialog could be coordinated by their association with other epigenetic factors including the ubiquitin-like containing plant homeodomain (PHD) and really interesting new gene (RING) finger domains 1 (UHRF1). Through its multiple functional domains, UHRF1 acts as an epigenetic reader of both DNA methylation patterns and histone marks. The present review discusses the epigenetic regulation of honeybee’s chromatin and how the early diets in honeybees can affect the DNA/histone modifying types of machinery that are necessary to stimulate the larvae to turn into either queen or worker. The review also looks at future directions in epigenetics mechanisms of honeybees, mainly the potential role of UHRF1 in these mechanisms.
Introduction
In a honeybee colony (Apis mellifera), royal jelly (RJ), produced by the hypopharyngeal and mandibular glands of young honeybees “nurse workers” is used for feeding bee larvae as well as the adult queen.1,2 All bee larvae are fed RJ during the first 3 days of larval development. Worker bees then switch to their special diet, that is, worker jelly composed of pollen, honey, and nectar, while queen bee larvae consume RJ in large quantities as a main food throughout their adulthood. 3 This differential feeding results in 2 different female castes with prominent physiological and morphological differences, a long-lived queen with fully developed ovaries and a short-lived functionally sterile worker.4 -6 To generate this phenotypic polymorphism from 2 identical genomes, honeybees utilize epigenetic mechanisms controlled by diet. These mechanisms include DNA methylation3,7 and histone post-translational modifications (PTMs), mainly histone methylation and acetylation.3,8,9
During honeybee development, methylation fidelity could be regulated by several molecular mechanisms including alterations in the expression of the methyltransferases DNMT1, DNMT3A, and DNMT3B. 10 The de novo methyltransferases DNMT3A and DNMT3B methylate the cytosine of non-methylated CpG sites on both DNA strands. On the other hand, hemimethylated CpG sites, generated during DNA replication, are specifically targeted and methylated by DNMT1, ensuring the maintenance of genomic methylation. In honeybees, DNA methylation may be differentially altered by nutritional input, leading to profound shifts in developmental fates, with massive implications for reproductive and behavioral development. 7 The implication of histone 3 acetylation at the lysine 27 (H3K27) was also identified as a key chromatin modification, with caste-specific regions in the honeybee. 9
As a result of inhibition of DNMT3 expression in larvae using RNA interference, 72% of the adult bees became queens with fully developed ovaries, which are identical to those of a queen reared on pure royal jelly in the hive. 7 These findings indicate that RJ has inhibitory effects on DNMT3, producing queens with fully developed ovaries. Interestingly, the fatty acid called 10-Hydroxy-2-decenoic acid (10HDA), the active ingredient in RJ was shown to inhibit the histone deacetylase HDAC3, allowing the acetylation of histones, stimulating the larvae to turn into queens.3,11 Based on these findings, it is suggested that RJ contains epigenetically active compounds that inhibit either DNMT3 7 or HDAC3. 11 Interestingly, RJ has several biologically active ingredients such as flavonoids and phenolic acids,1,12 which could act as epigenetic modulators.13 -15
Changes in chromatin accessibility affecting transcription are achieved through coordinated dialog between DNA methylation and PTMs. 16 Such dialog suggests the presence of a dynamic “epigenetic reader” which possesses a set of functional domains. The ability of ubiquitin-like containing plant homeodomain (PHD) and really interesting new gene (RING) finger domains 1 (UHRF1) to interact with DNMT3A, DNMT3B, 17 DNMT1,18 -21 histone deacetylases HDAC1, 22 HDAC3, 23 histone acetyltransferase TIP60,24,25 and histone 3 methyltransferase G9a, 26 give UHRF1 a role as sensor for both DNA methylation patterns and histone marks. UHRF1 gene is conserved in vertebrates, and it ensures together with DNMT1 the maintenance of 5-methylcytosine (5 mC) in their genomes during DNA replication.18,20,27 It is interesting to note that honeybees have UHRF1 protein. 28 Thus, UHRF1 could be a key regulator of the global epigenetic reprograming of early embryos in honeybees.
This review aims to discuss the role of epigenetic mechanisms in caste determination in honeybees, and how early diets have effects on their epigenome with significant impacts on queen development. The review also looks at future directions in epigenetics mechanisms of honeybees, mainly the potential role of epigenetic integrator UHRF1 in these mechanisms.
DNA Methylation and Gene Regulation in Honeybee
The honeybee Apis mellifera and humans share all 3 DNMT1, DNMT2, and DNMT3 enzymes29 -32 making the honeybee an ideal “epigenetic” experimental model for studying the function of DNA methylation in invertebrates, as well as for understanding the dialog between the types of DNMT enzymes in honeybee that may shed light on the increasing role of the nutrition in the epigenetic reprograming in human. In honeybees, feeding of newly hatched larvae destined to become queens with RJ results in changes in the expressional levels of a panel of ubiquitous genes due to epigenetic mechanisms, including DNA methylation.3,7 DNA methylation in honeybees may be differentially altered by nutritional input, leading to profound shifts in developmental fates, with massive implications for reproductive and behavioral development.7,33,34 Several molecular mechanisms have been proposed to regulate methylation fidelity during honeybee development, including the alterations in the expression of both DNMT1 and DNMT3 enzymes which regulate the efficiency of DNA methylation maintenance and de novo DNA methylation activities respectively during honeybee development. 10 In honeybees, DNA methylation is characterized by two major features. First, CG methylation in all developmental stages is targeted specifically to exonic regions. Second, DNA methylation patterns change across the entire gene sequence during honeybee development. 10 The honeybee genome encodes one DNMT3 de novo methyltransferase as well as two isoforms of DNMT1, namely DNMT1a and DNMT1b which are responsible for CG methylation maintenance. 35 All DNMTs are expressed and differentially regulated during honeybee development. High ex-pression levels of the two DNMT1 isoforms are detected in the embryo, while DNMT3 expression levels are the lowest in this stage but its expression consistently increases during development, especially in adult heads. 10 In the larva stage, there is a decrease in CG methylation levels because of a reduction in DNMT1 expression. As a consequence, the highest CG methylation level in the embryo corresponds with the increase in expression levels of both DNMT1s in this stage, while the highest CG methylation level detected in post-larva stages is correlated to increased DNMT3 expression levels. 10 Contrary to mammals, where DNMT3 expression is usually lower than that of DNMT1, 36 DNMT3 expression in honeybees is higher than that of both isoforms DNMT1. 10 In addition to CG methylation, there is an increase in DNMT3-depend non-CG methylation in adult heads, particularly in queen heads which express high levels of DNMT3. 10
DNA methylation patterns are established during development by the de novo methyltransferase DNMT3.35,37 -39 The depletion of DNMT3 using siRNA in newly hatched L1 larvae significantly decreased DNMT3 mRNA expression and this reduction was highly observed after 48 to 50 hours, 7 a critical time known as the “decision-making” period in larval development as it coincides with the larval transition from L2-to-L3. 40 Intriguingly, as a result of DNMT3 inhibition in larvae, 72% of the adult bees were queens possessing fully developed ovaries, like a queen reared on pure royal jelly in the hive. 7 These findings indicate that DNMT3 inhibition-induced alterations in the DNA methylation levels, and downstream events mimic the dietary effects of RJ on caste phenotype, namely altered the development of worker larvae into queens with fully developed ovaries.7,29 Although the overall DNA methylation is low, specific genes such as the dynactin p62 gene in selected tissues could be specifically targeted for methylation. 7 During development, the dynactin p62 gene is differentially methylated, and its methylation status and expression levels change in response to dietary alterations in Drosophila, highlighting the role of this gene in growth and feeding-dependent processes.7,34 Notably, a decrease in the methylation of the dynactin p62 gene was detected in the queen larvae compared to the worker larvae, suggesting that the methylation state of the dynactin p62 gene may play a role in the larva’s developmental trajectory. 7 Indeed, 79% of cytosines in ten CpG sites, located in exons 5, 6, and 7 across the dynactin p62 gene are methylated in the heads of worker-destined larvae. 7 On the other hand, when DNMT3 was silenced in queen-destined larvae, the rate of cytosine methylation decreased by 18% (from 79% to 63%). 7 Interestingly, for two individual CpG sites (from the ten sites), namely, 2 and 4 located in exons 5 and 6 respectively, the decrease in CG methylation between the worker larvae and queen larvae was more than 30%. 7 These findings suggest that methylation of individual CpG sites in honeybees by DNMT3 is used for transcriptional silencing of certain genes, including p62. They also suggest that RJ has an epigenetically active ingredient that serves as a DNMT3 inhibitor, allowing reprograming of larval transcriptomes which establish the DNA methylation state necessary to generate a queen from a worker. This notion is supported by the observation that feeding larvae with RJ for long periods (5 days) significantly decreased both activity and expression of DNMT3 along with a reduction in the overall methylation of the dynactin p62 gene 34 (Figure 1A). Increasing RJ feeding duration resulted in significantly lower rates of methylation in almost half of the CpG sites across the dynactin p62 gene. 34 As a consequence of these molecular changes, all emerged adults were queens (100%) suggesting that a complete switch-over could be induced by dietary manipulation. 34 These findings provide further evidence that methylation of a target gene by DNMT3 is specifically occurred at “individual CpG sites” rather than the overall amount of methylation of that gene and shed light on how a specialized diet affects the bee’s DNA methylation, causing the same genome of two larvae to be differently expressed. In this context, an analysis of microarray data revealed that 240 genes out of 6000 genes were differentially expressed between workers and queens during larval development, namely L3, L4, and L5S2. 40 While workers upregulated a high number of developmental genes compared to queens, a greater proportion of physiometabolic genes were upregulated in queens such as the TOR gene, a member of the insulin-signaling pathway. 40 Intriguingly, the upregulation of the TOR gene detected in the L4 queen is associated with an increase in the titer rates of juvenile hormone (JH). 40 The high levels of JH protected the ovaries from the cell death process, resulting in an extremely high number of ovarioles in adult queens.41 -43 This indicates that the RJ through its inhibitory effects on DNMT3 led to increasing the number of ovarioles in adult queens through increasing TOR pathway-mediated JH synthesis (Figure 1B).
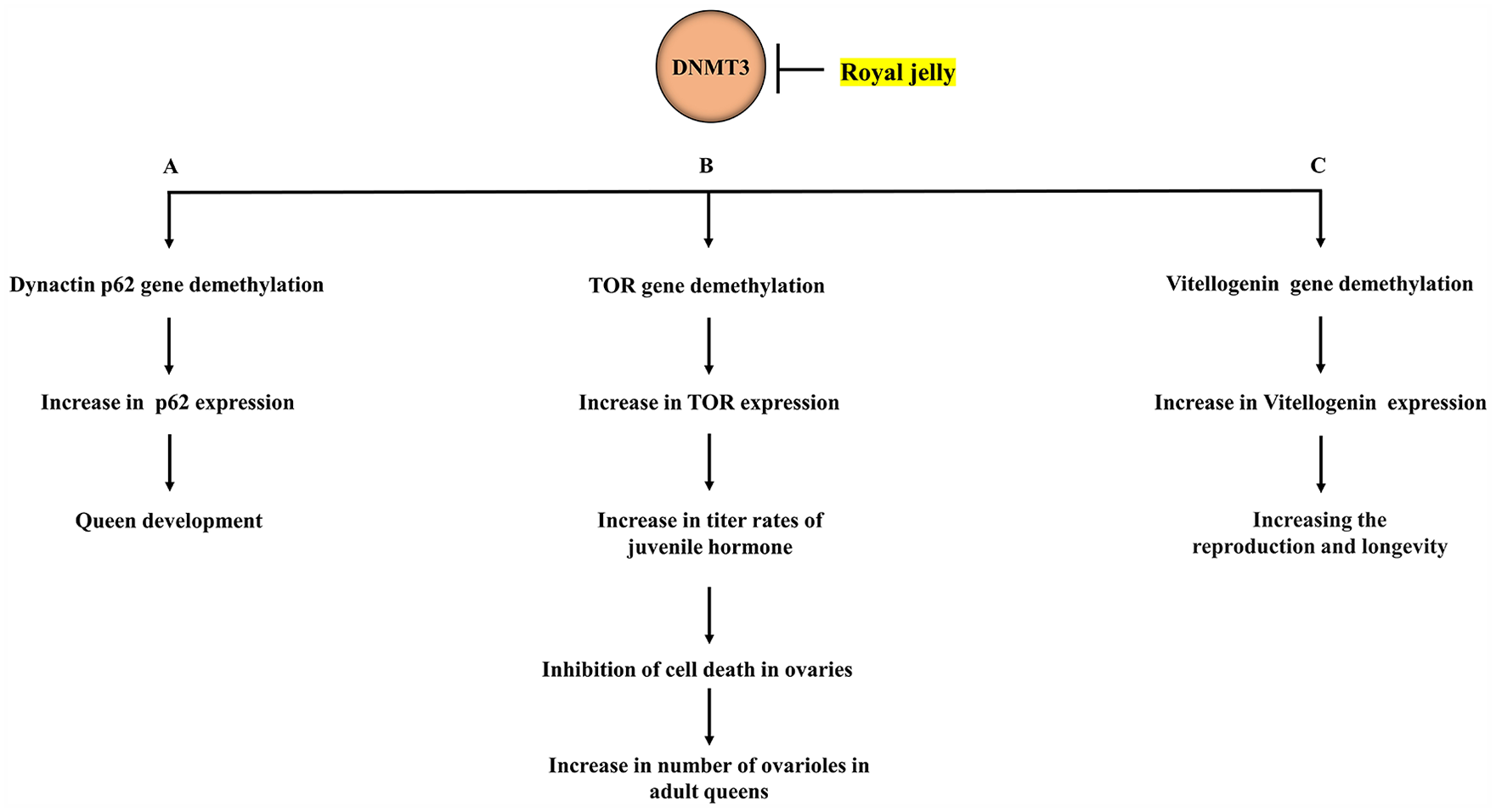
Schematic representation of royal jelly-induced DNA demethylation in honeybees and the related events. Royal jelly, through inhibitory effects on DNMT3, decreases the methylation of several genes such as the dynactin p62, mTOR, and vitellogenin which result in increasing their expression levels: (A) increased dynactin p62 expression levels play a role in queen development, (B) TOR upregulation increases the titer rates of juvenile hormone, which in its turn protects queen ovaries from cell death, highly increasing the number of ovarioles in adult queens, and (C) the sustained production of vitellogenin could increase both reproduction and longevity of the queen.
Contrary to human genome, which is heavily methylated, sequencing of whole genomes from the brains of worker and queen showed only a small and specific proportion of the honeybee genome are methylated (approximately 70 000 out of 60 million cytosines). 29 Methylation mainly occurs in conserved genes that play a crucial role in cell function. Almost all the methylated cytosines are in CpG sites in exons. Interestingly, over 550 genes had different methylation patterns in the brains of workers and queens, which could explain the profound behavioral differences between the 2 female castes. 29 One of those genes is GB15356 which is hypermethylated in workers with low methylation levels in queens, highlighting the possible role of DNA methylation in the regulation of caste-specific gene expression in the brain. 29 Another interesting example of the complex role of DNA methylation in caste-specific differences is the GB18602 gene which encodes a transmembrane protein with the YhhN domain. GB18602 gene has 2 spliced variants L (long protein) and S (short protein). 29 The expression level of transcript S, which encodes the short protein is significantly increased in the queen brain compared to the worker brain, whereas the brains of both queens and workers have the same expression levels of the L variant encoding a long protein. 29 The truncated protein encoded by the S transcript is caused by a clustering of methylation within parts of the GB18602 gene, containing a stop codon where splicing occurs, suggesting that the methylation in Apis could control which of the 2 variants of GB18602 is expressed by taking a decision on which exons are included in mature transcripts. 29 This data indicates that honeybees use gene body methylation (intragenic exonic regions) to modulate gene activity rather than inhibiting genes via promoter methylation.29,44,45 In support of this, molecular sequencing of brain methylomes of queen and worker bees shows a correlation between DNMT3-related DNA methylation and alternative splicing.29,44 In the same context, DNMT3 knockdown in abdominal fat tissue of honeybees inhibited DNA methylation, which affected approximately 2613 genes, including 524 genes that show significant differences in alternative splicing. 46 These studies indicate that DNA methylation through its effects on alternative splicing of mRNA transcripts in honeybees can increase the diversity and complexity of phenotypes generated by a specific gene. Another study also showed a significant increase in differentially methylated genes in the heads of queen and worker larvae compared to adult brains (2399vs 560). 44 In workers, more than 80% of larval differentially methylated genes (1967 out of 2399) were up-methylated 44 which could be explained by the fact that queens have lower levels of methylation as a result of feeding continuously on RJ and the inhibitory effects of this diet on DNMT3. 7 Intriguingly, the differentially methylation was found in genes known to be implicated in key metabolic pathways in honeybees such as the tricarboxylic acid (TCA) cycle, the inositol phosphate/TOR/insulin pathway, and the ubiquitin–proteasome pathway 44 which adds further support to the notion that nutritional signals epigenetically control larval development. One of the interesting genes of the TCA cycle which is differentially methylated in larvae is the ATPCL gene which code for the enzyme, ATP citrate lyase. 44 This enzyme converts citrate into acetyl-CoA, a substrate used by histone acetyltransferases to generate histone acetylation at lysine residues, so activating transcription.47,48 Through such differential methylation-related activity, 44 ATP citrate lyase acts as a key linker between nutrient metabolism and histone acetylation and thus gene expression. 48 Additionally, there was also either methylation or differential methylation of all genes implicated in the response to juvenile hormone (JH). 44
Intriguingly, brain methylomes of the newly emerged queens and workers revealed the presence of rare asymmetrically methylated CpGs (ie, hemimethylated sites). 49 In this context, CpG methylation in cabin-1 and nadrin-2 genes were rarely asymmetric, which suggests that those genes have consistently hemimethylated sites. 49 Moreover, queen-specific CpG sites in the cabin-1 gene showed less methylation compared to the worker-specific patterns suggesting that asymmetrically methylated CpGs in the brain are caste-specific. 49
Recently, methylation analysis of queen and worker larvae genomes at different periods 3, 4, and 5 days showed about 90 000 cytosines were methylated, out of approximately 49 million cytosines. 50 Ninety-nine percent of methylated cytosines were found at CpG sites, mainly occurring in exons. 50 Interestingly, a dynamic difference was observed in the methylation levels of worker and queen larvae between 3 and 5 days of age. 50 At 3 days of age, queen larvae had higher methylation levels than worker larvae. Methylation levels then shifted at 4 days of age to be higher in worker larvae than queen larvae. At 5 days of age, both castes had similar methylation levels. 50 The reverse shift in DNMT3-dependent methylation levels in worker larvae, from low at 3 days old to high levels at 4 days old suggests that this stage, that is, the larvae age of 4 days is a critical moment for the differentiation of the newly emerged larvae. 50 Unlike workers, queens sustain high rates of egg production over their long lifetimes, likely by continually producing the egg-yolk precursor, Vitellogenin protein.51,52 The sustained production of Vitellogenin could result from lifelong feeding of the queen with RJ which regulates both reproduction and longevity.53,54 These physiological changes could be the result of the effect of diet-related genomic DNA methylation status in honeybees on vitellogenin gene expression 55 (Figure 1C). In support of this idea, an analysis of DNMT gene expression in workers’ head thorax and abdomens revealed an increase in DNMT1a and DNMT3 expression and a decrease in DNMT1b and DNMT2 expression. 55 Interestingly, using RG108, a DNMT inhibitor51,52 for genome demethylation caused an increase in worker lifespan through mechanisms involving vitellogenin gene expression. 53
In addition to diet, the social environment is another factor that can influence the reproduction and longevity of adult workers. The queen releases a variety of pheromones such as the queen mandibular pheromone (QMP) which affect worker fertility by suppressing the activation of worker ovaries.56,57 Intriguingly, when workers fed RJ, the effect of QMP on worker fertility was suppressed through a mechanism involving DNMT3. 53
All these works suggest that DNA methylation is used in Apis for storing epigenetic information and this epigenetic mark is controlled and differentially altered by nutritional input as well as social environment to regulate gene expression and/or to affect alternative splicing which results in profound shifts in developmental fates and social behavior. However, other studies showed that methylation patterns are maintained during development in honeybees, standing up against the role of DNA methylation as a key in queen-worker differentiation and behavior in honeybees.10,58,59 One of these studies showed the lack of differences in methylation patterns between queens and workers and that the CG methylation levels in gene bodies fluctuate globally during honeybee development but without alterations in gene expression. 10 Another study indicated that the potential regulation of DNA methylation reprograming in honeybees is very limited. 58 A recent study showed an absence of DNA methylation reprograming during embryogenesis in honeybees, providing evidence that DNA methylation marks are intergenerational transferred, that is, parent to offspring in honeybees. 59 Other studies revealed that queens and workers do not exhibit statistically significant differences in DNA methylation 60 or have moderate levels of DNA methylation with the absence of a clear relationship to sociality level. 61 However, it is important to note these studies10,60,61 compared samples from adult queens and workers, but not from larvae stage during which the critical decision is made on larval development, that is, whether the larva is a worker or a queen. 62 In light of these findings, the intergenerational transfer of DNA methylation patterns in honeybees require further studies. 63
Histone Post-Translational Modifications in Honeybees
DNA methylation does not appear to be the sole epigenetic factor implicated in driving nutrition-controlled caste differentiation. Indeed, histone proteins in honeybees have been shown to undergo extensive PTMs indicating that histone changes, in addition to DNA methylation, are also involved in determining the developmental trajectory of the larvae in response to nutrition.8,9 In this context, acetylation/methylation at lysine residues has been shown to modify honeybee histone H3.1, H3.3, and H4. 8 A comparison between histone modifications in queen ovaries and larvae aged 96 hours revealed 23 similar profiles. A combination of methylation patterns at lysine 27 and lysine 36 of histone 3 (H3K27, H3K36) was more commonly observed in histones extracted from queen ovaries than larvae indicating that histone methylation can establish different gene expression patterns. 8 These findings suggest that histone-modifying machinery is implicated in stimulating the larvae to turn into queens (Figure 2). In agreement with this hypothesis, high expression levels of H3K4me3 and H3K27ac were detected around transcriptional start sites of genes in larval heads of worker and queen castes at 96 h post-hatching, whereas H3K36me3 levels were depleted. 9 Interestingly, in the queen genome, H3K27ac is located mainly within exons close to transcriptional regions, whereas in the worker genome H3K27ac is more ubiquitous, located mostly in introns. 9 Thus, histone acetylation at specific lysine residues can also contribute to caste differentiation. The treatment of NIH/3T3 K-ras cells with either pure 10HDA or a suspension of 5% RJ, containing 2% 10HDA inhibited the activity of histone deacetylase HDAC, mainly class I and class II, 3 allowing a histone acetylase to induce histones acetylation, so activating transcription (Figure 2A). Interestingly, royal jelly at 5% induced the reactivation of the Fas gene through epigenetic mechanisms involving an increase in histone acetylation without affecting DNA methylation, indicating that 10HDA acts as an inhibitor of HDAC but not of DNMT 3 (Figure 2A). Also, the treatment using fractionated RJ showed that only the low molecular weight fraction (<3 KDa) was able to induce the reactivation of the Fas gene, indicating that the RJ-induced epigenetic regulatory activity is triggered by the content of this diet in compounds with low molecular weight. 3 This idea was supported by the fact that the proteinase K treatment did not affect the activity of RJ, further indicating that the epigenetically active component of RJ is not from proteinaceous origin but from a small molecule of non-protein source. 3 The small fatty-acid phenyl butyrate, one of the ingredients of RJ 64 is known as an HDAC inhibitor that also targets class I and IIa HDAC such as HDAC1 and HDAC3,65,66 further supporting the role of RJ in queen development through the regulation of histone acetylation controlling gene expression (Figure 2A). Since the compound 10HDA represents between 2 to 6% of the wet weight of RJ, it is likely that 10HDA is the dominant small molecule in this diet which is responsible for most of the HDAC inhibitory activity in RJ. Class I and IIa HDAC are zinc-dependent enzymes. 67 Most HDAC inhibitors have zinc-binding groups and the binding of this group to zinc has a decisive role in HDAC inhibition.67,68 In this context, 10HDA was shown to inhibit the activity of HDAC3, and this mechanism is modulated by levels of metal cations, potassium, and zinc which, are present in RJ and worker jelly at varying levels. 11 During the first 3 days of larvae development, the content of zinc is significantly higher in RJ than in worker jelly. 69 These observations support the existence of a strong link between high zinc levels in RJ during the developmental stages and its inhibitory effects on HDAC 11 which allow the activation of a histone acetyltransferase to induce histone acetylation (Figure 2A). When HDACs are active, chromatin is more condensed and transcription is inhibited, whereas the induction of histone acetylation by histone acetyltransferases is associated with an increase in chromatin accessibility and transcription activation. Histones are acetylated by histone acetyltransferase enzymes by transferring acetyl group from the substrate acetyl-CoA to specific lysine residues.47,48 The Tat interactive protein 60 kDa (Tip60) is a histone acetyltransferase implicated in the acetylation of histone 2A al lysine 5 (H2AK5).25,70,71 The enrichment of Tip60 was also correlated with H3K27ac levels and chromatin accessibility. 72 Considering that H3K27ac is enriched at exons close to transcriptional regions in the queen genome, 9 such histone mark, that is, H3K27ac could be catalyzed by the activation of Tip60 in response to RJ’s ingredients, mainly 10HDA which has HDAC inhibitory activity (Figure 2A). Acetyl-CoA is the acetyl donor for histone acetyltransferase such as Tip60. Since the gene coding ATP citrate lyase, responsible for producing acetyl-CoA is differentially methylated in larvae, 44 the activation of ATP citrate lyase could result from the inhibitory effects of RJ on DNMT3 which allow ATP citrate lyase to convert citrate into acetyl-CoA (Figure 2B), a substrate for TIP60 to acetylate histone 3 at 27 lysine (Figure 2A). Taken together, all these findings suggest that components of RJ such as 10HDA regulate gene expression in larvae through a process involves the inhibition of HDAC3 and the activation of Tip60, the consequence is the acetylation of histone 3 at lysine 27, increasing chromatin accessibility and gene expression which stimulate the larvae to turn into queens (Figure 2).
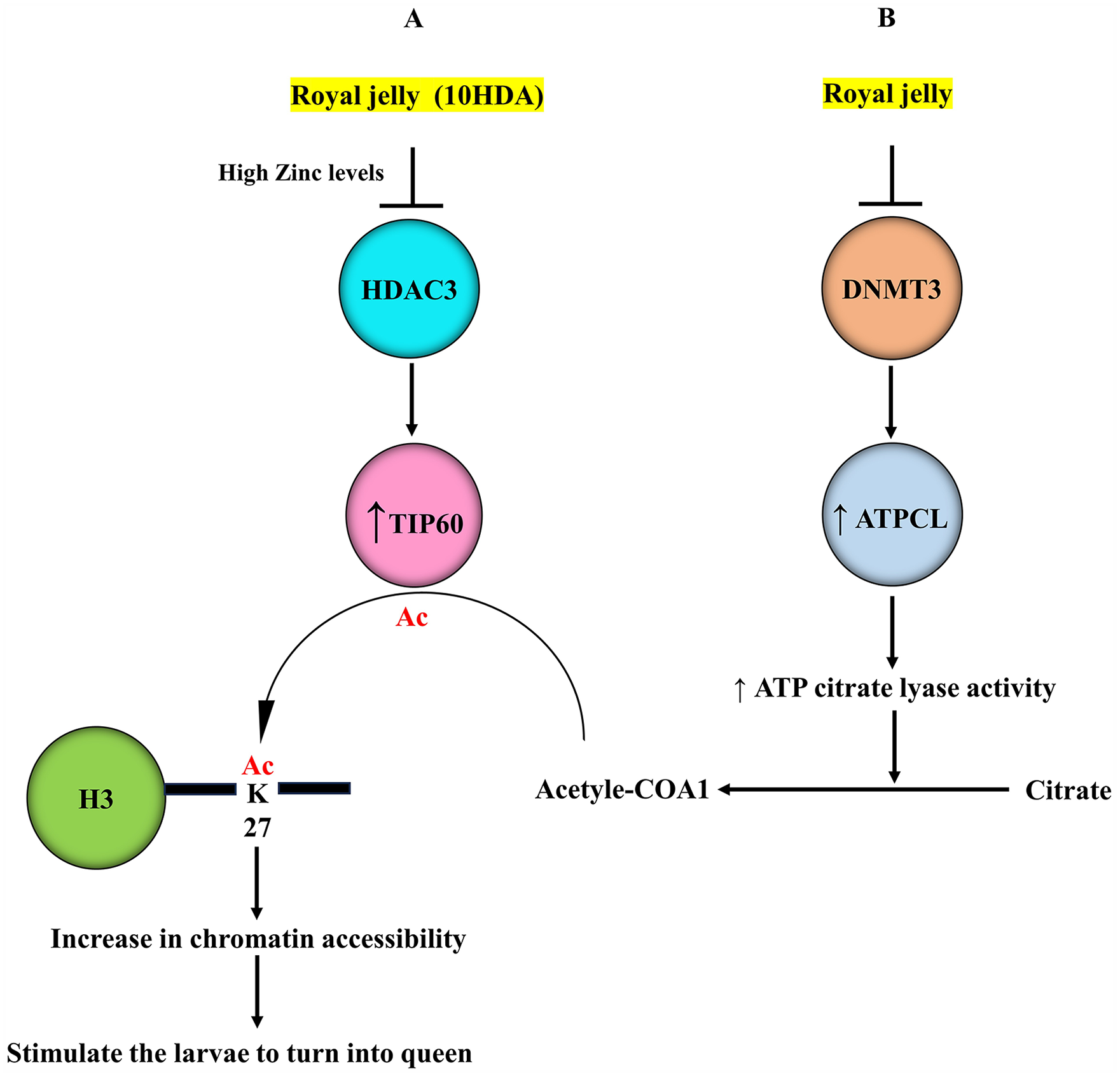
Schematic representation of royal jelly-induced histone acetylation and the related events. (A) The fatty acid called 10-Hydroxy-2-decenoic acid (10HDA), the active ingredient in royal jelly inhibits the histone deacetylase HDAC3 through a mechanism involving the high content of zinc in royal jelly. HDAC3 inhibition may induce the activation of the histone acetyltransferase TIP60. (B) Royal jelly, through its inhibitory effects on DNMT3, decreases the methylation of the ATPCL gene coding the enzyme ATP citrate lyase. This enzyme converts citrate into acetyl-CoA, a substrate for TIP60 to induce the acetylation of histone 3 at lysine 27 (H3K27ac). H3K27ac increases chromatin accessibility and gene expression, stimulating the larvae to turn into queen.
Histone methylation was also shown to play a role in larval development into worker bees. 73 In this context, monomethylation of histone H3 at lysine 4 (H3K4me1) has been reported to promote the development of honeybee larval toward worker bees. 73 Genes of both queen larvae and worker larvae exhibited H3K4me1 enrichment around transcriptional start sites. While workers’ H3K4me1 was predominantly localized in promoter regions, close to transcription initiation sites, queen-specific H3K4me1 was predominantly located in intronic regions. 73 Intriguingly, H3K4me1 enrichment significantly regulated the expression of several regulatory factors of caste differentiation in honeybees such as Juvenile hormone, vitellogenin, juvenile hormone acid O-methyltransferase (JHAMT), hexamerin 70a (Hex70a), and heat shock protein 90 (Hsp90). 73 Workers’ vitellogenin genes exhibited a significantly higher enrichment of H3K4me1 at stages 2 and 4, compared to queens’ genes. Additionally, differential H3K4me1 levels influenced the expression of genes involved in signaling pathways such as FoxO and TOR, 73 which are known to affect caste differentiation in honeybees.53,74 These findings suggest that an asymmetry in H3K4me1 modification patterns between queen and worker larvae is associated with developmental asymmetry. These studies indicate that histone post-translational modifications together with DNA methylation can regulate caste differentiation in honeybees.
The Epigenetic Integrator UHRF1 in Honeybees
DNA methylation and PTMs work together to influence chromatin accessibility, consequently the transcription. With its multiple functional domains, the epigenetic reader UHRF1 can serve as a sensor for both DNA methylation patterns and histone marks, thus enabling a coordinated dialog between several DNA/histone modifiers.27,75,76 The conservation of UHRF1 in vertebrates ensures the maintenance of 5 mC in their genomes during DNA replication.18,20,27 Interestingly, honeybees have UHRF1 protein. 28 The primary amino-acid sequence of honeybee (Apis Mellifera) UHRF1 has been reported (Genbank accession number NP_001229385.1. 77 Sequence alignment with human UHRF1 (Genbank accession number: NP_001276980.1) shows 45% identity and 62% homology (Personal observations obtained with BLAST alignment tool at NCBI). Analysis of the SET and RING-associated (SRA) domain sequences of human UHRF1 and honeybee UHRF1 revealed 76% identity and 84% homology, suggesting that the SRA domain of UHRF1 in honeybees ensures the same role as the human counterpart, that is, recognizing hemi-methylated DNA and recruitment of DNMT1 during DNA replication to faithfully reproduce the methylation patterns on the DNA daughter strands. 78 Since the SRA domain of UHRF1 is involved in the maintenance of DNA methylation and has a high degree of similarity to the SRA domain of honeybees, it is proposed that UHRF1 plays an important role in the development of honeybees via its SRA domain, thus making this domain a potential target for biologically active ingredients of royal jelly.
A Look at Future Directions in Epigenetics Mechanisms of Honeybees: Role of UHRF1
Epigenetic information, transmitted from one generation to the next is carried mainly by DNA methylation and PTMs. During germ cell development, these carriers of inherited epigenetic information exhibit dynamic patterns of regulation. Unlike somatic tissues which have significantly lower methylation fidelity, bee sperm contains high levels of methylation in complete exons suggesting that DNA methylation is transgenerational transmitted and could be a common feature of honeybee development. 10 The alteration in DNMT1 and DNMT3A and DNMT3B expression levels during honeybee development is one of the main molecular mechanisms involved in the regulation of methylation fidelity. 10 The de novo methyltransferases DNMT3A and DNMT3B methylate the cytosines of non-methylated CpG sites on both DNA strands, while DNMT1 specifically methylate hemimethylated CpG sites generated during DNA replication which, ensuring the maintenance of genomic methylation. This mechanism is considered the molecular basis for DNA methylation patterns inheritance. Thus, it is of immense importance to understand how DNA methylation patterns are established in the early embryo by de novo methyltransferases DNMT3A and DNMT3B and successfully maintained by DNMT1 across consecutive cell division. Several mechanisms have been suggested to explain how DNMT1 can recognize hemimethylated DNA generated during DNA replication. DNMT1 interacts with PCNA (the DNA polymerase processivity factor), 79 which may guarantee DNMT1 localization at the replication foci which, suggesting that DNMT1-mediated methylation maintenance includes other epigenetic players. One of the key questions to be addressed in honeybee methylome is how DNMT is guided and recruited to the right place, that is, individual CpG sites in internal exons of specific genes at the right time. Such a precise process suggests the presence of a key epigenetic player in chromatin structure regulation. Such a candidate is expected to have a preferential affinity for hemimethylated CpGs sites over symmetrically methylated DNA and to be located at the replication fork along with DNMT1, to recruit and guide this enzyme to its target (non-methylated CpGs) on the opposite DNA strand. In addition to its association with DNMT1, the potential candidate should also be able to bind the de novo methyltransferases DNMT3A and DNMT3B, for recruiting these enzymes to complete the methylation process after DNA has left the replication fork. The epigenetic reader UHRF1 is a potent candidate for playing such multiple roles in the epigenetics of honeybees since it has various functional domains which allow UHRF1 to be effectively implicated in the regulation of chromatin structure and replication of the epigenetic code to ensure DNA methylation fidelity during cell division. Indeed, many works showed that methylation fidelity could be regulated by UHRF1.27,80 -83 UHRF1 interacts with DNMT1,18 -21 playing an essential role in DNA methylation maintenance in mammalian somatic cells. During DNA replication, SRA domain of UHRF1 preferentially binds the hemimethylated CG sites, and via the same SRA domain, UHRF1 interacts with DNMT1, recruiting this enzyme to methylate the non-methylated CpGs sites on the opposite strand. 83 Moreover, UHRF1 interacts with DNMT3A and DNMT3B 17 and plays a role in de novo DNA methylation during the global epigenetic reprograming of oocytes and early embryos. 84 UHRF1 is associated with both DNMT1 and PCNA at replication foci in S phase, when DNA replication occurs, indicating a crucial role of UHRF1 in DNA methylation inheritance. 18 DNMT3A, DNMT3B, and DNMT1 are differently expressed in oocytes and early embryos, suggesting that the 3 DNMT enzymes may interact and work together to establish DNA methylation in oogenesis and embryonic development. 36 The DNMTs enzymes act in concert with HDACs. In the light of the interactions between UHRF1 and DNMT3A, DNMT3B, 17 DNMT1,18,24,83 as well as with several histone modifiers such as HDAC1, 22 histone acetyltransferase TIP6024,25 and histone 3 methyltransferase G9a, 26 UHRF1 is proposed to play a role as a sensor for both DNA methylation patterns and histone marks. Due to the high similarity of the SRA domain of honeybee UHRF1 to that of human UHRF1, this latter could be a main regulator of the global epigenetic reprograming of early embryos in honeybees. Thus, the differentiation of newly hatched females into queen and worker can be explained by epigenetic mechanisms, including DNA methylation and histone acetylation and methylation, all controlled by UHRF1. Since RJ contains epigenetically active compounds that act either as DNMT3 inhibitor 7 or HDAC3 inhibitor 11 and that UHRF1 is associated with both DNMT1 and HDAC3, it is hypothesized that UHRF1 is also inhibited by one or more of RJ’s ingredients (Table 1). The inhibition of UHRF1 by RJ may prevent its two partners, DNMT3 (Figure 3A) and HDAC3 (Figure 3B) from exerting their catalytical roles in DNA methylation and histone deacetylation respectively, which could have significant impacts on larval developmental trajectory (Figure 3). Multiple lines of evidence support this hypothesis. First, UHRF1 was found to be overexpressed in primary nonsmall cell lung carcinomas tissues and its high expression levels were correlated with DNMT1-related hypermethylation of the tumor suppressor gene RASSF1A. 85 UHRF1 inhibition in A549 lung adenocarcinoma cells resulted in lower methylation levels of RASSF1, indicating that UHRF1 plays an important role upstream in the regulation of DNMT1/RASSF1A pathway. 85 Interestingly, the treatment of A549 lung adenocarcinoma cells by RJ decreased DNMT1 expression and increased RASSF1A expression, 86 which indicates that the decrease in DNMT1-mediated methylation of RASSF1A gene promoter in response to RJ treatment could result of direct inhibitory effects of this diet on UHRF1 expression. Second, through its SRA and ubiquitin-like domains, UHRF1 interacts with DNMT3A, resulting in the epigenetic silencing of the cytomegalovirus promoter in embryonic stem cells. 17 Thus, it is suggested that the inhibitory effects of RJ on DNMT3 and the DNA methylation-related profound shifts in developmental fates in honeybees 7 are triggered by inhibiting UHRF1. Third, the polyphenol epigallocatechin 3-gallate decreased the association between UHRF1 and DNMT3A and HDAC3 in colon cancer cells, triggering the degradation of the two enzymes, 23 indicating that the HDAC3 inhibition by 10HDA, the active ingredient in RJ 11 could be also triggered by targeting UHRF. Fourth, UHRF1 overexpression in cardiomyocytes led to a dramatic decrease in the expression of the dynactin p62 gene, 87 known as a main target of DNMT3 activity in honeybees. 34 Because DNMT3 inhibition significantly decreased the methylation levels of dynactin p62 gene in larvae fed with RJ for long periods, 34 it is possible that the decreased levels of dynactin p62 gene methylation detected in the queen larvae compared to the worker larvae 7 could be explained by RJ-mediated inhibition of UHRF1 as a first event. These findings support the idea that one or more of the RJ’s epigenetically active compounds target UHRF1, and that UHRF1 inhibition would be sufficient to target both key epigenetic mechanisms in honeybee development, namely DNMT3-dependent DNA methylation and HDAC3-mediated histone acetylation (Figure 3).
Epigenetically active compounds in royal jelly.


Proposed mechanisms of royal jelly’s polyphenols-induced UHRF1 inhibition and the related events. Royal jelly’s polyphenols might directly inhibit UHRF1 activity and/or expression which results in the inhibition of its partners DNMT3 and HDAC3: (A) DNMT3 inhibition leads to DNA demethylation-dependent increase in the expression of ubiquitous genes including dynactin p62, TOR and vitellogenin and (B) HDAC3 inhibition induces the activation of the histone acetyltransferase TIP60 to generate histone acetylation. As a result of these molecular changes, larvae are stimulated to turn into queen.
Royal Jelly’s Polyphenols as UHRF1 Inhibitors
RJ has biologically active ingredients with potent inhibitory effects on the de novo methyltransferase DNMT3A and the histone deacetylase 3 HDAC3. Thus, the sustained consumption of RJ could be the decisive key in regulating the DNA/histone modifying machineries that are necessary to produce either queen or worker. Thus, it is of interest to identify the epigenetically active compounds in RJ, which regulate chromatin modifiers including UHRF1, thereby determining developmental fate. RJ is a source of flavonoids including luteolin and quercetin and phenolic acids such as ferulic acid and caffeic acid1,12 which could act as inhibitors of UHRF1. In support of this idea, extracts or juice rich in luteolin,88,89 derivatives of quercetin, 90 and ferulic acid, and caffeic acid 91 have been shown to exhibit inhibitory effects on UHRF1 expression in cancer. Luteolin decreased UHRF1 and DNMT1 expression in colorectal cancer cells as well as in human cervical cancer cells.88,89 Aronia melanocarpa juice, rich in derivatives of quercetin reduced UHRF1 expression and induced apoptosis in human leukemia cells. 90 Similarly, the root extract of Leonurus sibiricus, rich in ferulic acid and caffeic acid decreased UHRF1 expression in human glioma cells. 91 Based on these findings, RJ is suggested to contain polyphenolic compounds directly target UHRF1 protein (Table 1). Thus, inhibition of UHRF1 activity and/or expression by one or more of RJ’s polyphenols would be a sufficient event to inhibit several epigenetic players acting either as writers such as DNMT3 (Figure 3A) or erasers including HDAC3 (Figure 3B), and the consequence is significant impacts on queen development (Figure 3).
Conclusion
This review has discussed how epigenetic mechanisms have effects on caste determination in honeybees, and how early diets affect their epigenomes, with significant impacts on queen development. Producing queens with fully developed ovaries in DNMT3-depleted larvae represents strong evidence that RJ has epigenetically active ingredients able to create and maintain the epigenetic state necessary in the developing larvae to generate a queen. Intriguingly, the 10HDA, the active ingredient in RJ can inhibit the histone deacetylases including HDAC3, stimulating the larvae to turn into queens. However, it is still unclear how DNMT can recognize and methylate individual CpG sites in internal exons of specific genes in the honeybee genome and how HDAC can be recruited and enriched at these locations to form a condensed chromatin. To help answering these questions, the review have looked at the potential role of the epigenetic integrator UHRF1 as sensor for both DNA methylation patterns and histone marks in honeybees, recruiting a set of appropriate enzymes, namely DNMT3, HDAC3 and Tip60 to the right place at the right time. The review has also discussed how royal jelly’s polyphenols can regulate the DNA/histone modifying machineries through direct targeting of UHRF1 (Figure 3).
Footnotes
Funding:
The author received no financial support for the research, authorship, and/or publication of this article.
Declaration Of Conflicting Interests:
The author declared no potential conflicts of interest with respect to the research, authorship, and/or publication of this article.
Author Contributions
MA designed the review article, performed the research and designer the figures, and write the manuscript.