
Abstract
Background
Ictal epileptic headache is a very rare form of painful seizure that closely mimics several classic headache syndromes, including trigeminal neuralgia.
Case
Here, we present a woman who was treated as for treatment-refractory unilateral trigeminal neuralgia over 10 years, which was later deemed to be due to somatosensory seizures from the insular cortex. A cavernous haemangioma in the right insular cortex was identified as the causative lesion for these recurrent seizures and was successfully treated with resective surgery.
Conclusion
This case underscores the importance of considering lesional ictal epileptic headaches as a differential diagnosis of treatment-resistant neuralgiform headaches, which can be effectively managed by targeted surgical resection thereby inducing long-term headache or seizure remission.
This is a visual representation of the abstract.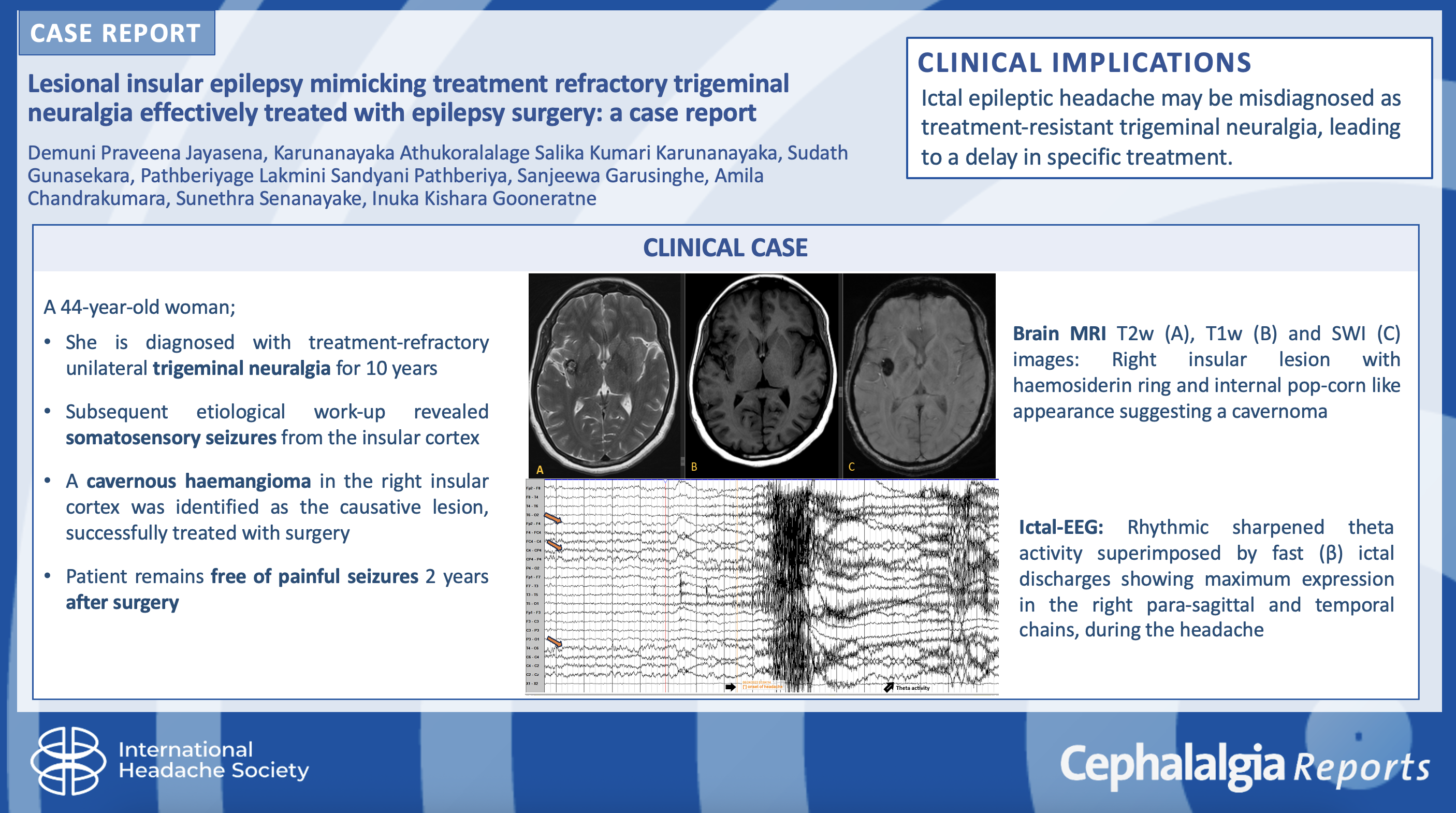
Introduction
Ictal epileptic headache (IEH) is an infrequent manifestation of a painful seizure, where the headache occurs simultaneously with a focal-onset seizure. 1 In IEH the headache occurs either ipsilateral or contralateral to the ictal discharging focus and resolves spontaneously following seizure termination. 1 The cephalalgia in IEH can arise at any location with variable intensity and duration.1,2 IEH is often misdiagnosed as a primary headache disorder when it presents as the predominant or the sole ictal symptom.1–3 IEH can rarely mimic trigeminal neuralgias (TNs) and trigeminal autonomic cephalalgias.1–3 The revised diagnostic criteria for IEH in accordance with the international classification of headache third edition (ICHD-3) predominantly specifies the duration and the site of the headache but also outlines 2 additional requirements.2,4 These include: evidence of lateralized epileptiform discharges on scalp electroencephalogram (EEG) coinciding with the headache and the immediate resolution of headache together with EEG abnormalities after antiseizure medication.2,4 Despite a well-timed ictal EEG, it is clinically challenging to differentiate IEH from pre-ictal and post-ictal headaches.1–3,5
The temporal association between headache including migraine and epileptic seizure is a complex phenomenon that has been a subject of debate. 6 The ICHD-3 has clearly defined migraine with aura-triggered seizures also referred to as migralepsy, as a typical epileptic seizure occurring during or within one hour after an attack of migraine with aura which is not accounted for by an alternative diagnosis.5,6 This phenomenon is distinct because the migraine aura is usually characterized by visual disturbances or sensory changes, and precedes the seizure, which is often focal and commonly originates in the occipital lobe. 6 The underlying mechanism involves cortical spreading depression (CSD) triggering abnormal neuronal activity, leading to a seizure, with additional roles of glutamate excitotoxicity and altered GABAergic inhibition.2,4 Patients with migralepsy may also experience a post-ictal migraine, further complicating differentiation from occipital epilepsy or epileptic headaches. 6 However pre-ictal headache refers to a non-migrainous headache that occurs before the onset of a seizure, typically lasting from minutes to hours.5,6 Clinically, pre-ictal headaches can serve as a warning sign for seizures and should be distinguished from inter-ictal headaches and migralepsy. 6 In contrast, post-ictal headache occurs within three hours after an epileptic seizure, usually resolving spontaneously within 72 h following seizure termination. 6 It is more commonly observed after generalized tonic-clonic seizures and can present as a migraine-like or tension-type headache. 6
There have been over 30 reported cases of IEH which have affected both sexes equally of all age groups.3,4 Herein we report a case of IEH which resembled a TN phenotype, secondary to a cavernous haemangioma in the insular cortex which was successfully treated with resective epilepsy surgery.
Patient information
A right-handed 44-year-old previously healthy woman was referred for the evaluation of treatment-resistant TN for 10 years. She had episodic right facial numbness affecting her teeth and right cheek, followed by an electric shock-like painful sensation of severe intensity in the same area with a salty taste in the mouth, hypersalivation, right facial and neck twitching (which was initially attributed to pain) lasting for up to 10 s. She had 4–5 attacks per day with preserved awareness throughout despite taking oxcarbazepine 750 mg bid, topiramate 25 mg bid and gabapentin 100 mg tid. She had no past history of epilepsy or other co-morbidities. She had no family history of chronic headaches or epilepsy. She had not undergone any neuroimaging previously for her treatment-refractory TN, as she was managed in a smaller peripheral hospital without access to magnetic resonance imaging (MRI) (Figure 1).

Timeline of our patient's care.
Clinical findings
On neurological examination, she had no focal neurological deficits. The rest of her systemic examination was unremarkable.
Diagnostic assessment
A 3-T MRI of the brain revealed a 1.4*1.15-centimeter size well-defined lesion with surrounding haemosiderin ring and internal pop-corn like appearance suggesting a cavernous haemangioma in the right middle and posterior short gyri of the insular cortex (Figure 2). She had continuous scalp EEG recording and video telemetry monitoring for over 24 h. The routine awake EEGs with hyperventilation and photic stimulation did not show any interictal epileptiform discharges. Prolonged video EEG telemetry was undertaken with a full array of 10–20 scalp electrodes and the later addition of centro-parietal electrodes in 10–10 positions after examining the first recorded seizure. This was a continuous in-hospital recording covering wakefulness and overnight sleep. No definite interictal epileptiform discharges were detected in the video EEG telemetry record either. However, four typical events were captured.

Magnetic resonance imaging (MRI) brain sequences (A) T2, (B) T1 and (C) SWI images show right insular well defined lesion with surrounding haemosiderin ring and internal pop-corn like appearance suggesting a cavernoma.
Consecutive EEG epochs during one of the events are shown in Figure 3(a)–(d). This event, starting in sleep shows a K complex followed by right fronto-centro-parietal changes with on-going fast activity becoming sharper, higher in amplitude, and later slowing in frequency. This is followed by theta activity in the same distribution. The patient awakens and reports headache about 15 s after the onset of the EEG changes. The EEG at this point shows myogenic and movement artifacts. However, right-sided theta activity is still visible. The headache lasts approximately 17 s. Finally, with the termination of headache, there is delta range activity in the right-sided leads, however, it is uncertain whether these are postictal changes or movement artifacts.
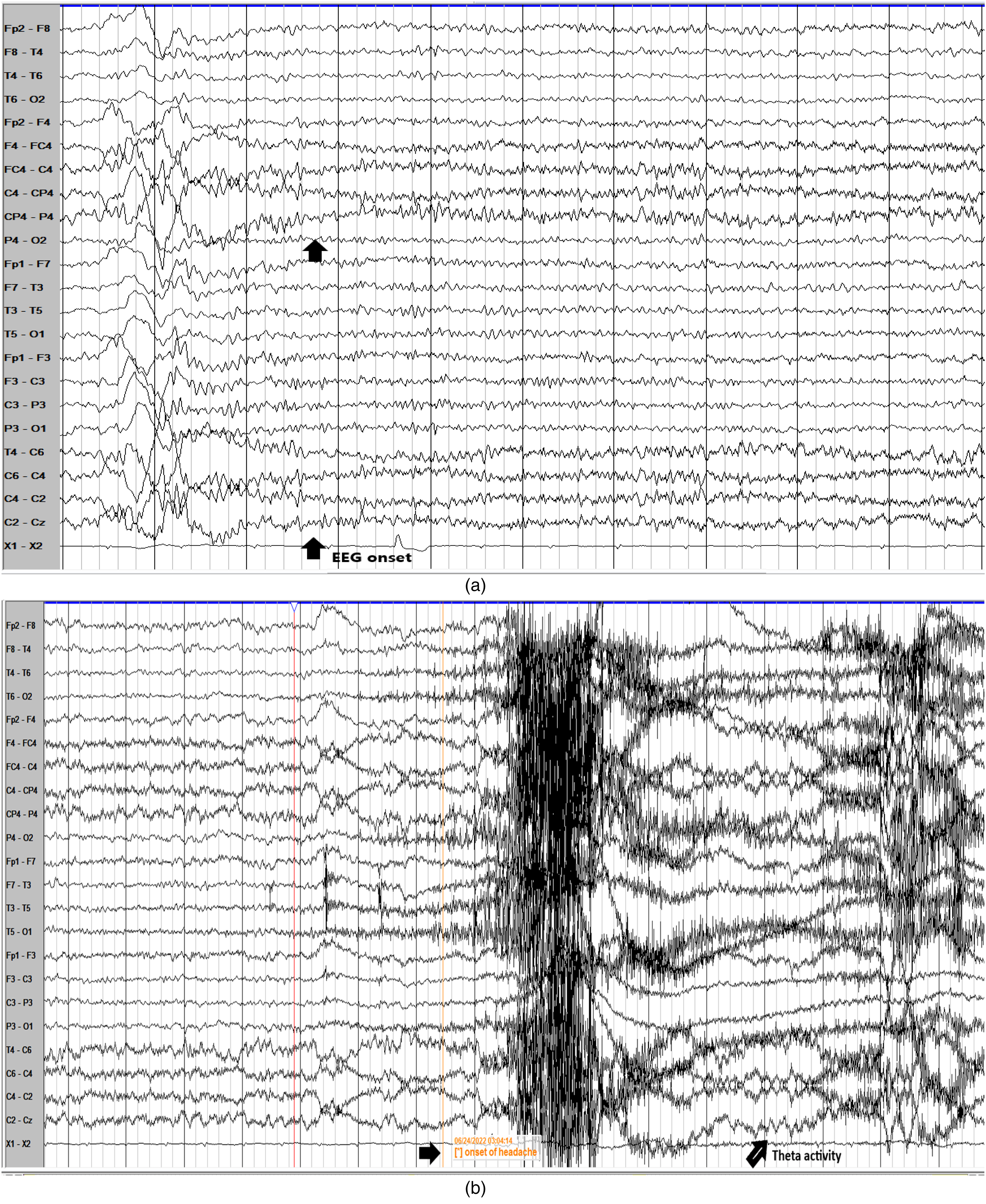
(a)–(d) Consecutive epochs of scalp video electroencephalogram (EEG) telemetry during a headache event showing electrographic seizure discharge lateralized to right side.
Therefore, her neuroimaging and neuro-electrophysiological studies were concordant with the diagnosis of IEH, as right-sided facial pain caused by the right insular cortical cavernous haemangioma.
The patient underwent a functional MRI which demonstrated left hemispheric language activation, suggesting that if the patient were to undergo surgery, the language function would be preserved.
Therapeutic intervention
As the patient fulfiled the criteria for drug-resistant epilepsy, she underwent standard lesionectomy of the cavernous haemangioma, including the surrounding haemosiderin ring over the right insular cortex. Post-operatively she developed left-sided hemiplegia due to an acute lacunar infarction in the right corona radiata. Her limb motor power improved markedly with intense rehabilitation.
Follow-up and outcomes
She remains free of painful seizures 2-year following surgery and takes levetiracetam 500 mg bid together with carbamazepine 200 mg bid. We have initiated a slow taper of the anti-seizure medications. She is independent in her activities of daily living.
Discussion
The ICHD-3 diagnostic criteria for IEH prioritize temporal and EEG characteristics, specifically requiring ictal EEG changes coinciding with headache episodes and their resolution with treatment.1,5,7 However, the criteria lack descriptions of the headache's intensity, localization or associated symptoms, making it challenging to distinguish IEH from primary headache disorders, particularly when the headache is the sole or predominant symptom. This limitation is evident in cases like ours, where IEH mimicked treatment-refractory TN, a rare clinical presentation.
Our patient's symptoms, including right-sided facial pain with a gustatory aura, hypersalivation, and orofacial twitching were initially diagnosed as TN. However, the combination of ictal EEG findings and neuroimaging revealing a cavernous haemangioma in the right insular cortex provided diagnostic clarity. The synchronization of ictal discharges with headache episodes was pivotal in diagnosing IEH, especially when pharmacological treatments failed, as it confirmed the epileptic origin of the patient's symptoms.2,3,5,7 The resolution of her symptoms post-surgery further affirmed the seizure-related nature of her pain.
The insula's role in pain modulation and its extensive connections with the thalamus, anterior cingulate cortex, primary (SI) and secondary somatosensory (SII) areas make it a key hub in the cortical pain network.1,8,9 This region processes nociceptive input and integrates sensory, emotional, and autonomic information.2,3,8,9 Lesions or epileptogenic activity in the mid-posterior insula, where somatosensory representation is highly organized, can produce pain syndromes resembling TN. Our patient's lesion was located more anteriorly in the insula thus the symptoms were more likely due to ictal propagation to this area given its proximity.8–10 Furthermore, our patient demonstrated an ipsilateral painful aura. Somatosensory auras originating from the primary somatosensory area (SI) tend to involve discrete parts of the body contralateral to the seizure focus. 11 The second somatosensory area (SII) can give rise to similar symptoms that are bilateral or ipsilateral to the seizure focus. Due to the widespread connections between the mid-posterior insular and SII, symptoms can be ipsilateral as in our patient. 11
Functional studies have shown that stimulation of mid-posterior insular evokes electric shock-like pain localized to the face, mirroring our patient's clinical presentation.8,9
Gustatory symptoms, hypersalivation, and orofacial motor signs are hallmarks of mid-dorsal insular epilepsy.8,9 These features localized the epileptogenic focus in our patient, further supported by MRI findings and ictal EEG data.8,10 Such symptoms underscore the specificity of the insular cortex in generating not only pain but also viscerosensory, autonomic and motor phenomena, which are critical for diagnosing epilepsy syndromes like IEH.8,9
The literature has revealed that most IEH cases are secondary to focal brain pathology.2,12 Interestingly, our patient uniquely satisfies IEH criteria and partially fulfils classical TN criteria as defined by the ICHD-3, where the latter fulfils criterion B but partially satisfies criterion A. Out of 30 previous case reports, only two cases reported by Charlesworth et al. 12 and Miro and Ortiz 13 described lesional epilepsy with a TN phenotype. 2 Charlesworth et al. 12 partially fulfils the characteristics of TN which was attributed to a lesion of the left fronto-parietal region. This was similar to our case as both were lesional epilepsies, however our patient's symptoms were distinctly linked to a right insular cavernous haemangioma identified on MRI, with ictal EEG showing concordant ictal discharges. The comparison Table 1 highlights similarities and differences between our patient with the aforementioned cases of focal epilepsy mimicking TN phenotype. Our patient achieved complete resolution of symptoms following surgical resection, an outcome reported only once in a previous case by Miro et al.12–14 Although the case titled trigeminalepsy had some similarities to our patient in terms of seizure propagation and transit through the same cortical pain network, it did not specify the site of the epileptogenic focus which further validates our case.12,13
Comparing our patient's clinical features with that of a patient I, case reported by Charlesworth et al. and patient II case reported by Miro and Ortiz.

EEG: electroencephalogram; MRI: magnetic resonance imaging.
As there was a failure to control our patient's seizures with adequate or tolerated doses of 2 or more anti-seizure medications, she fulfiled the criteria for drug-resistant epilepsy as per the ILAE definition. 15
Surgical intervention is a well-established treatment for refractory insular epilepsy but carries inherent risks due to the proximity of the insula to critical vascular and functional structures.
The lenticulostriate arteries, which supply the insular region, are particularly vulnerable during resection, as seen in our patient, who developed post-operative hemiplegia secondary to a lacunar infarction. 8 This complication highlights the surgical challenges in this area. However, with intensive rehabilitation, our patient regained motor function and achieved complete seizure and pain resolution.8,12,14,16
The surgical outcomes for insular epilepsy vary depending on factors such as completeness of resection, precise localization of the epileptogenic focus, and the use of advanced techniques to minimize complications.9,14,16 Long-term follow-up studies indicate that 50–70% of patients achieve seizure freedom (Engel Class I outcomes), with early post-operative seizure freedom being a strong predictor of sustained success.8,16 The resolution of our patient's symptoms over a 2-year follow-up aligns with these findings and supports the efficacy of surgical intervention in well-selected cases.
Conclusion
This case underscores the importance of recognizing IEH in patients presenting with refractory headache syndromes, especially those exhibiting atypical features. While the rarity of IEH and its overlap with other headache disorders can delay diagnosis, early consideration of IEH, along with EEG and neuroimaging, can guide effective treatment. The unique role of the insular cortex in integrating sensory, autonomic and motor functions further highlights its centrality in such cases, with targeted surgical interventions offering the potential for complete resolution.1,2,5,8
Clinical implications
IEH may be misdiagnosed as treatment-resistant TN, delaying specific treatment.
Neuroimaging and EEG are essential for diagnosing IEH and distinguishing it from primary headache disorders.
Resective epilepsy surgery can effectively treat lesional IEH, ensuring seizure and pain remission.
Insular cortex epilepsy can produce pain syndromes, which may mimic primary headaches such as TN.
Footnotes
Acknowledgements
The reported patient provided full consent for publication of her clinical history and imaging.
Declaration of conflicting interests
The authors declared no potential conflicts of interest with respect to the research, authorship, and/or publication of this article.
Funding
The authors received no financial support for the research, authorship, and/or publication of this article.
Informed consent
The reported patient provided full consent for publication of her clinical history and imaging.