
Abstract
Background/Hypothesis:
Atogepant, an oral calcitonin gene-related peptide receptor antagonist, is approved for the preventive treatment of migraine in adults. This manuscript characterizes the safety, tolerability, pharmacokinetics and pharmacokinetic/pharmacodynamic relationship of atogepant in healthy males.
Methods:
Data from two single-ascending dose phase 1 studies of atogepant were utilized to characterize pharmacokinetics and demonstrate proof of activity of atogepant in a capsaicin-induced dermal vasodilatation model and to determine the dosage(s) that results in 90% inhibition of capsaicin-induced dermal vasodilatation (effective concentration, EC90) over 24 hours.
Results:
Single (0.4−200 mg) doses of atogepant were generally well tolerated by healthy participants with no treatment-related study discontinuations. Atogepant was rapidly absorbed with peak plasma concentrations occurring 1–2 hours post dose and a mean elimination half-life of ∼11 hours. Based on the capsaicin-induced dermal vasodilatation and pharmacokinetic/pharmacodynamic models, atogepant has an estimated EC90 of 13.6 nM which was reached within 30 minutes at therapeutic doses and maintained for 24 hours at dosages of 60 mg once daily and 30 and 60 mg twice daily.
Conclusion/Interpretation:
Atogepant reached effective concentrations within 0.5 hours which were maintained for 24 hours at dosages of 60 mg once daily and 30 and 60 mg twice daily for the prevention of migraine.
Trial Registration:
Clinical Trial EudraCT Numbers: 2011-005020-18 (Study 1) and 2012-001192-36 (Study 2).
Keywords
Introduction
Calcitonin gene-related peptide (CGRP), a potent vasodilating neurotransmitter, was first discovered in 1982. 1 The subsequent 20 years of research on CGRP enabled our understanding that migraine is a neurological disease. Experiments demonstrating that migraine-like symptoms can be induced through intravenous infusion of CGRP and elevated levels of CGRP observed during a migraine attack can be normalized through antagonism of the CGRP pathway were key to linking CGRP to the pathophysiology of migraine. 2 –5 Since then, the CGRP neuropeptide and receptor have become validated targets for acute and preventive treatment of migraine attacks yielding the discovery and development of a diverse range of therapeutic agents. 6 The early small molecule CGRP receptor antagonists (i.e., gepants) were discontinued due to hepatic safety signals and formulation challenges. 7 –9 The development of the early gepants was therefore followed by the development of monoclonal antibodies (mAb) targeting the CGRP receptor or CGRP as a ligand. 10,11 In recent years, the development of oral gepants has been guided by their pharmacological properties that enable fast acting pain relief along with sustained prevention from migraine attacks and safety profiles that enable long-term use of the medication. With the approval of four mAbs (i.e., erenumab, galcanezumab, fremanezumab, and eptinezumab) and three oral gepants (i.e., rimegepant, ubrogepant, and atogepant) for either preventive or acute treatment of migraine attacks, a variety of treatment options are now available.
Atogepant (QULIPTA™), the third oral gepant to market, was recently approved by the US Food and Drug Administration, Health Canada, and Israel for the preventive treatment of migraine. Atogepant demonstrated statistically significant reductions in mean monthly migraine headache days across the 12-week pivotal clinical studies. 12,13 A 52-week long-term extension study demonstrated the long-term safety and tolerability of atogepant. Here, we report the data from two single-ascending dose (SAD) phase 1 clinical studies of atogepant evaluating its safety, tolerability, pharmacokinetics (PK), and pharmacodynamics (PD) utilizing a capsaicin-induced dermal vasodilation (CIDV) model. 14,15 With these data, an integrated population PK/PD model was developed to identify the dosage(s) of atogepant required to provide the targeted CGRP receptor coverage (90% effective concentration, EC90) for phase 2 clinical studies.
Methods
Ethics
Studies were conducted in accordance with Good Clinical Practice guidelines and the ethical principles that have their origin in the Declaration of Helsinki. All participants provided written informed consent before any study-related procedures were performed. The protocol and informed consent forms received approval from The Ethics Committee Research UZ/KU Leuven, Leuven, Belgium (Study 1) and the Ethics Committee Universitair Ziekenhuis Gent, Gent, Belgium (Study 2).
Study designs
Two randomized, double-blind, placebo-controlled phase 1 studies were conducted. Study schematics are shown in Figure 1. Study 1, the first-in-human study for atogepant, consisted of a SAD (Part I) and a PD study (Part II). This study initiated on December 12, 2011 and completed on May 25, 2012. In Part I, participants in two panels received alternating single rising oral doses of atogepant or placebo in five treatment periods with a minimum 7-day washout between periods. In each period, participants were randomized 6:2 to receive active drug or matching placebo after an overnight fast at doses of 1, 2.5, 5, 10, 20, 40, and 50 mg. In Study 1, atogepant was prepared as an oral solution at appropriate concentrations at the clinical site pharmacy using bulk supplied excipients (polyethylene glycol [PEG] 400, purified water, and Ora-Sweet® or Ora-Sweet® SF; see Figure 1 for more details). In Part II, a CIDV model was utilized to evaluate the effects of single doses of atogepant on dermal vasodilatation induced by capsaicin application in 16 participants in a four-period, crossover design. Participants received single doses of atogepant 0.4 mg, 2.5 mg, and 30 mg or placebo followed by a single topical dose of capsaicin solution on the volar surface of each forearm in each period. Further details on the PD assessment are described in the assessments and analytical methods section.
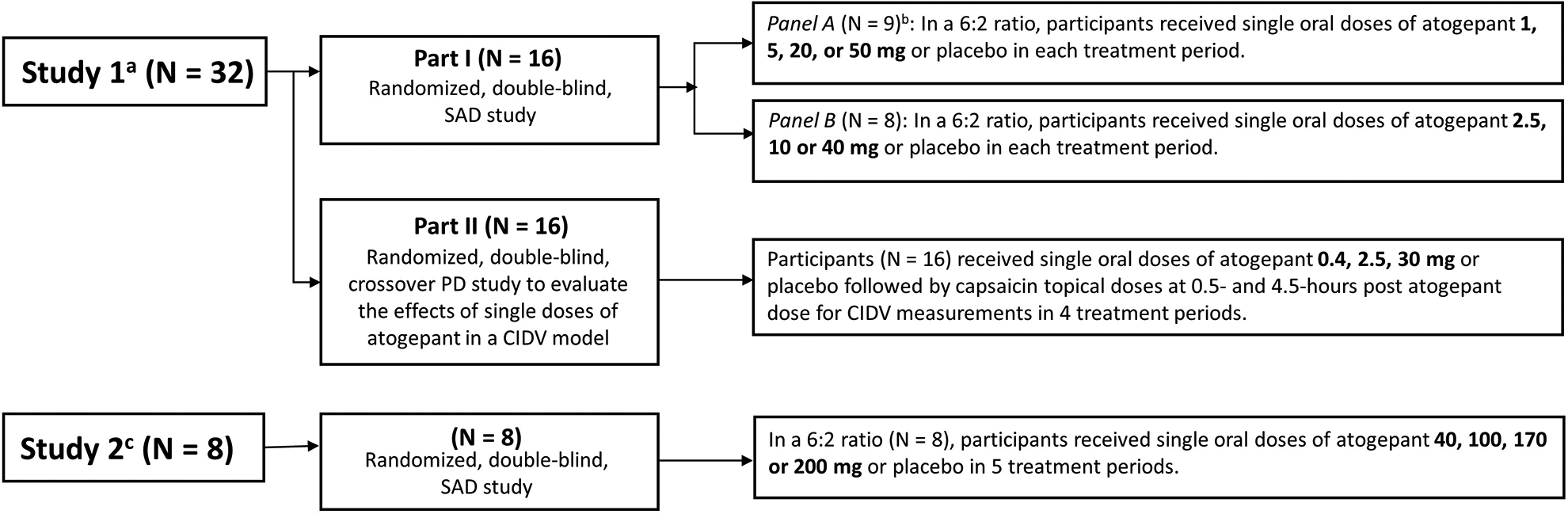
Atogepant phase 1 clinical study schematics and participant flow. Note: In Study 1 Part I, the 5 mg dose was assessed in Periods 2 and 5 in Panel A to allow for assessment of intra-participant variability in pharmacokinetics at the predicted clinical dose. In Panel B, the 10 mg dose was administered in Periods 2, 4 and 5. In Period 2, the 10 mg dose was administered as 10 mL of a 1 mg/mL solution followed by a 10 mL PEG vehicle wash; in Period 4, the 10 mg dose was administered as 10 mL of a 1 mg/mL solution with no PEG vehicle wash; and in Period 5, the 10 mg dose was administered as 0.5 mL of a 20 mg/mL solution with no PEG vehicle wash. CIDV: capsaicin-induced dermal vasodilation; N: number of participants; PEG: polyethylene glycol; PD: pharmacodynamic; SAD: single-ascending dose. aAtogepant was administered in Study 1 as an oral solution. bOne participant withdrew from the study for personal reasons following single doses of atogepant 1 mg and 5 mg in Periods 1 and 2 and was replaced with a new participant. cAtogepant was administered in Study 2 as an oral compressed tablet.
Study 2 consisted of an SAD study in which atogepant or placebo was administered as an oral compressed tablet (OCT) in combinations of 0, 10, or 50 mg tablets (see Figure 1). This study initiated on May 22, 2012 and completed on November 13, 2012. Participants received single rising oral doses of atogepant or placebo in a 6:2 ratio in five treatment periods with a minimum 7-day washout between periods. Doses included 40, 100, 170, and 200 mg and were administered after an overnight fast in Periods 1, 2, 3, and 5. Period 4 evaluated food effect and is not reported here. 16 In all periods, a different pair of participants was assigned to receive placebo.
Participants
For both studies, males aged 18−50 years, nonsmokers for at least 6 months, with body mass index ≤33 kg/m2 for Study 1 and ≤30 kg/m2 for Study 2, and in good health were eligible. Key exclusion criteria included history of clinically relevant medical conditions, estimated creatinine clearance ≤80 mL/min based on the Cockcroft–Gault equation, and requirement for concomitant medication. For the CIDV assessments, participants were required at screening to demonstrate an increase in forearm dermal blood flow of 100% or more after capsaicin application. Developmental and reproductive toxicity studies had not yet been conducted at the time of study initiation; therefore, only male participants were enrolled.
Assessments and analytical methods
Pharmacokinetics
In SAD studies, blood samples for PK were collected before dosing and at 20 min, 40 min, and 1-, 1.5-, 2-, 3-, 4-, 6-, 8-, 12-, 16-, 24-, 36-, 48-, and 72-hours after drug administration on Day 1 of each period. In the PD study, samples were collected predose and at 0.5-, 1-, 1.5-, 2-, 3.5-, 4-, 6-, 8-, and 24-hours post dose in each period. Plasma samples were generated and transferred to Merck Research Laboratories (West Point, Pennsylvania) on dry ice for atogepant quantification utilizing a liquid–liquid extraction for analyte isolation followed by liquid chromatographic-tandem mass spectrometric detection. 17 The lower limit of quantitation for the plasma assay was 0.1 ng/mL (0.1657 nM) when 200 µL of plasma was processed. Quantitation for the wide range of sample concentrations was obtained through cross validation of two linear calibration curves ranging from 0.1 to 100 ng/mL (0.1657 to 165.7 nM) and 1 to 1000 ng/mL (1.657 to 1657 nM). Intra- and inter-assay variability for the analyses herein were both <6% for Study 1 and <6% and <3%, respectively, for Study 2.
Pharmacodynamics
Pharmacodynamic measurements were obtained in all periods of Study 1 Part II (see Figure 1). Assessments of the CIDV were conducted prior to the study for participant inclusion, prior to atogepant/placebo dosing, and 1 and 5 hours post-atogepant/placebo administration. Participants received single topical doses of 300 µg/20 µL and 1000 µg/20 µL capsaicin (in ethanol/polysorbate 20/water [3:3:4]) in 10-mm rubber “O”-rings at two sites on the volar surface of participants’ left and right forearms as per a computer-generated allocation schedule. Laser Doppler scans were performed just prior to and 30 min following each capsaicin application.
Safety
Safety was monitored throughout both studies with clinical and laboratory evaluations including collection of adverse events (AEs), physical examination, vital signs, 12-lead electrocardiogram (ECG), and laboratory safety data. Predose liver function tests (LFTs) were conducted in both studies and reviewed prior to dosing. LFTs were also conducted after dosing was completed in the post-treatment follow-up (PTFU) period of both studies at 2 weeks, 1 month and 2 months. In Study 1, Columbia Suicide Severity Rating Scale and Columbia–Classification Algorithm of Suicide Assessment was also performed. Safety evaluations were performed at poststudy visits in the PTFU period for all participants in both studies. Any transaminase elevations were monitored until values were restored to within normal limits.
Pharmacokinetic analyses
For all PK analyses, atogepant plasma concentrations and actual post-dose sampling times were used to determine PK parameter values. Plasma concentration values below the assay limit of quantitation (BLOQ) were replaced with zero. PK parameters were calculated by non-compartmental analyses using the software Phoenix WinNonlin® Professional (Version 6.3) and included maximum and minimum observed plasma concentration (Cmax and Cmin, respectively), observed plasma concentration at 12- and 24-hours post dose (C12 hr and C24 hr, respectively), and the time to Cmax (T max). Area under the plasma time–concentration curve from zero to infinite time (AUCinf) was calculated using the linear trapezoidal method for ascending concentrations and the log trapezoidal method for descending concentrations. For each participant, terminal elimination rate constant (λz) was calculated by regression of the terminal log-linear portion of the plasma concentration-time profile, and the apparent terminal half-life (t 1/2) was calculated as the quotient of the natural log of 2 (ln[2]) and λz. At least three data points (excluding the Cmax) in the terminal phase were used for λz calculations. Dose proportionality of atogepant AUC and Cmax was explored through assessments of slopes from a power law model of observed PK data and dose. All available data were used for the analysis; no imputations were made for missing data.
Population pharmacokinetic model
A population PK analysis of atogepant based on data from five clinical studies (two studies [Studies 1 and 2] included in this publication and three other phase 1 studies [Studies 3–5], see Table S1) was developed using NONMEM® (version 7.2, ICON, Hanover, MD, USA). Three single dose and two multiple dose studies were included in this analysis. Various covariates of potential clinical interest (e.g., body weight, age, gender, race, pretreatment with famotidine, and drug formulation) were tested during model building in order to assess their respective impacts on atogepant exposure. The covariate selection was performed using a forward addition process followed by backward deletion (i.e., stepwise covariate method [SCM] search). The likelihood ratio test was used to evaluate the significance of incorporating or removing fixed effects into the population model. For forward selection and backward deletion, significance levels of 0.01 and 0.001 were employed, respectively. The final model was assessed through goodness of fit plots and prediction-corrected visual predictive checks. The final population PK model was comprised of first-order oral absorption of atogepant with an absorption lag time followed by two-compartment disposition.
Pharmacodynamic analyses
For CIDV analyses, least squares geometric means (GM) and 95% confidence intervals (CI) were calculated for each treatment, and mean perfusion values for capsaicin response were analyzed for each time point (1- and 5-hours post dose). Individual mean perfusion values were natural-log transformed and analyzed in a linear mixed effects model including fixed effects for period, treatment, panel, and site and a random effect for participant.
A test for first-order carryover was conducted at the 0.10 level and found to be statistically significant for the 1000 µg/20 µL capsaicin dose at both time points. Therefore, the final models for 1000 µg/20 µL capsaicin concentration included an adjustment for carryover by including three additional continuous factors (sum-to-zero parameterization) in the model.
For each atogepant dose level, a two-sided 90% CI (equivalent to a one sided upper 95% CI) for the true mean difference (atogepant–placebo) in log-scale capsaicin response was computed from the mixed effects model using the mean squared error and referencing a t-distribution. The CI was back transformed to obtain a GM and corresponding CI for the true treatment mean ratio in capsaicin response. The resulting GM ratio and corresponding confidence limits were then transformed to percent inhibition using the following formula: %inhibition = (1 − [GM ratio]) × 100%.
Pharmacodynamic and PK/PD models
A previously established and validated PK/PD model for CIDV 15 was expanded to incorporate data from five studies conducted for different compounds (i.e., MK-3207, 14 ubrogepant, 15 MK-2295, 14 telcagepant, 18 and atogepant; see Table S2) and used to evaluate the relationship between inhibition of CIDV and atogepant plasma concentration, and specifically, to estimate the EC90 of atogepant. The capsaicin-related increase in blood flow was described by an Emax model with competitive interaction between capsaicin and MK-2295. The effect of CGRP antagonists (MK-3207, telcagepant, ubrogepant, and atogepant) was included in the model as an additive reduction in blood flow related to CGRP receptor antagonist concentration by an Emax model. The final model structure is described by F = F0 + Fcaps • [1 − Emax • C/(EC50 + C)], where F is the observed blood flow measured by laser Doppler imaging, F0 the baseline blood flow, Fcaps the incremental blood flow due to application of capsaicin, Emax the maximal inhibition of capsaicin-induced blood flow by atogepant, C the plasma concentration of atogepant, and EC50 the plasma concentration of atogepant corresponding to 50% inhibition of the maximal CIDV effect.
The model was fit using NONMEM® VII (ICON Development Solutions, Dublin, Ireland) using a first-order conditional model with interaction. Parameter estimates for the PK/PD model are provided in Table S2. Model qualification was performed with model diagnostic plots to illustrate goodness of fit, while accuracy and robustness of the model was assessed by visual predictive checks and a non-parametric bootstrap analysis. The integrated population PK/PD model was utilized to perform simulations of dosing scenarios of 10, 30, and 60 mg QD as well as 30 mg and 60 mg BID and compared against the CIDV EC90 benchmark.
Results
Participant demographics and disposition
Participant demographics for both studies are summarized in Table 1. Study 1 was conducted from December 12, 2011 through May 25, 2012 with the first and last participant visit, respectively. In Study 1, 33 white males were enrolled into the study, and 32 completed the study. One participant in Part I Panel A withdrew for personal reasons after completing Periods 1 and 2 (1 mg and 5 mg atogepant, respectively) and was replaced with a new participant who began dosing in Part I Panel A Period 2 and completed the study. One participant in Part II did not complete Periods 3 and 4 due to a confirmed cytomegalovirus infection (see Safety section below). Study 2 was conducted from May 22, 2012 through November 13, 2012 with the first and last participant visit, respectively. In Study 2, all eight enrolled participants completed the study. The PK and safety analyses included all available data from all participants in each study.
Demographics of atogepant phase 1 studies in healthy adult males.
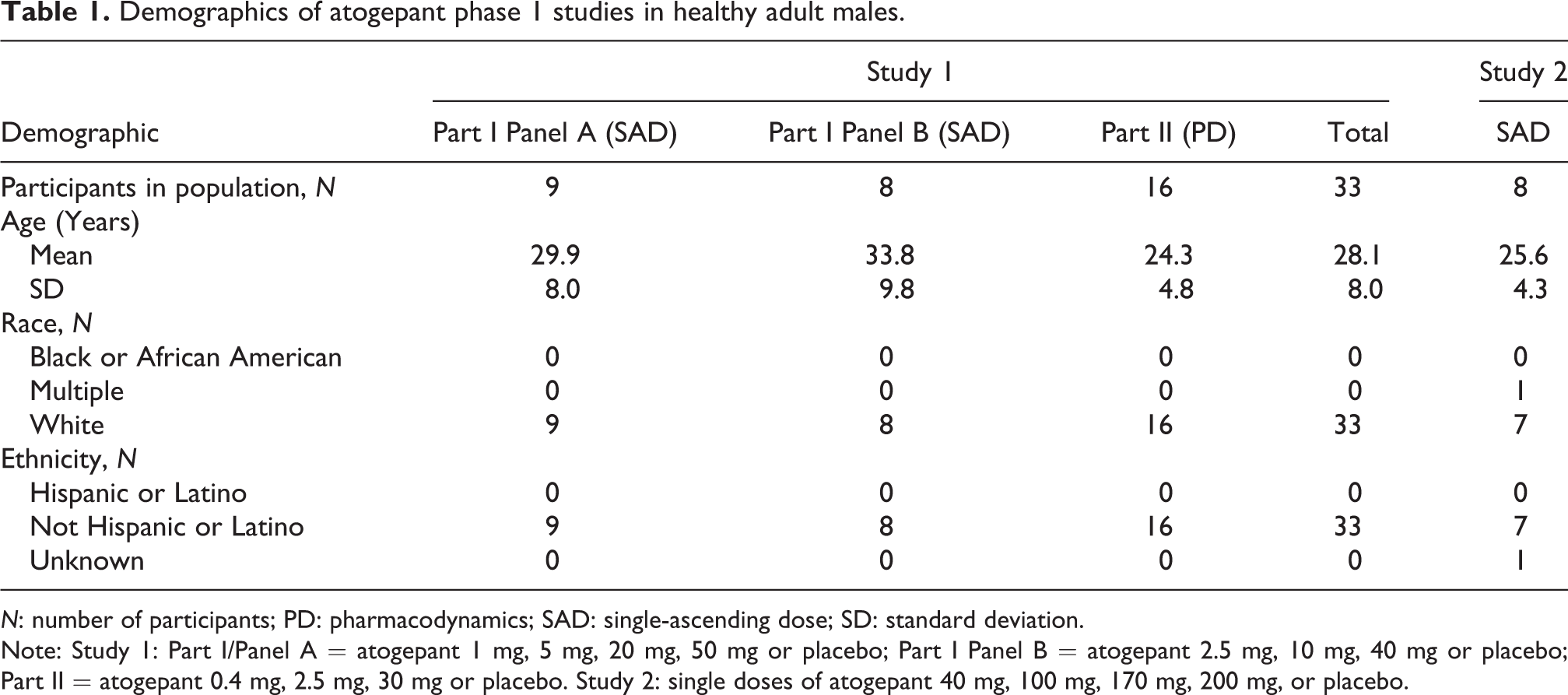
N: number of participants; PD: pharmacodynamics; SAD: single-ascending dose; SD: standard deviation.
Note: Study 1: Part I/Panel A = atogepant 1 mg, 5 mg, 20 mg, 50 mg or placebo; Part I Panel B = atogepant 2.5 mg, 10 mg, 40 mg or placebo; Part II = atogepant 0.4 mg, 2.5 mg, 30 mg or placebo. Study 2: single doses of atogepant 40 mg, 100 mg, 170 mg, 200 mg, or placebo.
Safety
In both studies, single doses of atogepant were generally well tolerated in healthy participants. There was no evidence of suicidality or suicidal ideation, no participant discontinued because of an AE, no treatment-related clinically significant abnormalities were found in routine clinical (i.e., physical examination, vital signs, ECG) or laboratory assessments, no case met the criteria for potential Hy’s law, 19 and there were no SAEs in either study.
Study 1
Thirty out of 33 participants reported a total of 93 treatment-emergent AEs (TEAEs) of which 20 TEAEs were deemed drug related and reported by 11 participants during the treatment period; all TEAEs were rated as mild (N = 87) or moderate (N = 6). The most frequent TEAEs reported were nasopharyngitis and headache. The most common drug-related TEAEs in participants receiving atogepant during the treatment period were headache (N = 5 participants), postural dizziness (N = 2 participants), and dizziness (N = 2 participants). Of the two participants with postural dizziness, each episode started 2 hours after receiving a 30 mg dose of atogepant and were mild with one lasting for 1 minute and the other for 2 hours with no significant changes in vital signs. The other drug-related TEAEs were reported by individual participants receiving atogepant and included nausea and vomiting. Drug-related TEAEs among participants receiving placebo during the treatment period were headache (N = 4 participants), diarrhea (N = 1 participant), feeling warm (N = 1 participant), and postural dizziness (N = 1 participant). There were no meaningful changes in LFTs related to treatment; there were no post-dose elevations of transaminases >1.5× upper limit of normal (ULN) except for one participant who had maximum alanine aminotransferase (ALT) and aspartate aminotransferase (AST) elevations of 2.1× and 1.7× ULN, respectively, after receiving 2.5 mg atogepant which was deemed by the investigator to be related to a confirmed cytomegalovirus infection and therefore probably not drug related.
Study 2
Seven out of eight participants reported a total of 33 TEAEs of which 12 were deemed drug related and reported by five participants during the treatment period; all TEAEs were reported as mild (N = 25) or moderate (N = 8). The most common TEAEs reported were nasopharyngitis and oropharyngeal discomfort; headache was reported by two participants in the placebo group. The most frequent drug-related TEAEs were mild postural orthostatic tachycardia syndrome (N = 1 participant, 4 occurrences) and orthostatic hypotension (N = 1 participant, 3 occurrences [two rated as mild and one rated as moderate]). These drug-related TEAEs resolved spontaneously the same day. The other five drug-related TEAEs were single occurrences reported by individual participants (three of whom received placebo during the treatment period) and included oropharyngeal discomfort, back pain, postural dizziness, hot flush, and fatigue.
Pharmacokinetics
Study 1: Atogepant oral solution single dose PK
Following single dose administration of atogepant in fasting healthy adult participants, atogepant was rapidly absorbed with median T max ranging from 1.0 to 2.0 hours (Table 2). The plasma concentration–time profile declined generally biexponentially post-Cmax with an apparent half-life of ∼10 to 16 hours in the dose range of 10 mg to 50 mg (see Figure 2). Both AUCinf and Cmax appeared to increase roughly dose proportionally following single oral doses from 1 to 50 mg. Moderate inter-participant variability was observed for AUCinf and Cmax, with %CV ranging from ∼20% to 40%. The pharmacokinetics of atogepant were not significantly affected by coadministration of a 10 mL PEG wash (data on file at AbbVie).
Geometric mean (95% CI) plasma atogepant pharmacokinetic parameters following single oral doses in healthy young adult males.
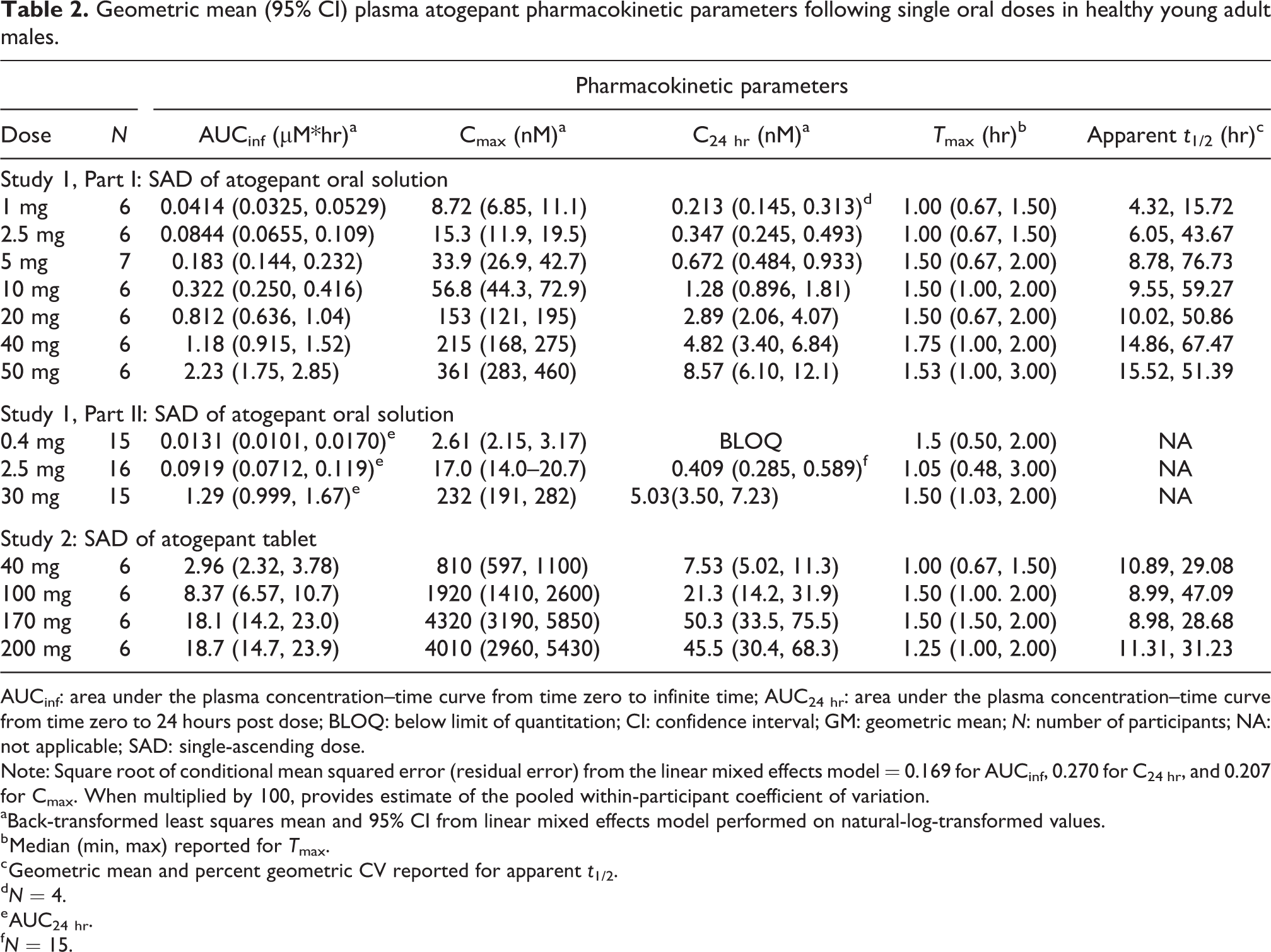
AUCinf: area under the plasma concentration–time curve from time zero to infinite time; AUC24 hr: area under the plasma concentration–time curve from time zero to 24 hours post dose; BLOQ: below limit of quantitation; CI: confidence interval; GM: geometric mean; N: number of participants; NA: not applicable; SAD: single-ascending dose.
Note: Square root of conditional mean squared error (residual error) from the linear mixed effects model = 0.169 for AUCinf, 0.270 for C24 hr, and 0.207 for Cmax. When multiplied by 100, provides estimate of the pooled within-participant coefficient of variation.
aBack-transformed least squares mean and 95% CI from linear mixed effects model performed on natural-log-transformed values.
b Median (min, max) reported for T max.
c Geometric mean and percent geometric CV reported for apparent t 1/2.
d N = 4.
e AUC24 hr.
f N = 15.

Study 1: arithmetic mean (±SD) plasma concentration versus time following administration of single oral doses (solution) of atogepant to healthy young male participants. Data for Part I is shown on a linear scale with an inset (a) and on a semi-log scale (b). Part II is shown on a linear scale (c) and a semi-log scale (d). Note: In Part I, N = 6/group except for the 5 mg dose group, which was N = 7, and in Part II, N = 15/group except for the 2.5 mg dose group, which was N = 16. N: number of participants; SD: standard deviation.
Study 2: Atogepant OCT single dose PK
Atogepant plasma concentration–time profiles following single dose administration of atogepant OCT in fasted healthy participants are shown in Figure 3. Following single dose OCT administration, atogepant was rapidly absorbed with median T max ranging from 1.00 to 1.50 hours and t 1/2 ranging from ∼9 to 11 hours over the 40–200 mg dose range. Systemic exposure of atogepant appeared to increase roughly dose proportionally following single oral doses from 40 to 200 mg (data on file at AbbVie). The plasma PK parameters and summary statistics for Study 2 are summarized in Table 2.
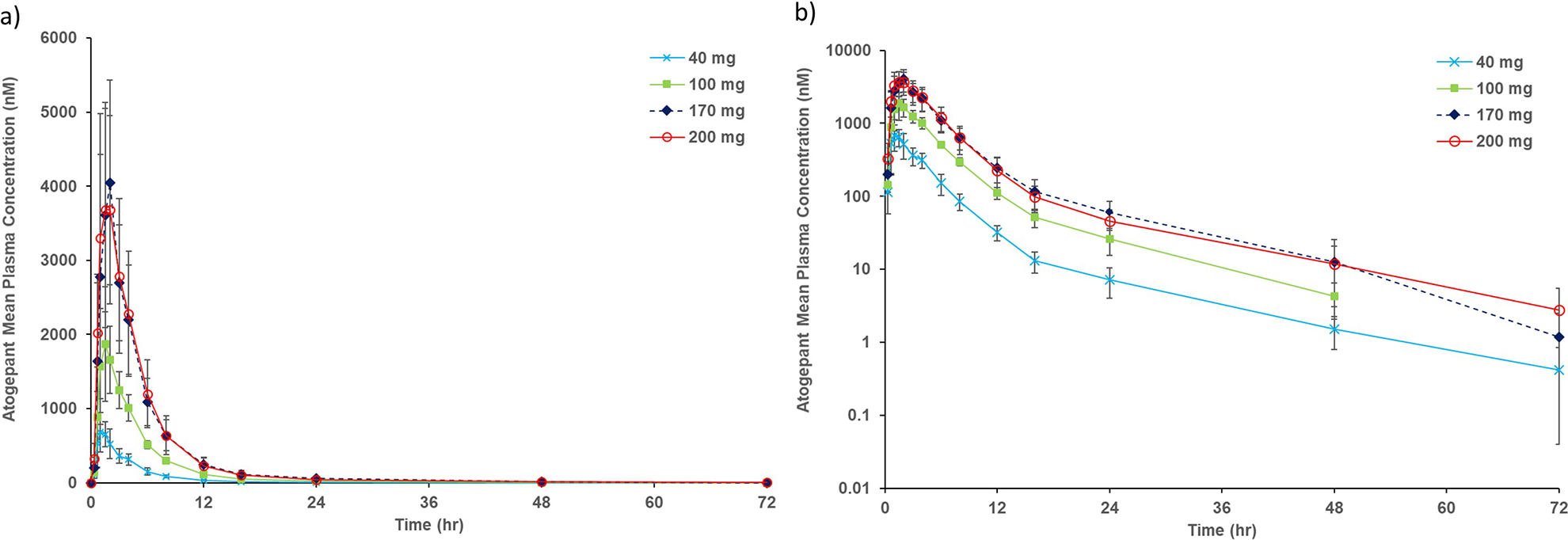
Study 2: arithmetic mean (±SD) plasma concentration versus time following administration of single oral doses (tablet) of atogepant to healthy young male participants. Data for Study 2 (N = 6 per dose group) is shown on a linear scale (a) and a semi-log scale (b). N: number of participants; SD: standard deviation.
PK comparison of atogepant oral solution and OCT formulations
During the course of these two studies, two formulations were employed and included the oral solution used in Study 1 and the OCT used in Study 2. Study 2 tested the 40 mg dose to enable direct comparison of these two formulations. AUCinf, Cmax, and T max were 1.18 µM*hr, 215 nM, and 1.75 hr for the oral solution, respectively, and 2.96 µM*hr, 810 nM, and 1.00 hr for the OCT, respectively. In this case, the solid oral dosage form (OCT) demonstrated higher rate and extent of absorption with increased AUC and Cmax and a shorter median T max. Based on these results, the OCT was chosen for further development of atogepant.
Pharmacokinetic/pharmacodynamic modeling
Summary of capsaicin-induced dermal blood flow (mean perfusion) following single oral administration of atogepant or placebo is shown in Table 3. The estimated EC50 and EC90 values for atogepant for inhibition of CIDV were 1.51 nM and 13.6 nM, respectively, with reasonable precisions. Time above the CIDV EC90 threshold for selected first-dose/steady-state dosing scenarios are shown in Figure 4. Concentrations from the 30 and 60 mg BID regimens show that coverage above the CIDV EC90 are maintained at almost all times after the first dose and at steady state. Similarly, the steady-state profile for 60 mg QD appears to achieve nearly complete CGRP receptor coverage.
Summary of capsaicin-induced dermal blood flow (mean perfusion) following single oral administration of atogepant or placebo.
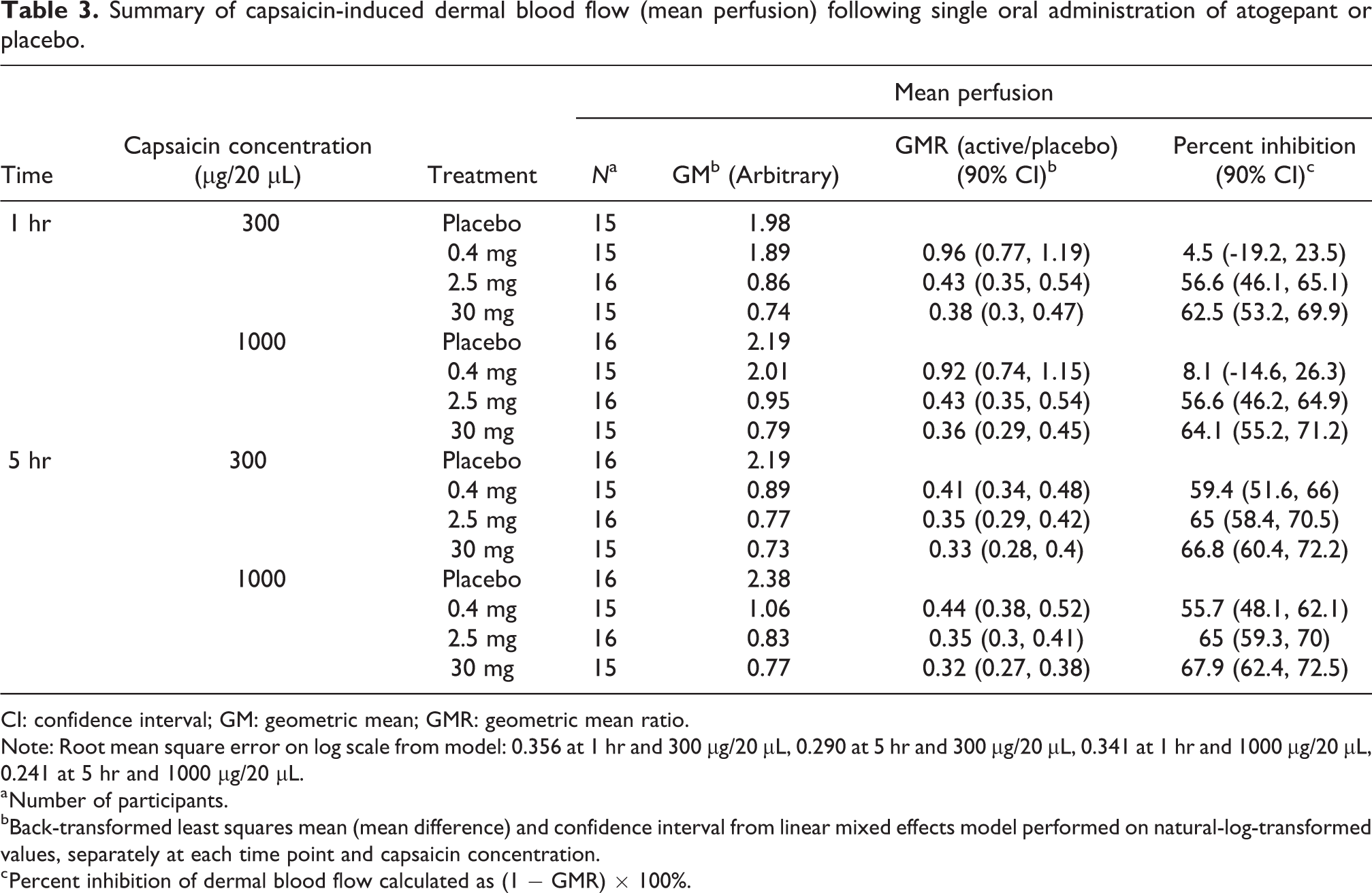
CI: confidence interval; GM: geometric mean; GMR: geometric mean ratio.
Note: Root mean square error on log scale from model: 0.356 at 1 hr and 300 μg/20 μL, 0.290 at 5 hr and 300 μg/20 μL, 0.341 at 1 hr and 1000 μg/20 μL, 0.241 at 5 hr and 1000 μg/20 μL.
a Number of participants.
bBack-transformed least squares mean (mean difference) and confidence interval from linear mixed effects model performed on natural-log-transformed values, separately at each time point and capsaicin concentration.
c Percent inhibition of dermal blood flow calculated as (1 − GMR) × 100%.

Time above the CIDV EC90 threshold for selected single-dose and steady-state dosing scenarios. Dots represent arithmetic mean and lines represent 90% confidence intervals. Concentrations from the 30 and 60 mg BID regimens are maintained above the CIDV EC90 at essentially all times after first dose and at steady state. BID: twice daily; CIDV: capsaicin-induced dermal vasodilation; EC90: 90% effective concentration; QD: once daily; SS: steady state.
Discussion
The primary goals of the phase 1 studies and analyses reported herein were to characterize atogepant safety, tolerability and PK after single doses in young, healthy adult males as well as determine the human EC90 value via PK/PD modeling with inputs from a CIDV model conducted at potential clinically effective dosages. Following single dose administration, atogepant was rapidly absorbed with median T max of 1.0–2.0 hours, and plasma concentrations declined biexponentially post T max with a mean apparent elimination t 1/2 of ∼11 hours at potential clinically relevant doses (note that the t 1/2 was shorter at lower doses because the plasma concentrations were below the lower limit of quantitation in the terminal elimination phase). Systemic exposure (AUCinf, Cmax, and C24 hr) increased approximately dose proportionally following single oral doses of 0.4 mg to 200 mg. Based on the safety, potency and observed half-life of atogepant, once daily dosing is adequate to maintain therapeutic levels for 24 hours.
The FIH study (Study 1) was also designed to evaluate proof of activity through the CIDV model and to characterize the PK/PD of atogepant in order to predict the range of doses anticipated to be efficacious for the preventive treatment of migraine. An established PD model based on data from earlier CGRP receptor antagonists (ubrogepant, MK-3207, and telcagepant) was used to estimate the EC90 for atogepant. Reduction in dermal blood flow as measured in the CIDV assay has been shown to be a predictor of efficacy for CGRP receptor antagonists. 14,15,18,20 –23 Specifically, doses of telcagepant which achieved drug concentrations at or above the EC90 in the CIDV assay were shown to be superior to placebo in measures of migraine pain relief, pain freedom, photophobia, phonophobia and nausea in a phase 2/3 trial. 24 The target plasma EC90 of 13.6 nM for atogepant was attained within 30 minutes at doses of 5 mg and higher. EC90 coverage at steady state over a 24-hour dosing interval for QD doses of 10, 30 and 60 mg atogepant was predicted to be ∼12, 21 and 24 hours, respectively. For preventive treatment of migraine, the primary clinical endpoint is a decrease in the incidence of migraine attacks, often measured as a reduction in the number of headache days per month. Thus, the ∼11-hour t 1/2 of atogepant along with the duration of EC90 coverage during a dosing interval may allow for significant inhibition of the CGRP receptor during the dosing interval with QD administration. Supporting evidence of dosages achieving EC90 was also observed in the phase 3 study of atogepant in which dosages of 10, 30, and 60 mg QD, determined to achieve EC90 concentrations in the work herein, all reduced mean monthly migraine days across the 12 weeks of treatment compared to placebo. 13
The Emax for inhibition of CIDV by CGRP receptor antagonists was estimated to be 92%, suggesting that CGRP is the primary, but not the only, contributor to CIDV. This is consistent with conclusions from previous CIDV studies, and the capsaicin responses observed in this study were generally consistent with those observed historically (similar Emax for capsaicin dose response for vasodilatation in prior studies).
14,15
The CIDV model contributed substantially to the development of both gepants and mAbs targeting the CGRP pathway by providing the dose and concentration of the antagonist needed to fully block CGRP-dependent vasodilation in the forearm. Although it is a very powerful tool that guides the dose selection for efficacy trials in migraine patients, it is primarily a target engagement biomarker and does not necessarily predict the clinically efficacious dose. Indeed, in general, the efficacy data demonstrated that the clinical dose is higher than the model-suggested dose needed to block the CGRP response in the forearm. This should not come as a surprise given the fact that the target tissue in migraine is completely different. Several potential reasons for such underpredictions for mAbs were recently reviewed by Al-Hassany et al.
25
Historically used CIDV models assessed the reduction in capsaicin-induced dermal blood flow in the forearm (far from trigeminal ganglia) of young males by investigational agents. Dose predictions could improve with a forehead (closer to trigeminal ganglia) model. Migraine being more prevalent in females of reproductive age, additional factors, such as age, sex (hormonal changes), and BMI may contribute to the variations in the CIDV predicted (in young males) versus clinically effective doses; however, population pharmacokinetic modeling and exposure-response analyses evaluating demographic covariates have not demonstrated any significance of race, age, or sex on the pharmacokinetics, efficacy, or safety of atogepant.
26
Further, for atogepant, time above the EC90 was relied upon for dose selection; however, the following need to be better understood: Do all CGRP receptors need to be blocked for prevention of migraine? Is continuous 24 h/day blockade of the CGRP receptors required for migraine prevention?
The safety goals of these studies were to evaluate the safety and tolerability of rising single doses of atogepant in young healthy male participants. In general, single doses of atogepant were safe and well tolerated. All AEs were mild or moderate in nature and did not show a trend of increasing incidence with escalating doses. No serious adverse events were reported in either study. Of the AEs possibly related to atogepant, only headache, dizziness, and postural dizziness (without measured changes in vital signs) occurred in more than one participant. Headache and postural dizziness were also recorded in participants receiving placebo. No clinically significant abnormalities were noted in hematology, ECG or physical examinations including blood pressure and heart rate. The potential for hepatotoxicity was evaluated based on experience with the CGRP receptor antagonists telcagepant and MK-3207 studied previously. 27 –29 Although for telcagepant significant elevations in aminotransferases were most closely related to duration of continuous daily dosing, for MK-3207 both single and multiple doses led to elevations in aminotransferases either during the treatment period or with a delayed onset up to 2 months post treatment. Based on this history, liver function tests were monitored in this study both during the study and during the post-study period. There was no evidence of drug-related increases in aminotransferases. With the continued development of other CGRP-targeting drugs such as monoclonal antibodies and “new” gepants, the totality of safety results demonstrates that safety challenges with early gepants were a compound specific issue, and not a class issue.
Conclusions
Atogepant was safely administered as single doses up to 200 mg and demonstrated pharmacologically active plasma concentrations, as determined with an integrated PK/PD model, within 30 minutes and sustained for up to 24 hours at 60 mg QD or 30 and 60 mg BID for preventive treatment of migraine.
Supplemental material
Supplemental Material, sj-pdf-1-rep-10.1177_25158163231191582 - Pharmacokinetic and pharmacodynamic assessments of atogepant in healthy male adults: Results from phase 1 studies
Supplemental Material, sj-pdf-1-rep-10.1177_25158163231191582 for Pharmacokinetic and pharmacodynamic assessments of atogepant in healthy male adults: Results from phase 1 studies by Ramesh Boinpally, Marleen Depré, Griet Van Lancker, Marissa F Dockendorf, Phung Bondiskey, Jean-Francois Denef, Tom Reynders, Catherine Zhou Matthews, K Chris Min, Jialin Xu, Joel M Trugman and Jan de Hoon in Cephalalgia Reports
Footnotes
Acknowledgments
The authors thank Stormy Koeniger, PhD, an employee with AbbVie for medical writing support for this manuscript.
Article highlights
Single (0.4−200 mg) doses of atogepant were generally well tolerated in healthy participants. Atogepant reaches pharmacologically active plasma concentrations within 30 minutes which is maintained for up to 24 hours at dosages of 60 mg once daily and 30 and 60 mg twice daily.
Authors’ note
KCM and JX were affiliated with Merck & Co., Inc., at the time they contributed to this work.
Data sharing statement
AbbVie is committed to responsible data sharing regarding the clinical trials we sponsor. This includes access to anonymized, individual and trial-level data (analysis data sets), as well as other information (e.g., protocols and Clinical Study Reports), as long as the trials are not part of an ongoing or planned regulatory submission. This includes requests for clinical trial data for unlicensed products and indications. This clinical trial data can be requested by any qualified researchers who engage in rigorous, independent scientific research, and will be provided following review and approval of a research proposal and Statistical Analysis Plan (SAP) and execution of a Data Sharing Agreement (DSA). Data requests can be submitted at any time and the data will be accessible for 12 months, with possible extensions considered. For more information on the process, or to submit a request, visit the following link: ![]() .
.
Declaration of conflicting interests
The authors declared the following potential conflicts of interest with respect to the research, authorship, and/or publication of this article: RB and JMT are AbbVie employees and may hold AbbVie stock or options. KCM and JX are former employees of Merck & Co., Inc. and may hold stock or options in the company. MFD, PB and CZM are employees of Merck & Co., Inc. and may hold stock or options in the company. JFD and TR are employees of Merck Sharpe & Dohme and may hold stock or options in the company. MD, GVL, and JDH declare no conflicting interests.
Funding
The authors disclosed receipt of the following financial support for the research, authorship, and/or publication of this article: The studies were funded by Merck before atogepant was in-licensed by Allergan, an AbbVie company. AbbVie and Merck contributed to the interpretation of data, writing, reviewing, and approving of this publication. GVL and JDH declare no funding support.
Supplemental material
Supplemental material for this article is available online.
References
Supplementary Material
Please find the following supplemental material available below.
For Open Access articles published under a Creative Commons License, all supplemental material carries the same license as the article it is associated with.
For non-Open Access articles published, all supplemental material carries a non-exclusive license, and permission requests for re-use of supplemental material or any part of supplemental material shall be sent directly to the copyright owner as specified in the copyright notice associated with the article.