
Abstract
Epigenetic changes may be biomarkers of health. Epigenetic age acceleration (EAA), the discrepancy between epigenetic age measured via epigenetic clocks and chronological age, is associated with morbidity and mortality. However, the intersection of epigenetic clocks with microRNAs (miRNAs) and corresponding miRNA-based health implications have not been evaluated. We analyzed DNA methylation and miRNA profiles from blood sampled among 332 individuals enrolled across 2 U.S.-based firefighter occupational studies (2015-2018 and 2018-2020). We considered 7 measures of EAA in leukocytes (PhenoAge, GrimAge, Horvath, skin-blood, and Hannum epigenetic clocks, and extrinsic and intrinsic epigenetic age acceleration). We identified miRNAs associated with EAA using individual linear regression models, adjusted for sex, race/ethnicity, chronological age, and cell type estimates, and investigated downstream effects of associated miRNAs with miRNA enrichment analyses and genomic annotations. On average, participants were 38 years old, 88% male, and 75% non-Hispanic white. We identified 183 of 798 miRNAs associated with EAA (FDR q < 0.05); 126 with PhenoAge, 59 with GrimAge, 1 with Horvath, and 1 with the skin-blood clock. Among miRNAs associated with Horvath and GrimAge, there were 61 significantly enriched disease annotations including age-related metabolic and cardiovascular conditions and several cancers. Enriched pathways included those related to proteins and protein modification. We identified miRNAs associated with EAA of multiple epigenetic clocks. PhenoAge had more associations with individual miRNAs, but GrimAge and Horvath had greater implications for miRNA-associated pathways. Understanding the relationship between these epigenetic markers could contribute to our understanding of the molecular underpinnings of aging and aging-related diseases.
Keywords
Introduction
Epigenetic markers are mitotically heritable yet potentially reversible modifications to DNA that regulate gene expression without affecting the genetic code itself. While epigenetic markers are critical to biological functioning and controlled and regulated by complex biologic processes, they have also been shown to be responsive to environmental exposures.1,2 Epigenetic marks are also considered important factors in the development of age-related diseases, including cancers. 3 As a result, epigenetic markers have been proposed as biomarkers of effect from toxic exposures as well as biomarkers of health and early markers of certain diseases. For example, among firefighters, we have previously observed longitudinal epigenetic alterations in both DNA methylation and microRNA (miRNA) in mid-life, that are consistent with specific health outcomes overrepresented among firefighters, including cancer.4 -6
DNA methylation is an enzyme catalyzed process involving the addition of a methyl group to the 5-carbon of cytosine residues at CpG dinucleotides. Sometimes, methylation of CpG sites can result in the regulation of nearby genes. 7 Knowledge about methylation patterns has been utilized recently to estimate cellular or biological aging via epigenetic clocks. Epigenetic clocks are estimated based on DNA methylation at multiple CpG sites that change with chronological age. Epigenetic age acceleration (EAA) is a measure of discrepancy between epigenetic age and chronological age, where a positive value suggests more advanced biological aging compared to chronological age. Studies have reported associations between EAA and risk of age-related health endpoints (eg, cardiovascular health loss, cardiovascular disease, Alzheimer’s disease, cancers), age-related clinical phenotypes (eg, physical functioning, walking speed, grip strength, Mini Mental State Examination, Montreal Cognitive Assessment), and all-cause mortality.8 -11 Additionally, there is growing evidence linking environmental and occupational exposures to EAA.1,12 -14 Incumbent firefighters, for instance, have greater epigenetic age acceleration than new recruit firefighters, 14 and some species of per- and poly-fluoro alkylated substances (PFAS), a chemical that firefighters are commonly exposed to, is also associated with greater epigenetic age acceleration among firefighters. 1
Another type of epigenetic marks include microRNA (miRNA) expression profiles. MiRNAs are small, endogenous, non-coding RNAs that regulate gene expression at the post-transcriptional level by targeting messenger RNA for degradation or by inducing translational repression. 15 Recognition of miRNAs as important regulators of aging and longevity has grown, as research has revealed that the miRNA regulatory network is extensive and miRNA-mediated regulation plays an important role in developmental timing, apoptosis, senescence, and proliferation.16 -18 Additionally, dysregulated miRNA expression has been shown to play a role in the age-related pathologies of cardiovascular diseases, neurodegenerative diseases, and cancer.16,19 -22 Studies of circulating levels of miRNAs in humans have identified associations between dysregulated miRNAs and increased risk for cardiovascular aging, heart failure, hypertension, atherosclerosis, atrial fibrillation, as well as diabetes. 19 MiRNAs have also been suggested to be a key regulator of homeostasis in neurons, and some studies have demonstrated that their dysregulation may contribute to neurodegenerative diseases including Alzheimer’s disease, Parkinsons’s disease, and Huntington’s disease.20,21 Recent research has also suggested that miRNAs can regulate tumor development by acting as tumor suppressors or oncogenes.23,24
A growing area of research indicates that epigenetic markers can work together in various combinations and can cross-regulate each other.25,26 One such interaction has been identified between miRNAs and DNA methylation, where miRNA expression is regulated by DNA methylation at miRNA promoters, and aberrant methylation patterns at these promoters have been associated with aging-related diseases, including cancer and Huntington’s disease, among other diseases.26 -29 However, it is not yet understood if EAA, measured via epigenetic clocks, is associated with miRNA expression.
Firefighting has been shown to be associated with dysregulated miRNA, epigenetic age, epigenetic profiles, and aging-related diseases.1,4 -6,14,30 -32 miRNAs have also been associated with aging-related diseases, although currently no summary measure equivalent to the DNAm clocks exist for miRNA. By identifying those miRNAs that are associated with epigenetic DNAm clocks, which may function as a subacute summary indicator of overall health, we can further identify molecular drivers of disease and biological targets for prevention and treatment. With this in mind, in our current study, we integrated data on DNA methylation and miRNA expression profiles of blood samples collected from 2 U.S.-based occupational cohorts to determine if EAA was associated with miRNA expression, to explore if these associations varied across different epigenetic clocks, and to identify miRNA-based health implications. Research that helps address these topics could provide evidence that biological aging influences miRNA expression and broaden our understanding of cancer and other aging-related disease processes.
Materials and Methods
Study population and data collection
Our analysis included participants from 2 studies investigating disease and cancer risk factors among United States (U.S.) firefighters as a convenience population to address our study questions. The first study was conducted by the University of Arizona in partnership with the Tucson Fire Department. The second study, the Fire Fighter Cancer Cohort Study (FFCCS) (https://www.ffccs.org/), is an ongoing, prospective, multi-site study including fire departments across the U.S. and collaboration with multiple academic and research institutions. Study protocols and materials were reviewed and approved by institutional review boards (IRBs) (University of Arizona IRB (approval No. 1509137073) and University of Miami IRB (approval No. 20170997)) and all participants provided their written informed consent. At the time of enrollment, participants provided blood samples as well as demographic and occupational information (fire department, total years of service as a firefighter) via a survey.
Eligibility criteria in the current analysis included enrollment in the University of Arizona study between 2015 and 2018, or enrollment in the FFCCS between 2018 and 2020, and having both miRNA and DNA methylation data that met quality control standards. For individuals who had enrolled in both studies (n = 47), only the earlier study enrollment was considered. The final study sample in our current analysis included 332 firefighters: 143 (43%) from the University of Arizona study and 189 (57%) from the FFCCS.
MiRNA expression processing and measurement
Sample collection and processing have been reported previously.5,6 Briefly, whole blood samples were collected using Tempus™ Blood RNA tubes (Applied Biosystems, Foster City, CA), shaken and aliquoted into 5 mL cryogenic tubes (VWR International, Radnor, PA, Cat. # 89094-820) and stored frozen until further processing (at −20°C temporarily and at −80°C for long-term storage). RNA was isolated following the manufacturer’s recommendations using MagMAXTM for Stabilized Blood Tubes RNA Isolation Kit (Life Technologies, Carlsbad, CA, Catalog #4451893) and total RNA was measured using the NanoDrop 1000 Spectrophotometer (Thermo Fisher Scientific, Wilmington, DE).
MiRNA expression was measured using the nCounter® Human v3 miRNA expression panel (NanoString Technology Inc., Seattle, WA), a profile of 798 curated and clinically relevant human miRNAs from miRBase v21 as well as 5 housekeeping genes and 20 assay controls (6 positive, 8 negative and 6 ligation controls) in 5 batches analyzed in 2016, 2017, 2018, 2019 and two batches analyzed in 2021. This panel accounts for almost all (>95%) observed sequencing reads in miRbase 21 (Dennis 2015 white paper). Raw counts of each gene were normalized against background genes and the overall assay performance was assessed through the evaluation of the positive controls.
To further normalize and remove unwanted variation, or batch effects, from our miRNA expression data, we utilized the Removing Unwanted Variation-III method (RUV-III) via the “ruv” R package, 33 as previously described. 5 This method makes vital use of pseudo-replicates and control genes. 34 In our miRNA expression measurement, we purposely measured 3 to 4 samples in all batches, serving as pseudo-replicates. RUV-III (i) takes the residuals from the replicate expression measurements and estimates one aspect of the unwanted variation; (ii) takes the results of (i) together with the expression of the negative controls of the NanoString platform and estimates another aspect of the unwanted variation; and (iii) estimates total unwanted variation (results of (i) and (ii) combined) and subtracts this value from the data. Relative log expression (RLE) plots by batch were used to visualize and confirm batch correction.
DNA methylation data for epigenetic clocks
Collection and processing of DNA methylation data have been previously reported.1,35 Blood samples were collected using 6 mL dipotassium ethylene diamine tetraacetic acid tubes for isolation of DNA (BD, Franklin Lakes, NJ). DNA was isolated from blood leukocytes, quantified using a QuantiFluor dsDNA System (Promega, Madison, WI), and bisulfite converted using Zymo kits prior to Infinium MethylationEPIC array analysis. 36 Samples were randomized across array chips, hybridized, and scanned in batches at the University of Utah DNA Sequencing and Genomics Core Facility or the University of Michigan Advanced Genomics Core. The “minfi” R package 37 was used to read in raw image files and “ENmix” 38 was used to perform quality control, background correction via noob, and dye bias correction with regression on logarithm of internal control (RELIC) probes followed by quantile normalization.39,40
We then used the EPIC data to estimate the proportion of blood cell types (CD4+ T cells, natural killer cells (NK cells), B cells, monocytes, granulocytes; relative abundance of plasma blasts, CD8+CD28-CD45RA- T cells, and naive CD8+ T cells) using established algorithms.10,41 We also computed seven widely used and validated epigenetic age biomarkers, or epigenetic clocks, with the New Methylation Age Calculator (https://dnamage.genetics.ucla.edu/new). 10 These included the multi-tissue Horvath clock, 10 the Hannum et al clock designed for blood samples, 42 and the skin-blood clock designed for skin, blood, or saliva samples. 43 Second generation clocks—PhenoAge and GrimAge—were also computed.8,44 In statistical analyses, we used measures of EAA, residuals after regressing each clock on chronological age. We also estimated intrinsic epigenetic age acceleration (IEAA) and extrinsic epigenetic age acceleration (EEAA), markers of intrinsic cellular aging and immune system aging, respectively. 45
Statistical analysis
Descriptive statistics were generated for demographic variables, including age (years), BMI (kg/m2), sex, race/ethnicity (non-Hispanic White, Hispanic White, Black, and Other), smoking status (current, former, never), state, incumbent or recruit status, and years as a firefighter. In regression models, race/ethnicity was categorized as non-Hispanic White, Hispanic White, and other (due to small numbers of participants). The exposures of interest included the 7 measures of EAA (5 based on the Horvath, skin-blood, Hannum, PhenoAge, GrimAge clocks, as well as IEAA and EEAA) and the outcome of interest was differential expression of the 798 miRNAs. In our main analysis, separate linear regression models of each measure of EAA were fit for each miRNA and adjusted for sex, chronological age, and race/ethnicity. We initially considered adjusting for smoking status, however, due to the high degree of missing values (61%) and sparseness of the data across the variable categories, chose not to include this variable in our final models. Because we used data from firefighter study participants as a convenience population to address questions concerning EAA and miRNA expression, we considered incumbent or recruit status as a potential confounder as we hypothesized that incumbents would have increased cumulative occupational exposures and we have previously shown that miRNA expression can differ between incumbents and recruits 6 and that incumbents have higher EAA compared to new recruits. 14 However, we chose to not include this variable in our final models since model fit (expression levels of miRNAs) did not improve after adjustment, 46 within the top 10 miRNAs (ordered by p-value) for the measures of EAA. Five of the 7 EAA measures (Horvath, skin-blood, Hannum, PhenoAge, and GrimAge) were also adjusted for cell type estimates (granulocytes, plasmablasts, CD8+ naive T cells, and CD8+CD28-CD45RA- T cells). A 5% false discovery rate (FDR, q-value < 0.05) was used to identify miRNAs associated with EAA.
For measures of EAA that were found to be associated with differential miRNA expression, we also performed a separate pathway enrichment analysis as a secondary analysis to investigate potential downstream effects of miRNAs. The pathway analysis was performed using the “rbioapi” package 47 and the miRNA Enrichment Analysis and Annotation Tool (miEAA). 48 Specifically, we performed a gene set enrichment analysis adapted for miRNAs (miRNA enrichment analysis) where the test set consisted of the full array list of miRNAs (n = 798) ranked by effect size. The enrichment categories were limited to miRNA-disease annotations (MNDR v2.0) and miRNA-pathway annotations (Gene Ontology (GO) cellular components, GO molecular functions, GO biological processes; miRPathDB 2.0),49,50 and a q-value threshold of 0.05 on the pathways was used for statistical significance. The MNDR v2.0 database contains 28 144 predicted miRNA-associated entries for Homo sapiens and miRPathDP 2.0 database contains 28 352 miRNA targets and 16 833 miRNA pathways for Homo sapiens.49,50 Separate miRNA enrichment analyses were performed for each measure of EAA considered. All statistical analyses were performed using R (version 4.1.0). 51
Results
A total of 332 participants were included in our analysis. Median values for chronological age and BMI were 38 years old (interquartile range (IQR) 31,48) and 26.9 kg/m2 (IQR 24.4, 29.0) (Table 1). The majority of study participants were male (88%) and non-Hispanic white (75%), which is reflective of current estimates of the fire service in the United States. 52 Approximately 10% were Hispanic white, 5% were Black, and 13% reported other race. Of participants who reported information about their smoking status (n = 131, 38%), the majority reported being never smokers (n = 127). Participants were distributed mostly across the western United States, with over 60% of participants recruited from fire departments in Arizona and California, and most were incumbents (85%) within their departments rather than recruits (15%). The median reported work duration was 12 years (IQR 3, 19 years).
Characteristics of study participants, n = 332.
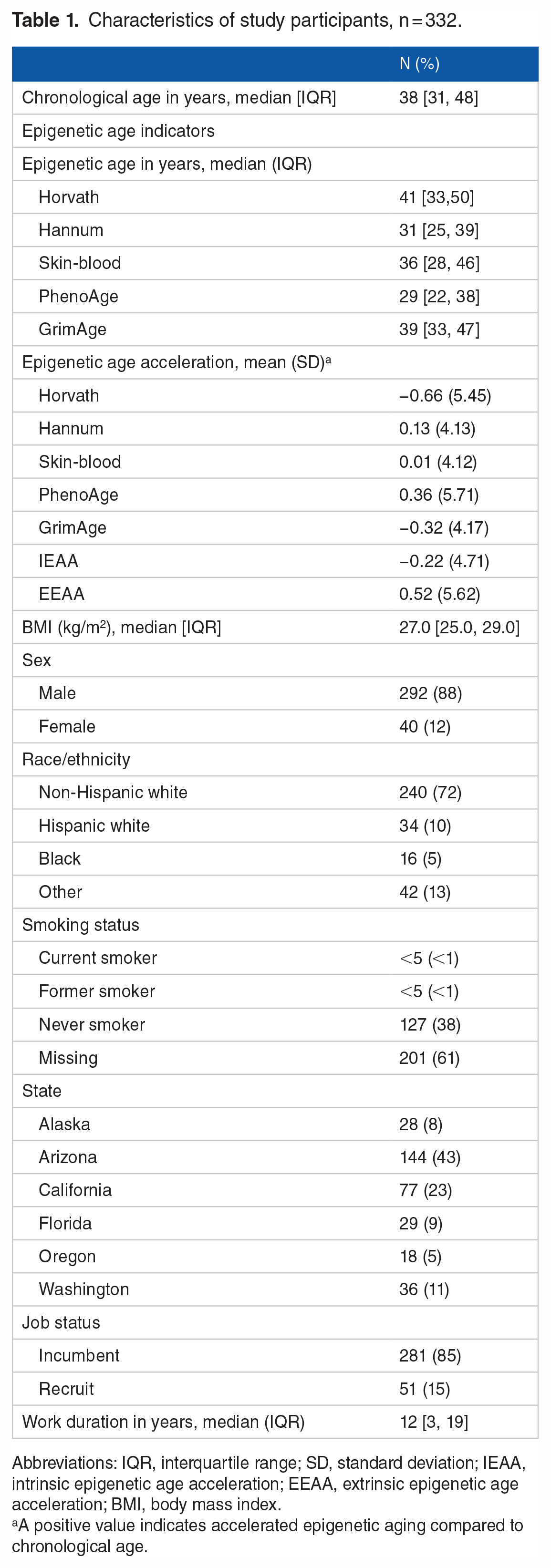
Abbreviations: IQR, interquartile range; SD, standard deviation; IEAA, intrinsic epigenetic age acceleration; EEAA, extrinsic epigenetic age acceleration; BMI, body mass index.
A positive value indicates accelerated epigenetic aging compared to chronological age.
EAA varied based on the epigenetic clock used. Averages of 3 of the 7 measures of EAA (based on the Hannum and PhenoAge clocks, and EEAA) were positive, indicating that on average, study participants had accelerated epigenetic aging compared to their chronological age, while 3 (based on the Horvath and GrimAge clocks, and IEAA) were negative (Table 1). The average value of EAA based on the skin-blood clock was positive but null (0.01, standard deviation 4.12), suggesting no difference between epigenetic and chronological age. The correlation between the 7 measures of EAA ranged from 0 to .94 (Supplemental Table 1). Measures of EAA that had the highest correlation included Hannum and EEAA (r = .94), Horvath and IEAA (r = .86). All other estimated correlations were less than 0.50.
In our analysis of EAA and miRNA expression, we identified a total of 183 miRNAs that were significantly associated with at least one of 4 measures of EAA (PhenoAge, GrimAge, Horvath, and skin-blood) (Supplemental Table 2). Most miRNAs were associated with PhenoAge (n = 126), followed by GrimAge (n = 59), Horvath (n = 1), and skin-blood (n = 1). There was little overlap between miRNAs associated with the 4 measures of EAA, and no miRNA that was significantly associated with more than 2 of the 4 measures of EAA (Table 2). PhenoAge and GrimAge had the greatest overlap, with 4 miRNAs in common (miR-1180-3p, miR-193a-3p, miR-3144-3p, and miR-4421) and 2 (miR-3144-3p and miR-4421) that were expressed in the same direction (Table 2). PhenoAge also had one miRNA in common with skin-blood (miR-510-5p) while miR-873-5p was associated with both GrimAge and Horvath. The remaining measures of EAA (Hannum, IEAA, and EEAA) were not significantly associated with miRNA expression.
Comparison of miRNAs associated with epigenetic age acceleration. a

Abbreviations: miRNA, microRNA; q-value, Benjamini Hochberg FDR adjusted P-value.
Bolded text denotes miRNAS with q-value < 0.05.
6 of 183 total miRNAs from models adjusted for sex, chronological age, race/ethnicity, and cell type composition were associated with more than one measure of epigenetic age acceleration, and q-value < 0.05. Epigenetic age acceleration was calculated with 4 different epigenetic clocks (PhenoAge, GrimAge, Horvath clock, and skin blood clock). Beta values represent log(count) of miRNA expression.
A miRNA enrichment analysis was performed for each measure of EAA identified above (PhenoAge, GrimAge, Horvath, and skin-blood). We sorted the 798 miRNAs by their effect size, based on regression models including adjustment for sex, chronological age, race/ethnicity and cell type composition. Out of 9218 total miRNA annotations for Homo sapiens from the MNDV v2.0 (n = 1197) and miRPathDB 2.0 databases (8021) 48 considered, we found that there were 425 annotations significantly enriched for miRNAs associated with Horvath, consisting of 205 diseases and 220 pathways (156 GO biological processes, 21 GO cellular components, 43 GO molecular functions) (Supplemental Table 3). Among these disease annotations, the majority were cancer related. Some of the most prominent cancers included cancers of the brain, lung, blood, and to a lesser extent, cancers of the gastrointestinal system, bladder, liver, and thyroid. Several cancers of the female reproductive system were also significantly enriched among miRNAs, including cervical, endometrial, and uterine cancer. Common non-cancers included other age-related diseases such as metabolic, cardiovascular, and related disorders (eg, type 2 diabetes, obesity, non-alcoholic fatty liver disease, stroke, coronary artery disease, acute coronary syndrome), as well as muscular disorders, and psoriasis. Identified pathways were largely related to the regulation of metabolic processes, protein modification, cellular responses to stress and DNA damage, and gene expression.
There were 81 annotations significantly enriched for miRNAs associated with GrimAge, consisting of 15 diseases and 66 pathways (39 GO biological processes and 27 GO molecular functions) (Supplemental Table 4). More than 75% of these annotations (n = 61) were also significantly enriched for miRNAs associated with Horvath (Supplemental Table 5) and the top shared diseases and pathways are shown in Tables 3 and 4. These top shared diseases included cancers of the brain, blood, lung, as well as uterus. Non-cancer disease pathways also included stroke, non-alcoholic fatty liver disease, and muscular disorders. Similar to our enrichment analysis of Horvath-associated miRNAs, again, pathways enriched for miRNAs associated with GrimAge were mostly related to metabolic processes, protein binding and modification, cellular response to DNA damage stimulus, and gene expression. We also identified one GO molecular function pathway (ATPase activity) significantly enriched for miRNAs associated with PhenoAge (14 miRNAs, q-value = 0.021) that was not associated with either Horvath or GrimAge. No pathways were significantly enriched for EAA derived from the skin-blood clock.
Top shared miRNA-disease annotations significantly enriched for miRNAs associated with Horvath and GrimAge-derived measures of epigenetic age acceleration. a

Abbreviation: miRNA, microRNA; q-value, Benjamini Hochberg FDR adjusted P-value.
MiRNAs ranked by effect size using models adjusted for sex, chronological age, race/ethnicity, and cell type composition for gene set enrichment analysis adapted for miRNAs. MiRNA enrichment analysis performed using miRNA Enrichment Analysis and Annotation Tool (miEAA) and q-value < 0.05.
Curated miRNA-disease annotations obtained from MNDR v2.0.
Enriched (absolute maximal deviation from zero is positive and miEAA assumes an enrichment at the top of the ordered list of miRNAs) or depleted (inverse enrichment, the absolute maximal deviation from zero is negative and miEAA assumes an enrichment at the end of the ordered list of miRNAs).
Observed number of miRNAs significantly enriched for a given pathway.
Top 10 of 205 disease pathways, ranked by p-value, displayed here.
Top 10 of 15 disease pathways, ranked by p-value, displayed here.
Top shared miRNA-pathway annotations significantly enriched for miRNAs associated with Horvath and GrimAge-derived measures of epigenetic age acceleration. a
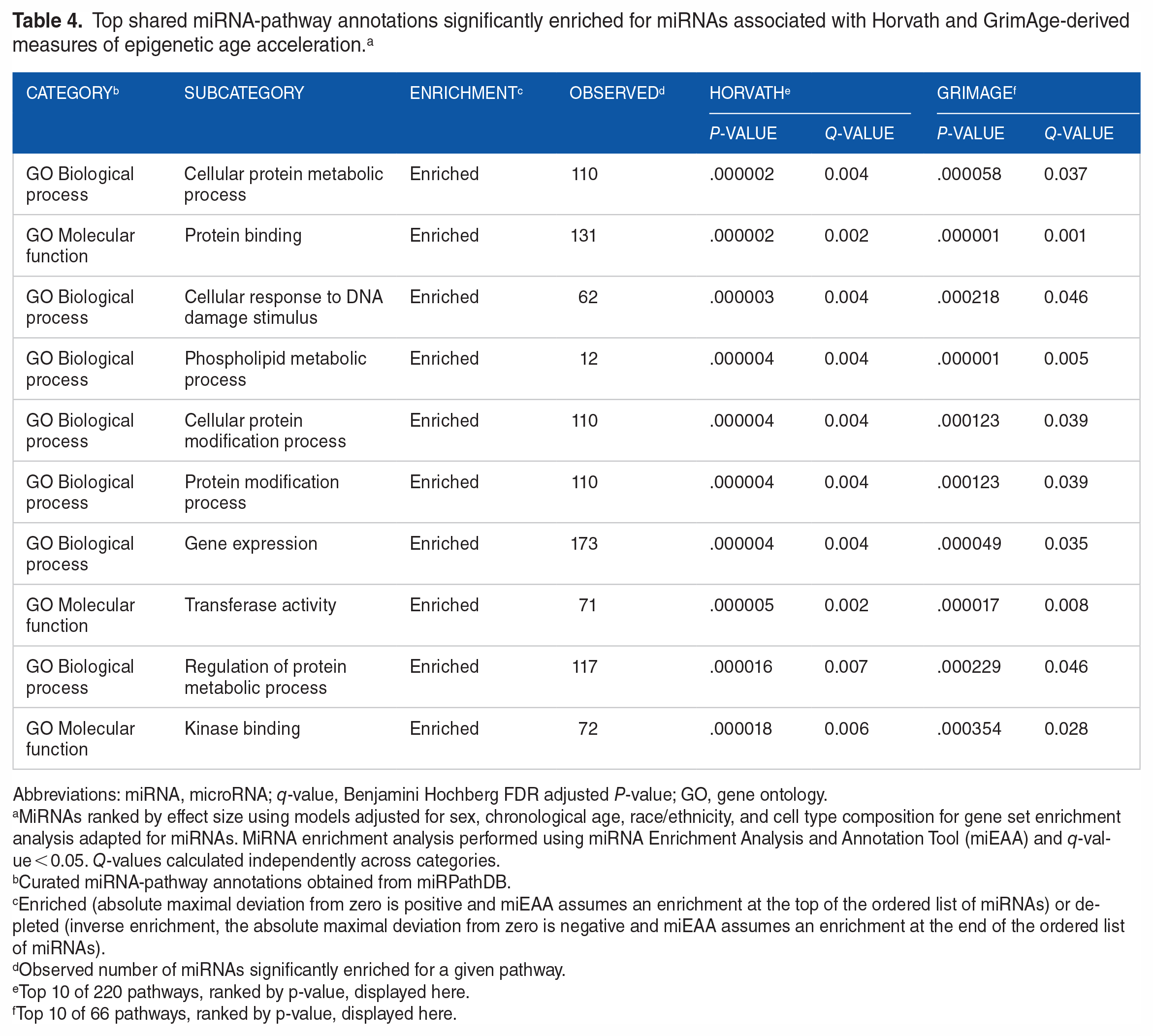
Abbreviations: miRNA, microRNA; q-value, Benjamini Hochberg FDR adjusted P-value; GO, gene ontology.
MiRNAs ranked by effect size using models adjusted for sex, chronological age, race/ethnicity, and cell type composition for gene set enrichment analysis adapted for miRNAs. MiRNA enrichment analysis performed using miRNA Enrichment Analysis and Annotation Tool (miEAA) and q-value < 0.05. Q-values calculated independently across categories.
Curated miRNA-pathway annotations obtained from miRPathDB.
Enriched (absolute maximal deviation from zero is positive and miEAA assumes an enrichment at the top of the ordered list of miRNAs) or depleted (inverse enrichment, the absolute maximal deviation from zero is negative and miEAA assumes an enrichment at the end of the ordered list of miRNAs).
Observed number of miRNAs significantly enriched for a given pathway.
Top 10 of 220 pathways, ranked by p-value, displayed here.
Top 10 of 66 pathways, ranked by p-value, displayed here.
Discussion
In this study, we examined associations between epigenetic age and miRNA expression across multiple epigenetic clocks and performed pathway analyses to investigate miRNA-based health implications. We identified several miRNAs significantly associated with 4 of the 7 EAA measures considered in our analysis: EAA derived from the PhenoAge, GrimAge, Horvath, and skin-blood clocks.
The Horvath clock is a widely used pan-tissue clock developed in 2013, that was based on methylation at 353 specific CpG sites and developed to predict chronological age across all sources of DNA. 10 However, due to the suboptimal performance of the Horvath et al clock to estimate fibroblast age, the skin-blood clock was developed to improve accuracy in estimation in fibroblasts and other cell types used in ex vivo studies.10,43 PhenoAge and GrimAge are among some of the recently developed blood-based clocks, that account for age-related and disease phenotypes in addition to chronological age. Nine age-related biochemical measures, including lymphocyte percentage, serum glucose, and albumin were used when designing PhenoAge. 8 PhenoAge has also been shown to capture tobacco-related methylation changes, a driving factor in mortality associated predictive methylation changes, which does not influence more traditional clocks such as Horvath and Raj.53,54 The GrimAge clock directly accounts for history of smoking, using methylation-based biomarkers for smoking pack-years, and estimators of plasma proteins. 44 PhenoAge and GrimAge have been shown to have improved ability to predict all-cause mortality, cancer, and other health outcomes and clinical phenotypes compared to other epigenetic clocks.8,9
The number of significantly different associations that we observed between EAA and miRNAs varied between epigenetic clocks used, with little overlap. The majority of statistically significant miRNAs that we identified were associated with either PhenoAge (n = 126) or GrimAge (n = 59). Given the use of whole blood samples in our study, the PhenoAge and GrimAge clocks, trained using whole blood methylation, may be especially well-suited for this analysis. Of the 6 miRNAs that were associated with at least 2 measures of EAA, 4 miRNAs (miR-3144-30, miR-4421, miR-510-5p, and miR-873-5p) are of particular interest because their estimates of effect were of the same direction of expression (increased or decreased) and were consistently associated with biological aging across multiple epigenetic clocks. To our knowledge, no previous studies have identified differential expression of these miRNAs in whole blood samples, though examinations of other tissues have identified potential associations with age-related health outcomes such as diseases affecting the eyes, Parkinson’s disease,55 -57 and multiple cancers, which supports known associations of EAA.8 -10,44
MiRNAs identified in our study that were associated with increased measures of biological aging were also generally linked with increasing cancer and other age-related disease risk in previous studies. We found that EAA derived from PhenoAge and GrimAge were both associated with decreased expression of miR-3144-3p and increased expression of miR-4421. High levels of miR-3144-3p have been shown to inhibit cell growth and promote apoptosis in HPV 16-positive cervical cancer cells in vitro, suggesting that decreased expression could be associated with oncogenic effects. 58 Separate studies have also reported that differential expression of miR-3144-3p was associated with glaucoma and psuedoexfoliation syndrome (a systemic, age-related, disorder of the extracellular matrix), both diseases that can affect the eyes.55,56 While the function of miR-4421 has not been fully investigated, its expression has been reported to be associated with telomere length and a genetic variation in telomerase reverse transcriptase (TERT), important factors in aging and cellular senescence. 59 Differential expression of miR-4421 in patients with Parkinson’s disease has also been reported to be involved in the regulation of genes associated with neuronal survival and the FoxO and PI3K-AKT signaling pathways, which are processes closely related to Parkinson’s disease. 57 Another study found that miR-4421 downregulated ERP29 expression and may alter risk and prognosis of oropharynx squamous cell carcinoma. 60 We also observed that PhenoAge and skin-blood were both associated with decreased expression of miR-510-5p. Studies have suggested that miR-510-5p has tumor suppressive effects in multiple cancers, including liver, renal, lung, and ovarian, and decreased expression was shown to be associated with poorer prognosis and overall survival.61 -64 Both the GrimAge and Horvath derived measures of EAA were associated with increased expression of miR-873-5p, which was previously reported to be upregulated in non-alcoholic fatty liver disease, lung cancer, and liver cancer.65 -67
In our secondary analysis we used a comprehensive miRNA enrichment tool to perform miRNA enrichment analyses, a threshold free analysis similar to a gene set enrichment analysis, 68 to examine potential downstream effects of miRNAs. We observed almost no enriched pathways associated with PhenoAge, which was unexpected given the number of differentially expressed miRNAs identified in the main analysis. The one pathway that was enriched for miRNAs was ATPase activity, which is involved in muscle contractions and has been implicated to decrease through complex mechanisms during aging.69,70 In comparison, we observed a greater number of enriched pathways associated with GrimAge and Horvath derived EAA, with many shared pathways. Based on ranking by statistical significance, the top ten shared enriched disease pathways included multiple cancers (Burkitt lymphoma, medulloblastoma, mesothelioma, uterine cancer, and blood cancers) and age-related diseases (stroke, muscular disorders, and non-alcoholic fatty liver disease). A recent evaluation by the International Agency for Research on Cancer has published that occupational exposure as a firefighter is carcinogenic on the basis of sufficient evidence of cancer in humans for mesothelioma and bladder cancer as well as limited evidence for colon cancer, prostate cancer, testicular cancer, melanoma, and non-Hodgkin lymphoma. 32 There is also a well-documented increased risk for morbidity and mortality due to cardiovascular diseases among members of the fire service,30,71,72 which some have suggested could be a result of occupational exposures to toxins such as particulate matter, stress, or other lifestyle factors. Whether exposure-induced biological aging—reflected in EAA and altered miRNA expression—could also affect this risk is unknown. However, there is some evidence that could support an underlying link between exposure-induced biological aging and risk of liver injury and disease. A cohort study published in 2023 of cancer and disease risk among Scottish firefighters found that there was increased risk of kidney and liver disease among firefighters. 72 Another recently published review of rodent studies and epidemiological studies found that there may be a link between exposure to per- and polyfluoroalkyl substances and markers of liver injury and disease, 73 while one of our previous analyses determined that PFAS levels in firefighters were associated with EAA and differential methylation. 1
The top pathway annotations were mostly related to proteins and protein modification (cellular protein metabolic process, protein binding, cellular protein modification process, protein modification process, gene expression, regulation of protein metabolic process, transferase activity, kinase binding) as well as cellular response to DNA damage stimulus. MiRNAs assessed in our study were taken from a curated array of clinically relevant miRNAs, so there may be some bias for enrichment of certain diseases and pathways. However, the miRNA array utilized in this study has been used broadly in biological research, including cancer biology, infectious disease, immunology, and others,74 -77 and not only did the majority of our results fall within the categories described above, but the diseases and pathways identified are consistent with studies reporting associations between epigenetic clocks and age-related outcomes.10,44 The breadth of these diseases additionally supports the notion that biological aging is the most important contributor to almost all diseases.
The reason for the large number of overlapping enriched diseases and pathways of miRNAs associated with GrimAge and Horvath-derived EAA is not immediately known. There is little overlap in CpG sites used to develop different epigenetic clocks, which could explain why some clocks are more strongly associated with some outcomes than others. 78 GrimAge and Horvath clocks were correlated among our study population, firefighters (Spearman’s r = .823). Previous studies among non-firefighting populations, that have directly compared GrimAge and Horvath clocks, have noted relatively weak correlations and that GrimAge outperforms when predicting age-related clinical phenotypes and all-cause mortality.9,44 However, future studies are required to investigate this finding. Additionally, groupings of functional annotations and pathways associated with GrimAge (eg, immune functions, adipocytokine signaling pathway, lipid function, and epigenetic regulation of gene expression) and Horvath clock (eg, cell death/survival, cellular growth/proliferation, organismal/tissue development, and cancer) have been reported previously.10,44 In our current analysis, we noted a number of cancers that were significantly enriched as well as pathways involved in gene expression and protein modification and regulation.
Our convenience sample of firefighters was composed of mostly (85%) incumbents, and relatively few recruits (15%). The relatively small number of new recruits prevented our ability to examine if relationships between EAA and miRNAs differ by occupational exposure. During our analysis we evaluated potential confounding by incumbent or recruit status, but observed generally small differences in model fit 46 and assumed that incumbent or recruit status was likely not a meaningful confounder on the association between EAA and miRNA expression. In 2 previous studies we found that that firefighting is associated with differential expression of 18 total miRNAs across both studies.5,6 Four of these miRNAs (miR-494-3p, miR-422a, miR-26a-5p, and miR-548h-5p) were also implicated in the current analysis and had estimates of a similar direction and magnitude. We have also previously shown that concentrations of certain per- and polyfluoroalkyl substances were associated with increased EAA in firefighters. 1 Taken together this may support that there is a previously undescribed relationship between occupational exposures, epigenetic aging, and miRNAs, which could support mechanistic evidence linking firefighting exposures with increased cancer and disease risk. 32 A second possibility is that there are unaccounted for occupational factors affecting these relationships. In this case, future studies of this occupational cohort should evaluate potential confounding by more descriptive or quantitative measures of occupational exposure than those that were available during our current analysis. Regardless, additional research is required to not only determine to what extent our findings may be relevant to the general adult population, but also to identify what exposures may affect these epigenetic age-miRNA relationships, and potentially risk of diseases. Investigating the relationship between these markers could help us further our understanding of biological processes related to age-related diseases.
In our study, we investigated the association between EAA and miRNA, given the potential importance of both for biological aging and disease processes. DNA methylation and miRNA influence gene expression on the transcriptional and post-transcriptional level, respectively.79,80 Epigenetic clocks are composite biomarkers of DNA methylation at subsets of CpG sites selected based on their ability to predict age and/or measures of healthspan; these loci are not necessarily associated with the regulation of critical genes for aging, carcinogenesis, or other disease processes. Thus, associations between EAA and miRNAs with known cancer and disease associations could instead be due to the fact that these miRNAs are broadly changed with biological aging. Still, it is worth further investigation given that aberrant methylation patterns of miRNA promoters have been associated with cancer.26 -28 There is also evidence that miRNAs also have a regulatory role over DNA methylation. Several studies have reported that miRNAs regulate the expression of enzymes such as DNA methyltransferases and 10 to 11 translocation enzymes, which regulate DNA methylation machinery.81,82 Because of this complex interplay between miRNAs and other epigenetic mechanisms, studies investigating the implications that miRNA expression may have on EAA should also be considered.
To support advances in developing more effective ways to diagnosis, treat, and prevent age-related diseases, a comprehensive understanding of the underlying mechanisms and regulators that drive these different diseases is needed. Biological aging, reflected by EAA and miRNAs are examples of such mechanisms. Our findings contribute to a growing amount of literature investigating the interactions between different mechanisms of epigenetic regulation with the goal of improving our ability to predict diagnosis and clinical outcomes of age-related diseases,26,27,83 though to our knowledge, this is the first study to focus on the relationship between EAA and miRNAs. Our findings suggest that biological aging has some influence on miRNA expression though it is uncertain if this is due to direct regulation or if the miRNAs we identified are broadly changed with biological aging. Overall, our study results support previous studies linking EAA with cancer and other age-related diseases,8,10,44 while also suggesting that the underlying aging and disease pathways that the clocks represent may involve protein modification and regulation. We also observed that our results varied by epigenetic clock, reinforcing findings from other publications that each clock has varied utility for predicting disease risk and/or aging. This could be useful to support biomarker selection in future studies. Based on our results, studies interested in identifying risk factors for aging-related diseases may consider the GrimAge clock and associated miRNAs as potential biomarkers; additional studies will be required to inform whether the biomarkers identified here are generalizable to other populations or occupational groups.
Supplemental Material
sj-docx-1-gae-10.1177_25168657231206301 – Supplemental material for Associations Between Epigenetic Age Acceleration and microRNA Expression Among U.S. Firefighters
Supplemental material, sj-docx-1-gae-10.1177_25168657231206301 for Associations Between Epigenetic Age Acceleration and microRNA Expression Among U.S. Firefighters by Alesia M Jung, Melissa A Furlong, Jaclyn M Goodrich, Andres Cardenas, Shawn C Beitel, Sally R Littau, Alberto J Caban-Martinez, John J Gulotta, Darin D Wallentine, Derek Urwin, Jamie Gabriel, Jeffrey Hughes, Judith M Graber, Casey Grant and Jefferey L Burgess in Epigenetics Insights
Footnotes
Acknowledgements
We would like to acknowledge the firefighter participants for graciously volunteering their time and samples to be in the study. We also acknowledge the partnering fire departments and their locals for their contribution to this research. The University of Michigan Advanced Genomics Core and the University of Utah DNA Sequencing and Genomics Core Facility completed the EPIC analyses.
Funding:
The author(s) disclosed receipt of the following financial support for the research, authorship, and/or publication of this article: This research was supported by grants awarded by the National Institute of Environmental Health Sciences (NIEHS) (grant no. P30 ES006694, P30 ES017885, R01 ES031259, R00 ES028743, and T32 ES007091), the U.S. Federal Emergency Management Agency (FEMA) (grant no. EMW-2014-FP-00200, EMW-2015-FP-00213, and EMW-2018-FP-00086), as well as Disaster Relief Funds from the U.S. Centers for Disease Control and Prevention (CDC).
Declaration Of Conflicting Interests:
The author(s) declared no potential conflicts of interest with respect to the research, authorship, and/or publication of this article.
Author Contributions
AJ performed the statistical analyses and did the write up. MF supervised the analyses and the writing. JG and AC provided guidance on appropriate use of epigenetic data and modeling. SB and SL managed the data collection and handling and storage of all laboratory based biospecimens. ACM, JMG, JB, CG supervised data collection procedures and study design of the parent study. JG, DW, DU, JG, and JH contributed fire service input and resources to promote firefighter recruitment, and also assisted in creation of study design materials.
Data Availability
The data that support the findings of this study are available on request from the corresponding author, MF. The data are not publicly available due to restrictions [eg, containing information that could compromise the privacy of research participants].
Supplemental Material
Supplemental material for this article is available online.
References
Supplementary Material
Please find the following supplemental material available below.
For Open Access articles published under a Creative Commons License, all supplemental material carries the same license as the article it is associated with.
For non-Open Access articles published, all supplemental material carries a non-exclusive license, and permission requests for re-use of supplemental material or any part of supplemental material shall be sent directly to the copyright owner as specified in the copyright notice associated with the article.