
Abstract
Background:
Methylation of two CpG sites related to neuronal pentraxin II protein (NPTX2) and SH2 domain containing 5 protein (SH2D5), corresponding to two neuroplasticity genes, has been associated to headache chronification. We aimed to investigate the epigenetic modification of these two genes in chronic migraine (CM).
Methods:
We conducted a case–control study in which the DNA of 305 age- and sex-matched subjects classified according to the International Classification of Headache Disorders version beta (ICHD-III β) in CM (109), episodic migraine (EM; n = 98), and healthy controls (HC; 98) was analyzed. Real-time quantitative methylation-specific PCR was performed using specific methylation primers for two representative CpG sites within these genes.
Results:
We found no significant differences in methylation level between CM, EM, and HC in the first exon of the NPTX2 gene nor in the 5′ upstream region of the SH2D5 gene. Methylation level in the first exon of the NPTX2 showed a low correlation with age (r = 0.266; p < 0.005).
Conclusion:
We did not find methylation level differences in analyzed regions related to NPTX2 and SH2D5 in our CM sample. Despite the potential relevance of neuroplasticity genes in headache chronification, we conclude that CM is a more heterogeneous clinical diagnosis than desired and that an epigenetic marker remains elusive.
Introduction
Migraine is a common and disabling neurological disease with a global prevalence of approximately 15%, 1 –3 the pathophysiology of which is not completely clear. The mechanisms by which migraine becomes chronic, as occurs in around 2% of the general population, 4 are even less known.
There is evidence of an important genetic component in migraine 5 with tens of genetic variants associated with common forms of the disease. 6 Most replicated variants confirmed several loci at PR Domain-Containing 16 (PRMD16), 7 –10 Myocyte Enhancer Factor 2D (MEF2D), 9,11 Transient Receptor Potential Cation Channel Subfamily M Member 8 (TRMP8), 7,9,11 Transforming Growth Factor Beta Receptor 2 (TGFBR2), 9,11 Phosphatase And Actin Regulator 1 (PHACTR1), 9,11 Metadherin (MTDH), 8,9,11 Astrotactin 2 (ASTN2), 9,11 and LDL Receptor Related Protein 1 (LRP1), 7,9 but effect sizes were discrete (odds ratio lower than 2 in all cases). In particular, susceptibility genes for chronic migraine (CM) remain unknown.
Environmental factors could play an important role in migraine, not only as triggers of an acute attack but inducing chronic changes in the brain that make it more susceptible to migraine. 12 Epigenetic mechanisms have the potential to link environmental influences with changes in gene expression that might lead to disease phenotype. 13 Thus, epigenetics is an emerging field in the study of some complex disorders such as migraine. Through epigenetic mechanisms, some environmental factors such as stress, female sexual hormones, and neuronal activity could cause changes in gene expression in brain pathways involved in migraine. 12 Interestingly, a high frequency of attacks, due to high neuronal activity, has been shown to produce changes in brain epigenome in genes implicated in neuronal plasticity 14 reducing the threshold for suffering new bouts, 12 which suggests the potential relevance of epigenetics in migraine chronification.
Despite this body of evidence, epigenetic studies in migraine are still scarce. 15,16 The first epigenome-wide association study (EWAS) performed in chronic headache (CH), published by Winsvold et al. in 2017, 17 found that the two 5’-cytosine-p-guanine-3’ (CpG) sites differentially methylated with strongest association to headache chronification were related to two brain-expressed genes involved in the regulation of synaptic plasticity: SH2 domain containing 5 protein (SH2D5) and neuronal pentraxin II protein (NPTX2), supporting a role of neuroplasticity pathways in the chronification process.
Our aim was to investigate if these two genes were epigenetically modified in a CM sample, strictly defined by the 3 rd edition of the International Classification of Headache Disorders version beta (ICHD-III β) criteria, 18 as a headache suffered by the patient for 15 or more days a month, for (at least) the last 3 months, and in which at least 8 of these days have to meet strict criteria for migraine, and in the other 7 days any of the criteria.
Methods
Design
We designed a case–control study conducted in the Headache Clinic of the University Hospital Marqués de Valdecilla in the Northern Spain. The study was approved by the Ethics Committee of Cantabria.
Participants
A total sample of 545 migraine patients and 390 healthy controls (HC) were identified from 2015 to date in our Headache Unit database belonging to three outpatient clinics. From this pool of subjects, a selection process followed a prespecified scheme of 1-1-1, age- and sex-matched for CM, episodic migraine (EM), and HC. A total of 305 subjects, aged 18–65 years, were finally gathered in the study after signing informed consent. Although sample was slightly unbalanced, with an excess of migraine patients, age and sex matching was always accomplished. All migraineurs were classified according to the ICHD-III β in CM and EM. CM patients fulfilled criteria even when they were on Onabotulinum toxin treatment, or under other preventatives; EM patients had never been diagnosed as CM, and HC had never had primary headaches. HC were selected among healthy family members (only eight males’ spouses) or friends, and staff volunteers.
For all participants, clinical and demographic variables and comorbidities were collected, including sex, age, age of onset of migraine, presence or absence of aura, total days of headache/90 days, medication consumption, migraine disability assessment scale (MIDAS) and six-item headache impact test (HIT-6) scale, alcohol consumption, smoking habit, hypertension, hyperlipidemia, and contraceptive use.
Procedure
Peripheral blood samples were drawn in ethylenediaminetetraacetic acid (EDTA) vacutainer tubes from subjects’ antecubital veins. DNA was isolated from EDTA whole blood using a REALPURE “SSS” Kit (Durviz SL, Valencia, Spain) following the manufacturer’s protocol, aliquoted and stored at −20°C until use. DNA concentrations were quantified using a Nanovue Plus spectrophotometer from GE HealthCare (Connecticut, USA). After quantification, bisulfite conversion was performed with “EZ DNA Methylation-Gold” ™ Kit (Zymo Research, Irvine, California, USA) in accordance with the manufacturer’s protocol, thereby converting all unmethylated cytosines to uracils but leaving methylcytosines unaltered, thereby allowing us to design specific methylation primers. Real-time quantitative methylation-specific PCR (RT-QMSP) amplification was then carried out with TB Green™ Premix Ex Taq™ II (TaKaRa Bio Inc., Japan) following the manufacturer’s instructions using 5-ng template DNA. CpG sites were determined by the European Molecular Biology Open Software Suite (EMBOSS) CpG plot tool (https://www.ebi.ac.uk/Tools/seqstats/emboss_cpgplot/) using the gene sequence obtained from the National Center for Biotechnology Information (NCBI) database (https://www.ncbi.nlm.nih.gov/gene). For these CpG sites, methylation-specific primers were designed on the MethPrimer tool (https://www.urogene.org/methprimer/). 19 Each PCR plate included patient samples (analyzed by age- and sex-matched triplets of CM, EM, and HC), serial dilutions of completely methylated DNA for constructing calibration curves, positive controls, and two wells with water blanks used as negative controls. Amplification conditions were as follows: denaturalization at 95°C for 10 min, followed by 40 cycles of 95°C for 30 s, 57°C for 30 s and 72°C for 30 s, and finally 72°C for 7 min. These reactions were run in duplicate on a StepOnePlus System (Applied Biosystems, San Francisco, California, USA) to confirm reliability of the results. DNA methylation levels were calculated by the ΔΔCt method 20 using StepOne Software v2.3 (Applied Biosystems).
Selected CpG sites
NPTX2
We analyzed a 662 bp CpG site located on chromosome 7 between positions 98617333 and 98617995 in NPTX2 gene exon 1. The primers selected by the MethPrimer software allowed amplifying a region of 109 bp within this CpG site.
SH2D5
We analyzed a 222 bp CpG site located on chromosome 1 between positions 20732592 and 20732369 of the 5′ UTR upstream of the SH2D5 gene. The primers selected by the MethPrimer software allowed amplifying a region of 203 bp within this CpG site. This fragment includes an open reading fragment which could lead to an alternative protein.
Statistics
For all statistics, we used SPSS (IBM Corp., v.21.0. Armonk, New York, USA). Data are presented as mean ± standard deviation for normal variables, and median and interquartile range for non-normal variables. Categorical variables were compared using χ 2 method or Fisher’s exact tests (if cell n < 5). Quantitative variables were tested for normal distribution using the Kolmogorov–Smirnov test and compared using t-test for independent samples, under normal distribution; otherwise, the Kruskal–Wallis method or Mann–Whitney U test for comparison of two samples were used. Bivariate correlations of normal variables were performed with Pearson’s r-test or Spearman’s rho test for non-normal variables. We also conducted a multinomial regression modeling using clinical categories as dependent variable (HC as reference subgroup), and methylation degree of SH2D5 and NPTX2, age and sex as independent variables. Regression estimates were given as regression coefficients, odds, and 95% confidence interval. For all tests, α = 0.05; a two-sided p < 0.05 was defined as statistically significant after Bonferroni correction for multiple testing. As there were not similar reports using our methodology, we could not perform sample size calculation.
Results
We recruited 305 subjects: 109 CM (mean age = 42.2 ± 10.6 years), 98 EM (mean age = 41.6 ± 10.9 years), and 98 HC (mean age = 41.6 ± 10.6 years). Clinical characteristics are summarized in Table 1. We found that CM patients trended to be slightly younger at age of onset of migraine than EM patients (−2.63 years for the difference, p = 0.088). CM patients were clearly more disabled than EM according to the results of impact tests MIDAS and HIT-6, number of headache days, and proportion of medication abuse headache. The proportion of patients having vascular risk factors was similar among clinical groups, except for the use of contraceptives, something more frequent among EM females (see Table 1).
Demographics and clinical characteristics for the different groups.
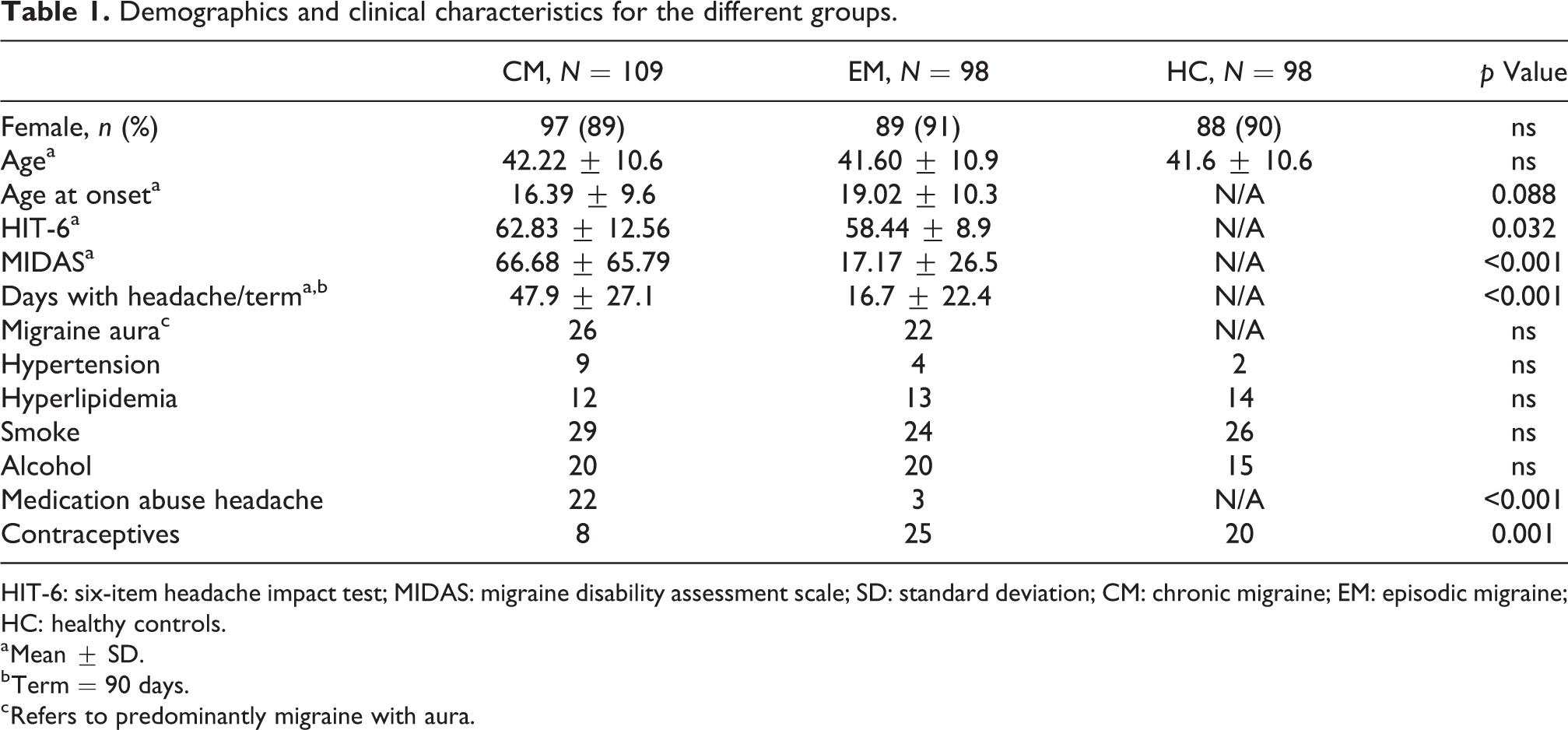
HIT-6: six-item headache impact test; MIDAS: migraine disability assessment scale; SD: standard deviation; CM: chronic migraine; EM: episodic migraine; HC: healthy controls.
a Mean ± SD.
b Term = 90 days.
c Refers to predominantly migraine with aura.
We found no significant differences in methylation level among CM, EM, and controls in the 5′ upstream region of the SH2D5 gene and in exon 1 of the NPTX2 gene (Table 2).
Methylation degree (ΔΔCt) of the 5′ region upstream the SH2D5 gene and in the first exon of the NPTX2 gene with respect to clinical groups.

IQ: interquartile range; SH2D5: SH2 domain containing 5 protein; NPTX2: neuronal pentraxin II protein; CM: chronic migraine; EM: episodic migraine; HC: healthy control.
We did, however, find a low correlation between methylation level in exon 1 of the NPTX2 gene and age (r = 0.266; p < 0.005) (Figure 1(a)). Correlation between methylation level of 5′upstream region of SH2D5 gene and age was not significant (r = 0.098; p = 0.135) (Figure 1(b)). Since age could be correlated with methylation level for exon 1 of NPTX2, age and methylation level of these two genes were analyzed in a multinomial logistic regression model using clinical groups as dependent variables, and NTPX2, SH2D5, and age as covariates. Under this model, none of the covariates differentiated clinical categories (see Table 3). Additionally, we also explored the influence of sex (female as reference) in the model. Both goodness-of-fit (Pearson’s r = 0.330) and pseudo-R 2 (McFadden = 0.003) were identical in both models as well coefficient estimates (data not shown).

Scatterplots showing (a) no correlation between the methylation level in the 5′ upstream region of the SH2D5 gene and age (r = 0.098; p = 0.135) and (b) low correlation between the methylation level in the first exon of NPTX2 and age (r = 0.266; p < 0.005). SH2D5: SH2 domain containing 5 protein; NPTX2: neuronal pentraxin II protein.
Regression coefficients (ß) and odds (OR) and 95% CI corresponding to the multinomial regression model.a

SE: standard error; OR: odds ratio; CI: confidence interval; SH2D5: SH2 domain containing 5 protein; NPTX2: neuronal pentraxin II protein; CM: chronic migraine; EM: episodic migraine.
a Independent variables SH2D5, NPTX2, and age were introduced under full factorial model.
b Interception.
Discussion
In the present study, we performed a DNA methylation analysis, the best known epigenetic mechanism. DNA methylation occurs mainly in the CpG dinucleotides (CpGs) 13 that are underrepresented in the human genome, and appear mostly methylated, except in the so-called CpG islands (CpGi), regions with a high density of such CpGs, highly enriched in gene promoters and exons. 13 Differential methylation among individuals and tissues is to be found in these CpGi. Methylation function in promoter CpGi is related to gene silencing by preventing the access of transcriptional machinery, while methylation found in CpGi located in the gene body has been linked to increased gene expression. 21 Despite incipient data suggesting that some single CpG dinucleotide methylation correlates with gene expression in some cellular types, 22 DNA methylation function has typically been related to the density of methylated CpGs in the region. 16 Therefore, analysis of the average methylation level of a CpG rich region (CpG site) could be expected to be informative of the degree of epigenetic regulation. To this end, RT-QMSP has shown to be a sensitive and specific technique. 23
We conducted a case–control study in a sex- and age-matched sample of CM, EM, and HC, in which we analyzed the methylation level of two CpGi related to SH2D5 and NPTX2 genes, as had been suggested by Winsvold et al. 17 to play some role in headache chronification, and we found no differential methylation among clinical categories. Winsvold et al. 17 conducted a follow-up study, comparing 36 females who progressed from episodic to CH over a period of 11 years and 35 controls who remained episodic. This type of study would be more appropriate to suggest epigenetic mechanisms of disease than our cross-sectional study. However, the questionnaire-based assessment they used could not accurately enable distinguishing cases of CM from other types of CH. Although the most prevalent group within CH was CM, 4 an investigation of a possible epigenetic modification of these two genes in a clearly defined sample of CM was necessary. In addition, it must be taken into account that aging itself could influence the methylation level of a gene. 24 In order to minimize this confounding factor, we used an age-matched sample while a multinomial logistic regression model was carried out using age and methylation levels as covariates.
Our strengths are an accurate clinical diagnosis, made by two skilled neurologists (AO and VGQ), conforming to the International Headache Society criteria, 18 and our sample consisted of three sex- and age-matched groups. We applied a different methodology, which could support additional evidence of association in case of positive replication.
Our study also has some limitations: First, given that a gene expression study was not carried out, we cannot know the functional status of these two genes, so we can only suggest an absence of differential epigenetic markers in the analyzed regions in chronic migraineurs versus episodic or HC cases. Second, since it is a cross-sectional study and epigenetic markers are dynamic, 12 we cannot exclude a possible epigenetic change in some evolutionary phase of the disease. Finally, as with all epigenetic studies in neurological diseases based on peripheral blood samples, some controversy exists concerning the correlation with epigenetic changes in brain tissue. Although, ideally, samples of the most relevant tissue for the disease under study should be used, analyzing brain tissue in humans is a well-known difficult task. Hence, the use of accessible tissues such as peripheral blood is acceptable, all the more reason in systemic diseases where epigenetic variations could more likely transcend the brain tissue. 24 Furthermore, interindividual variation in DNA methylation patterns seems to have a much smaller contribution to global differences than variation among tissues, so peripheral blood samples are accepted for comparisons among individuals. 25
Conclusions
Despite the potential involvement of neuroplasticity genes in CM, we found no differential methylation in the first exon of the NPTX2 gene nor in the 5′upstream region of the SH2D5 gene in our CM sample. CM is a more heterogeneous clinical diagnosis than desired for which an epigenetic marker remains elusive. More studies are needed to assess functional pathway markers that could be epigenetically modified in CM due to their potential diagnostic and therapeutic implications.
Key Findings
We found no significant differences in the methylation level in the first exon of the NPTX2 gene nor in the 5′upstream region of the SH2D5 gene between CM, EM, and HC.
CM is a more heterogeneous clinical diagnosis than desired for which an epigenetic marker remains elusive.
Standardization of the design of methylation studies is required in order to investigate the existence of possible epigenetic biomarkers of CM.
Footnotes
Declaration of conflicting interests
The author(s) declared no potential conflicts of interest with respect to the research, authorship, and/or publication of this article.
Funding
The author(s) disclosed receipt of the following financial support for the research, authorship, and/or publication of this article: This work was financially supported by the Carlos III Health Institute, Madrid, Spain (grant ISCIII-FISS PI15/01285) and the Marqués de Valdecilla Research Institute (IDIVAL), Santander, Spain.