
Abstract
Human genome contains many variations, often called mutations, which are difficult to detect and have remained a challenge for years. A substantial part of the genome encompasses repeats and when such repeats are in the coding region they may lead to change in the gene expression profile followed by pathological conditions. Structural variants are alterations which change one or more sequence feature in the chromosome such as change in the copy number, rearrangements, and translocations of a sequence and can be balanced or unbalanced. Copy number variants (CNVs) may increase or decrease the copies of a given region and have a pivotal role in the onset of many diseases including cardiovascular disorders. Cardiovascular disorders have a magnitude of well-established risk factors and etiology, but their correlation with CNVs is still being studied. In this article, we have discussed history of CNVs and a summary on the diseases associated with CNVs. To detect such variations, we shed light on the number of techniques introduced so far and their limitations. The lack of studies on cardiovascular diseases to determine the frequency of such variants needs clinical studies with larger cohorts. This review is a compilation of articles suggesting the importance of CNVs in multitude of cardiovascular anomalies. Finally, future perspectives for better understanding of CNVs and cardiovascular disorders have also been discussed.
Introduction
Copy number variants (CNVs) have gained a lot of interest over the past few years due to the complexities they offer. Apart from altering the expression and phenotype, CNVs are responsible for genetic diversity in population as well. Copy number variants (shown in Figure 1) are sequence of DNA of 50 base pairs (bp) or larger compared with its reference genome and collectively represent, duplications, complex multisite variants, and smaller elements called insertion and deletions (indels). 1 Terminal deletions occur at the terminal region of the chromosome and are combined with neuropsychiatric complications, retarded growth, and dysmorphic facial features as shown in Cri-du-Chat syndrome 2 ; micro-deletions are deletions up to 5 Mb which occur at 16p11.2 in Autism 3 ; duplications include micro-duplications, for example, in 1q21.1 micro-duplication syndrome. 4 One of the classic examples is duplication of an entire chromosome: chromosome 21 in Down syndrome. 5 Another type of variants is multisite variants that contribute to the genetic diversity between individuals. Other than mentioned, inversion that is flipped or reversed orientation of chromosomal segments has been established in mutation in Factor VIII gene, which plays a critical role in coagulation and leads to hemophilia A; this mutation is found to be responsible in 43% of the individuals suffering from hemophilia. 6 Translocation is shifting of large segments of DNA, for example, t(1;11) translocation is considered to have an association with increased risk of psychopathology due to decreased glutamate levels and altered cortical structure and function in the left temporal lobe of the brain. 7 Microdeletions, a type of interstitial deletions, are deletions of a very small portion around 1 to 2 Mb of the chromosome, whereas microduplications are comparatively rare, but are prominent reason behind neurodevelopmental genetic diseases. 8 Copy number variants, when not tolerated, have been identified as a potent genetic variation, regulating both genotype and phenotype in an individual’s susceptibility for a disease condition. The risk of changes in phenotype and occurrence of the disease is more of a de novo event, as CNVs are the rearrangements which may not present in the parent karyotype but offspring may carry the changes. Such variants are common among healthy individuals, however, but not necessarily affect the health and phenotype, for example, both deletions and duplications are found in HLA class III genes encoding complement proteins, C2 and C4, which are relatively common. 9 Apart from the genome-wide CNVs, there are mitochondrial DNA copy numbers (mtDNA-CNs), which decide the number of mitochondria and mitochondrial genomes per mitochondrion per cell and variation which may lead to mitochondrial dysfunction. 10 The mtDNA-CNs have been studied for predicting type 2 diabetes in a cohort that included 8 years of follow-up. 11 Also, leukocyte telomere length and mtDNA-CN and deletions have been proposed as risk markers for breast cancer in a European Prospective Investigation into Cancer and Nutrition (EPIC) study. 12 Interestingly, other species have also been under the influence of CNVs such as rat, mouse strains, and rhesus macaque, but human diseases such as genomic and monogenic disorders, infectious diseases, autoimmune diseases, and carcinomas have well-known links with CNVs.13–16 They affect the expression level by altering the transcription via disturbing the cis or trans-regulatory regions and other functional regions. 17 The number of copies of a gene present in the coding region is directly proportional to the number of transcript, thus facilitating more translation or vice versa. 18 However, studies have also advocated the parallel effect of copy number alterations in genes of single chromosome leading to detrimental phenotypes. 19 Figure 1 represents human chromosome, for example, with a segment as reference region, followed by different variation occurring in the reference region.

Copy number variation in the chromosome showing normal (reference) and CNV carrying chromosome regions.
Cardiovascular diseases (CVDs) include the diseases of heart and blood vessels including both arteries and veins. Cardiovascular diseases are leading cause of mortality and morbidity worldwide. Cardiovascular diseases include a broad range of disorders including heart and associated vascular system. Most commonly appeared CVDs are coronary artery disease (CAD) and stroke; other than these two, there are myocardial infarction (MI), congenital heart disease, cardiomyopathy, cardiac arrhythmia, heart valve disease, and other vasculature-related diseases such as atherosclerosis, venous thrombosis, pulmonary embolism, and peripheral artery disease. A report from World Health Organization has stated that around 31% of the total deaths in all over the world were caused by CVDs. Different epidemiologic studies on various continents have conducted to assess the early onset in the population, progression of the disease, prevalence, treatment, socioeconomic background of the individuals, and other factors contributing to CVDs.20–22 As the risk of CVDs is increasing significantly by the time, the research has been shifted to establish major aspects contributing to the disease, whereas most of the efforts have been put to understand the mechanism of the disease and the common predisposing factors; the global health care services in the field of cardiovascular research are yet to be improvised. 23 However, despite all the research attempts to identify the disease progression during early stages, increased risks have been a major barrier to decrease the global burden of CVDs as they account for one-half of all the deaths caused by noncommunicable diseases worldwide. 24
Having discussed the severity and prevalence of the CVDs and their known risk factors, including hypertension, cholesterol levels in blood, obesity, smoking, diabetes, and others, a major survey on the genetic risk factors is essential to complement the research. 25 Major advancements in the field of genetics have proposed genetic variants accountable for predisposing the CVD risk, among which single-nucleotide polymorphisms (SNPs) have majorly been studied due to the improvised technologies for detection, identification, and characterization of annotated as well as novel SNPs. Genome-wide association studies (GWAS) have been considered the most significant studies so far to detect a number of genetic risks for CVDs. These proposed risk factors could be considered as established genetic markers for risk stratification; however, they require more experimental proofs and validation.
Mechanism of Copy Number Variation in Genome
Alterations in the gene copy number are basically changes in the position of a chromosomal region in the chromosome itself; the segments could be of different sizes depending on the type of variation occurred. Mechanism of copy number changes is well studied in model organisms such as Drosophila, Saccharomyces cerevisiae, and Escherichia coli. The reason behind copy number changes in the chromosomes is basically recombination of 2 types: homologous recombination which occurs on more identical sequences or sequences with higher similarity, whereas nonhomologous recombination, on the contrary, requires very limited or microhomology or no homology at all; such recombination occur during replication mechanism as reviewed in a recent study. 26 As described in a recent article, when there is extensive similarity in the sequence of the chromosomal segments, nonallelic homologous recombination (NAHR) occurs; the segments with no homology or very less similarity have nonhomologous end-joining and microhomology-mediated end-joining recombination, respectively, for the origin of CNVs. In addition, other common replication errors may also lead to CNVs, such as fork stalling and template switching and microhomology-mediated break-induced replication and transposable element–mediated mechanisms. 27 Among all, NAHR are best known to contribute toward recurrent or frequently occurring CNVs; the nonrecurrent-type CNVs are produced by replication-based errors. 28 Although the knowledge regarding the origin and development of CNVs have been cited in various studies, mechanisms including damaged DNA followed by its repair mechanism having higher incidences of nonhomologous recombination-induced copy number changes are still unclear.
History of Copy Number Variations and Links with CVDs
A brief study published in 2004 strongly recommended the global role of large-scale copy number variations in human genome from heritable gain and loss of DNA for the first time; previously, there was no such breakthrough in the field of structural variants. 29 The study was conducted with 55 healthy individuals and showed the genetic differences in their genome in the form of structural variants. Going back to the discovery of CNVs in 1987, Nakamura et al 30 proposed variable number of tandem repeats (VNTRs), which are heritable regions in the chromosome where short DNA sequence motifs are present in repeats in random variable number, as the name suggests. The complex mutation pattern of one of the multifaceted disorders related to VNTR can be found in dopamine receptor D4 gene exon3 VNTR association in attention deficit hyperactivity disorder in which European and Asian ethnicities display 7- and 2-repeats, respectively. 31 After VNTRs, in 1989, microsatellite came into light for their distinguished role in complex multifactorial disorders and later they were used as a biomarker for gene map studies in humans; furthermore, these microsatellites were used widely to make the linkage map of all human chromosomes.32,33 In a recently published article, authors have provided the evidence that microsatellites may act as enhancers and regulate the expression of genes. 34 In later years, these genomic rearrangements, both balanced and imbalanced, were characterized as mutational changes in genome responsible for various ailments. 18 A classic example for such rearrangements is of a muscular atrophy called Charcot-Marie-Tooth disease type IA, in which the CNV responsible for the disease originated in the upstream of peripheral myelin protein 22 (PMP22) gene, which earlier was not documented in the human structural variants database. 35 After a long leap, in 2002, after almost 10 years, structural variants came into light as paralogous sequence variants (variants in few nucleotides in closely related gene families).36,37 One of the most prominent discoveries in this field was done by Redon et al, 38 identifying 1447 copy number variable regions in the genome of 270 healthy individuals from 4 different ethnicities, which suggested that around 12% part of the human genome has CNVs.
Despite the significant improvements in the technologies to identify CNVs, the proposed genetic risk factors for CVDs are not considered clinically significant so far. Notably, a study has challenged the notion for CADs that classical risk factors are responsible for over 50% of the cases, whereas a careful analysis of the articles published in the past has indicated that in fact over 80% of CAD cases have come up with nonclassical risk factors. 39 A few diseases with cardiovascular anomalies have been registered previously with smaller CNVs or large cytogenetic changes as the underlying cause. One of the pioneer studies in early 1980s on congenital polyvalvular disease (CPVD) suggested that the presence of an extra copy of chromosomal part of chromosome 18, called trisomy-18 syndrome, leads to congenital anomalies, especially CPVD. 40 Such diseases resulted after larger changes in the chromosomes, which could be visualized through microscope. Another study from late 1980s karyotyped 41 cases of trisomy-18, among which almost in all autopsy samples different forms of cardiac anomalies were pronounced including pulmonary, tricuspid, aortic and mitral valve anomalies, polyvavular disease, and so on. 41 Since 1990s, there were comparatively more studies regarding structural variations in the chromosome linked with cardiac abnormalities.42–44 However, the problem was to find out clinically significant CNVs and to link them with the cardiac diseases because many variations are found in healthy population as well, which required improvement in the detection and identification technologies with experimental validation.
Role of Copy Number Variations as Compared With SNP in CVDs
Single-nucleotide polymorphisms on the contrary to the CNVs occur due to a variation in a single nucleotide in the gene, which may influence the transcription by affecting promoter activity or messenger RNA stability; as a consequence, they are reported to have association with several diseases in humans. 45 Previously, candidate gene studies to identify presence or absence of common genetic variants were used for disease association analysis which lacked the reproducibility and it was practically not possible to test higher number of polymorphisms for larger cohorts. Genome-wide association studies have emerged as one of the most powerful tools to investigate the whole genome for SNPs in particular. 46 Genetic studies have led to upsurge the understanding of CVD association with SNPs. For instance, the clinical data suggest that protein levels of Apolipoprotein E (ApoE) in plasma are strikingly associated with the risk for coronary heart disease; hence, common polymorphisms affecting this gene in turn affect the protein level leading to the disease. 47 Common variants in ATP-binding cassette transporter 1 (ABCA1) gene, low-density lipoprotein receptor gene (LDLR), and others are reported to have association with cardiac diseases.48,49 Over the past few years, SNPs have been considered as a potential risk factor for genetic diseases in humans; CNVs, however, were detected lately but their potency to alter the genomic functions and phenotype was characterized in no time.
Copy number variants span more than 50 bases in the genome and are responsible for more variations in the chromosome, leading to disruption of the functional large genomic segments than do SNPs. 10 Copy number variants present in the coding region directly influence the copies of a gene during transcription, hence contributing to the progression of diseases, whereas if CNVs occur in the noncoding region of a gene, they may not influence the copy numbers. Around 4 Mb of CNVs is present in the whole human genome, whereas SNPs account for 2.5 Mb, suggesting CNV accountability for most of the genetic variations in humans. 50 Many CVDs have been identified having association with SNPs, but the share of CNVs in increasing the complexity of human genome and link with the diseases cannot be overlooked. A review suggesting implications of CNVs in CVDs has underlined majorly monogenic disorders suggesting more investigation in this field. 51 A genome-wide case-control study of CNVs has suggested a potent role of rare variants in large cohort responsible for dilated cardiomyopathy, a heart disorder leading to various cardiovascular complications. 52 Congenital heart disease (CHD) is a primary birth defect leading to early death of newborns worldwide. Reports are available on rare CNVs present in heterotaxy: a type of CHD and also in syndromic and nonsyndromic congenital heart defects on a cohort of 46 nonsyndromic individuals.53,54 Copy number variants play a major role in various blood disorders; for example, CNVs in the α-globin genes of hemoglobin lead to α thalassemia, a very common blood disorder, in which deletion in the α-globin gene makes rare disease onset, whereas duplication in the same gene causes severe phenotypic change in β-thalassemic carriers. 55 Furthermore, CNVs influence important regulatory sites in the gene from distance, leading to dynamic changes in the expression pattern. 56
CNVs in Cardiovascular Disorders
Cardiovascular diseases have widely been studied on the basis of their origin, type of disorder, organ, and vessels affected and their genetic inheritance, but very few studies are available corroborating the link between CNVs and CVDs. Table 1 represents different CVDs associated with CNVs and different methods to detect those in the genome.
Different cardiovascular disorders associated with CNVs and its detection via comprehensive arrays.

Abbreviations: Array-CGH, array comparative genomic hybridization; CNVs, copy number variations; FISH, fluorescence in situ hybridization; GWAS, genome-wide association study; HSP, heat shock protein; LPA, lipoprotein(A); MLPA, multiplex ligation–dependent probe amplification; qPCR, quantitative polymerase chain reaction; SNP, single-nucleotide polymorphism.
Congenital Heart Disease
Congenital heart disease is one of the major birth defects, leading to cardiac malformations in around 7 in every 1000 live births, resulting in higher incidences of mortality in infants. 63 Children with nonsyndromic CHD had a higher risk of potentially pathogenic CNVs when compared with healthy ones and found to be at higher risk of mortality. 64 Another study to determine pathogenic chromosomal abnormalities was conducted in fetuses diagnosed with CHD by microarray analysis. The results of the study concluded that a significant percentage of cases were found to be associated with different chromosomal anomalies supporting other same studies. 65 Furthermore, CNVs have also been implicated in congenital left-sided cardiac lesions. A recent study observed large CNVs were in 33 probands (∼3%), out of 1139 probands. 66 Also, global rare copy number variants and de novo mutations in histone-modifying genes have been linked to the risk of sporadic CHD, earlier.67,68 High-resolution genome-wide analysis have shown that coarctation of the aorta (CoA), which accounts for 5% to 8% of all CHDs, is due to abundance of large CNVs on the X chromosome of affected men. 69 Also, in patients with CoA, CNVs alter the expression of genes regulated by FOXC1. 70
Myocardial Infarction
Myocardial infarction is a prolonged condition of lack of oxygen to heart muscles. A GWAS has identified major SNPs as well as common and rare CNVs for possible association with early-onset MI in larger cohorts. The study tested 554 common CNVs and SNPs at 8 loci in patients, as compared with controls. 60 Another study has observed 7 CNV segments associated with the patients of hyperlipidemia (a major cause of MI) and MI suggesting the potential role of structural variants in MI. 71
CAD and Hypertrophic Cardiomyopathy
Coronary artery disease also called ischemic heart disease is presented by the patients with MI and stroke, making it one of the leading causes of deaths worldwide. A study investigated association between CNV in lipoprotein(a) (LPA) in CAD cases. Higher serum concentration of LPA is associated with increased risk of CAD; however, in this study, a protective role of 1 copy number of the LPA gene has been identified in southern Han Chinese population majorly in women and the elderly. 61 Hypertrophic cardiomyopathy (HCM) is a condition in which the myocardium becomes abnormally thick (hypertrophied), without an obvious reason. People have used high-throughput sequencing technique to screen CNV in genes associated with cardiomyopathy. 72 Also, people have studied minor genes and CNVs in HCM. 73
Essential Hypertension
Essential hypertension, a multifactorial disease, possesses substantial genetic risk factors including CNVs. Deletion of the CNVs esv27061 and esv2757747 on the chromosome 1p13.2 was found to be significantly higher in patients with hypertension identified by droplet digital polymerase chain reaction (PCR) technique. 74 Another study on different strains of rats has confirmed the pivotal role CNVs in hypertension by whole genome array. 75
Stroke
Stroke is the third leading cause of mortality worldwide after cancer and other heart diseases. A study has investigated the role of CNVs in modulating risks for ischemic stroke. The analysis showed 187 insertions (76%; 135 heterozygous, 25 homozygous duplications or triplications, 2 heterosomic) as well as 60 deletions (24%; 40 heterozygous deletions, 3 homozygous deletions, 14 heterosomic deletions) in the patients when compared with controls; among these, reported CNVs were previously reported. 62 A very recent detailed description of CNVs influencing stroke probability in individuals has reported and confirmed the positive association of stroke with CNVs. 76 Evidence are also available for microduplications involved in stroke. 77
Diseases Other than Cardiovascular Types and their Association With CNVs
Several studies have been done to link the cord between diseases other than cardiovascular types and their phenotypes with CNVs. A number of diseases having a background of structural variations are listed in Table 2. Moreover, an increase or a decrease in the copy numbers of one gene may lead to different diseases as shown in Table 2. A population study has suggested the role of segmental duplication encompassing the gene chemokine (C-C motif) ligand 3-like 1 (CCL3L1) increasing the susceptibility toward HIV-associated immune deficiency or AIDS. 90 Furthermore, enhanced expression of the same gene has been underlined as a risk factor for rheumatoid arthritis in a different case-control study. 79 Moreover, there are major structural rearrangements in the genes which are considered to be rare phenomena but they have their share of clinical significance as shown in Table 1. Among these, some changes or rearrangements were so large that they were cytogenetically visible, for example, more than 4 to 5 copy numbers of gene β-defensin showed significant risk of psoriasis, whereas the low copy numbers of the same gene are considered as a risk for Crohn disease.83,84 Besides duplication, deletions in the late cornified envelope 3B and 3C genes have been identified in case of patients with psoriasis. 85 Despite having an established role of CNVs in the development of autism, the de novo germline mutation has been characterized in recent past as a risk factor for the disease. 91
Copy number variations in different genes causing diseases, an increased or decreased copy of the same gene can cause different diseases.
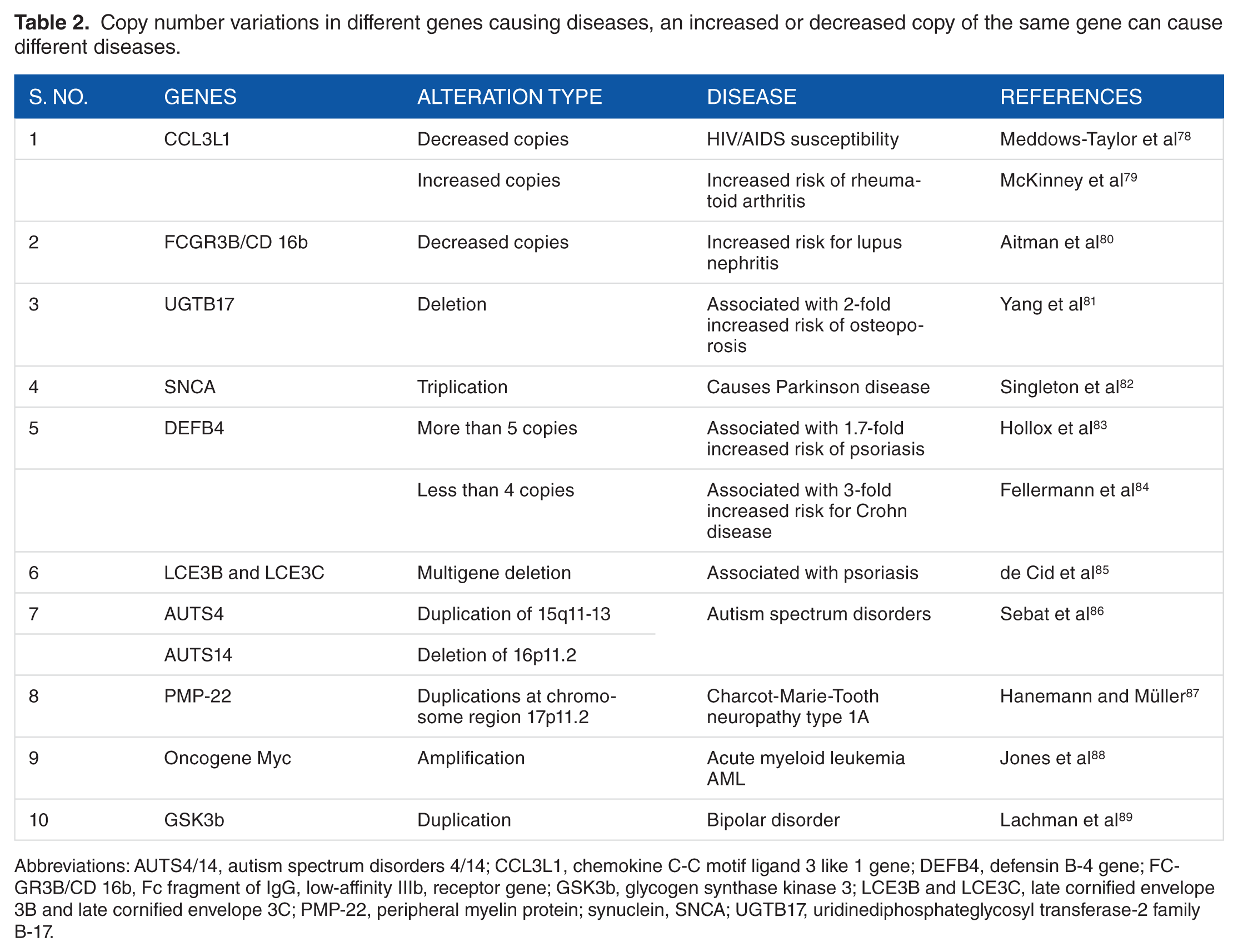
Abbreviations: AUTS4/14, autism spectrum disorders 4/14; CCL3L1, chemokine C-C motif ligand 3 like 1 gene; DEFB4, defensin B-4 gene; FCGR3B/CD 16b, Fc fragment of IgG, low-affinity IIIb, receptor gene; GSK3b, glycogen synthase kinase 3; LCE3B and LCE3C, late cornified envelope 3B and late cornified envelope 3C; PMP-22, peripheral myelin protein; synuclein, SNCA; UGTB17, uridinediphosphateglycosyl transferase-2 family B-17.
The effects of these CNVs are at times combined with the environmental factors as well, which may increase or decrease the severity of the disease, for example, in GSTM1 and GSTT1 genes in cancer susceptibility. 92 Within recent years, multiple evidence have led to a conclusion that CNVs play a crucial role in disease susceptibility and development. Also, large-scale studies have been conducted to augment the indications. For example, recently, a study with a cohort of 144 children (patients only) with absence epilepsy subtypes (a type of epileptic disorder found mostly in children) was conducted. The study highlighted duplications and deletions in the gene regions 1p36.33, 1q21.1, 22q11.2, and Xp22.31 increasing the potency to develop the disease. 93
Techniques to Identify Copy Number Variations
Before array hybridization technologies, imbalanced chromosomal rearrangements were detected and analyzed by cytogenetic techniques. For large and imbalanced rearrangements, the gold standard Giemsa banding was popular which gives a visible karyotype of all chromosomes, 94 whereas for molecular cytogenetics, fluorescent in situ hybridization (FISH) and comparative genomic hybridization (CGH) techniques were used. 95 These techniques come up with a nonnegligible percentage of error which reduces the authenticity of the results. So, an improvised version of FISH was introduced as 3D FISH but it also lacked the speed and the specificity required for high-level comprehensive analysis. 96 However, because of low resolution and less specificity, these methods were overpowered by the advancements in the detection techniques.
There are 2 classes of chromosomal abnormalities: microscopic, which can be seen by a microscope, and submicroscopic, which are rare and cannot be detected by microscope alone. 97 For submicroscopic variants, hybridization techniques like CGH and their modified versions like microarray-based CGH (array-CGH) to identify very rare variants were introduced. 98 Earlier, the arrays were introduced with larger sequence segments of DNA for probing; however, now, increased number of spots and shortened segment size on the array to improve the resolution are used. Different oligonucleotide arrays, PCR products, clone arrays, genotyping arrays, and others are in use. 99 A number of array platforms have been recommended in recent past for the detection of complex CNVs with nonexpressive phenotypes. For example, bacterial artificial chromosome arrays and oligo CGH (oligo-CGH) associated with sequencing are improvised versions of CGH, whereas SNP arrays are used to differentiate between 2 SNP alleles. Array techniques have been preferred over conventional karyotyping for diagnosis of fetal and prenatal abnormalities. 100 Customized arrays are also available to choose platforms and target genes and regions according to the need of the research. Furthermore, genome-wide analysis of CNVs can be done by multiplex PCR-based methodologies such as PCR followed by restriction fragment length polymorphism, real-time quantitative PCR for shorter length fragments, multiplex ligation–dependent probe amplification, and multiplex amplicon quantification techniques. 101 Such techniques are majorly used for screening and validation of CNVs. Also, whole genome sequencing, exome sequencing, and targeted exome capture followed by sequencing are widely accepted sequencing techniques to determine copy number variations in human genome.102–104
The advent of next-generation sequencing technologies has enabled accurate and rapid analysis of CNVs with high resolution per base. 105 Moreover, high-resolution array platforms such as NimbleGen (Madison, WI, USA) and Agilent (Santa Clara, CA, USA), pioneer to develop CGH arrays with differently labeled target and reference genome with fluorescent tags, are currently being used for CNVs. 91 Next-generation sequencing platforms such as Illumina, Roche 454 Life Sciences, and SOLiD from Applied Biosystems have provided more accurate CNVs for both genome-wide analysis and targeted gene analysis.106–108 Also, a number of computational tools are available which in combination with the sequencing techniques improve the analysis. 109 For better understanding of next-generation techniques for identifying CNVs, comparative studies have been comprehended and more sensitive tools and accurate algorithms have been introduced for CNVs from the big data sets. Flow diagram in Figure 2 shows different CNVs and techniques for detection methods. The CNVs are classified on the basis of detection techniques employed.
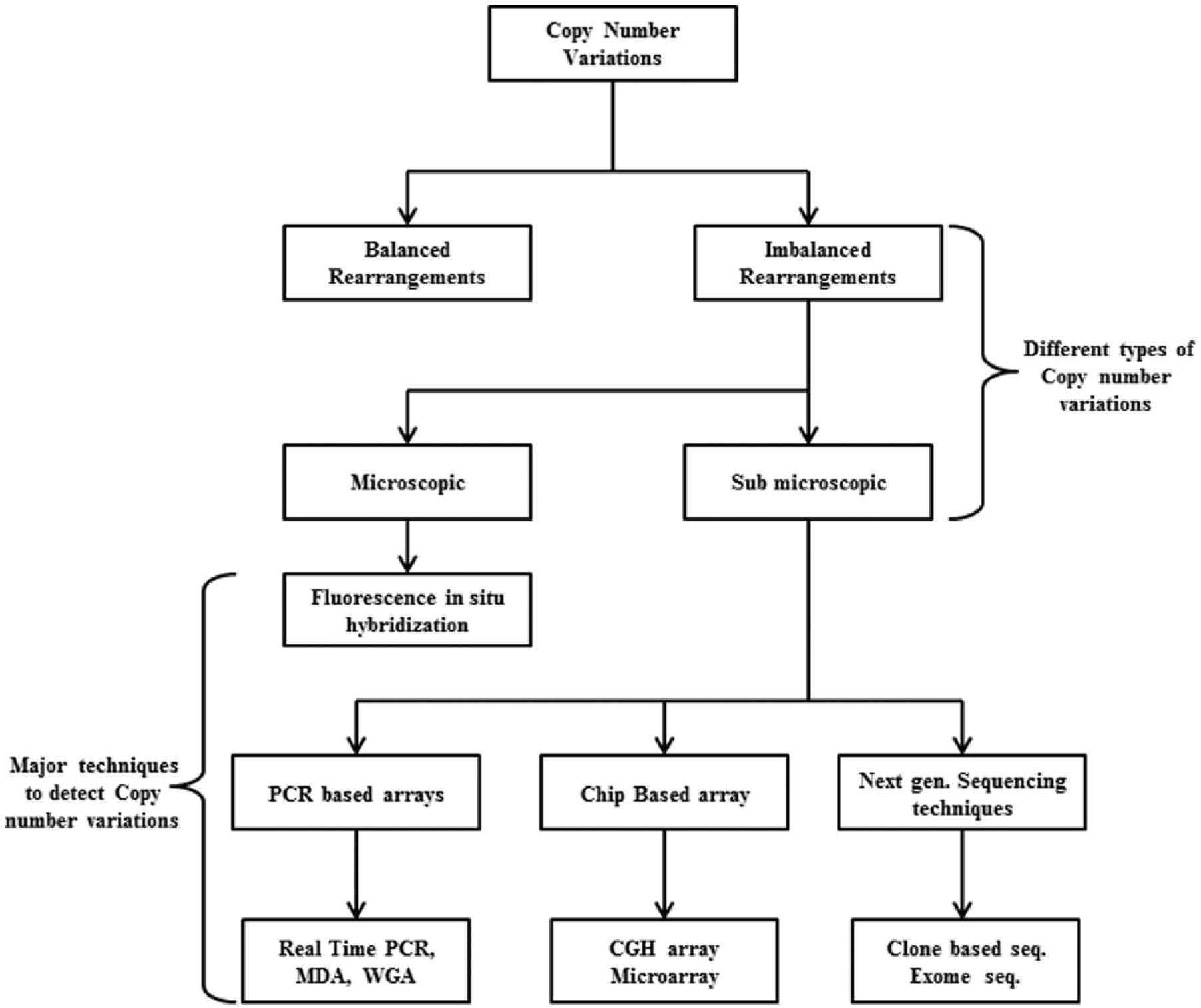
Classification of copy number variants based on the techniques employed for identification.
Future Perspectives
Although current platforms are able to detect clinically significant CNVs in whole genome with better specificity, there are technical challenges that remain to be directed. The paramount implementation of such techniques is early/prenatal detection of structural variants for diagnostic purposes. The low sample size of human subjects and limitations of high-throughput data analysis and interpretation tools are major bottleneck. Over the past years, CNVs have emerged as one of the possible reasons underlying multifaceted diseases, as the mechanisms of many complex diseases are yet to be elucidated. Although literature evidence are available for different CVDs, their association with CNVs suggests a not-alone role of single-nucleotide variations (SNPs) as genetic variants regulating human diseases and health. More investigation is required to culminate the magnitude of the effect in various CVD subtypes. The current techniques and developments have enabled us to elucidate genomic variations in humans; yet, the complexity itself requires a platform to delineate the neutral or negative effect of any variant on any individual. Current platforms to identify CNVs need improvisation as they need to optimize the threshold for CNV detection and minimize the noise created by other genetic variants. Although genomic maps for CNVs have suggested their potential effects on the healthy individuals, hence opening up a new obscured area of research, epidemiologic studies with larger cohorts and comprehensive methodologies are prerequisite for a conclusive statement.
Footnotes
Acknowledgements
The authors are extremely thankful to Director, DIPAS, Defence Research and Development Organization (DRDO). A.V. is a DST-INSPIRE fellow.
Declaration of conflicting interests:
The author(s) declared no potential conflicts of interest with respect to the research, authorship, and/or publication of this article.
Funding:
The author(s) received no financial support for the research, authorship, and/or publication of this article.
Author Contributions
MZA and AV conceived and designed the article. AV wrote the manuscript. IG and MZA edited the manuscript.