
Abstract
Background:
Ubrogepant is a novel, oral calcitonin gene–related peptide receptor antagonist approved by the US Food and Drug Administration for acute treatment of migraine with or without aura in adults.
Objectives:
To assess potential pharmacokinetic (PK) drug–drug interactions in healthy participants and inform the safety and tolerability of ubrogepant alone and in combination with acetaminophen or nonsteroidal anti-inflammatory drugs (NSAIDs) in healthy participants and participants with migraine.
Methods:
Two phase 1, three-way crossover studies randomized healthy adults to 100 mg ubrogepant alone, 1000 mg acetaminophen or 500 mg naproxen alone, and 100 mg ubrogepant plus 1000 mg acetaminophen or 500 mg naproxen. Geometric mean ratios (GMRs) and 90% confidence intervals were calculated based on statistical comparison of maximum plasma drug concentration (C max) and area under the plasma drug concentration–time curve (AUC) for treatment in combination versus alone. Two phase 3 randomized trials included adults with migraine. Treatment-emergent adverse events (TEAEs) were evaluated.
Results:
Time to C max and terminal elimination half-life for all treatments were unchanged when coadministered. Ubrogepant C max and AUC increased by approximately 40% when coadministered with acetaminophen. Acetaminophen C max decreased by 24% (GMR = 0.76) when coadministered with ubrogepant. There were no significant PK interactions between ubrogepant and naproxen. TEAE rates in the acetaminophen and NSAID rescue medication groups were similar to ubrogepant alone.
Conclusions:
Coadministration of ubrogepant and acetaminophen resulted in a statistically significant increase in ubrogepant exposure and a decrease in acetaminophen C max; however, these changes were not clinically relevant. No statistically or clinically relevant changes in PK were associated with ubrogepant and naproxen coadministration. No safety concerns were identified for ubrogepant alone or in combination with acetaminophen or NSAIDs.
Keywords
Introduction
Migraine is a highly prevalent, chronic, neurological disease that affects approximately one in seven individuals globally. 1 Characteristics of migraine include moderate or severe pain accompanied by nausea, vomiting, photophobia, and/or phonophobia. 2,3 The symptoms of migraine can be disabling, and repeated attacks can have significant negative impacts on patients’ quality of life, including familial relationships, social interactions, finances, and employment. 4 –6
Current evidence-based guidelines state that nonsteroidal anti-inflammatory drugs (NSAIDs), nonopioid analgesics, and acetaminophen (all of which can be used in combination with caffeine) are preferred for the acute treatment of mild to moderate migraine attacks. 3,7 –9 Migraine-specific agents, including triptans and dihydroergotamine (DHE), are recommended to treat moderate or severe attacks. If these first-line treatments do not provide relief, rescue therapy can be considered using parenteral formulations of triptans, DHE, corticosteroids, antiemetics, NSAIDs (e.g. ketorolac), or magnesium sulfate. 3 However, despite the availability of numerous therapies for the acute management of migraine, treatment is often poorly optimized and many patients fail to meet the treatment goal of adequate pain relief. 10 –13 When patients achieve only a partial response or experience dose-limiting adverse events (AEs), combining acute therapies is a commonly used treatment strategy. 3,14
Whenever multiple drugs are combined, there is a potential for drug–drug interactions (DDIs) that can alter therapeutic effectiveness or increase the likelihood of treatment-related side effects. 15 DDIs are an under-recognized problem in pharmacotherapy that can be associated with adverse clinical outcomes including increased rates of hospitalization and overall patient morbidity. 16
Naproxen and acetaminophen are among the most commonly used over-the-counter treatments for the management of migraine pain, and these medications are frequently used in combination with other medications. 14,17 Naproxen and acetaminophen are generally well tolerated and are modestly efficacious for the relief of migraine attacks of mild-to-moderate severity. 17 However, these treatments may be less appropriate for individuals with more severe migraine attacks. 17
Ubrogepant is an oral calcitonin gene–related peptide (CGRP) receptor antagonist approved by the US Food and Drug Administration for the acute treatment of migraine with or without aura in adults. 18,19 CGRP is a neuropeptide that is highly expressed in pain-sensitive trigeminal neurons, and CGRP levels are increased during migraines. 20 Ubrogepant binds to the human CGRP receptor with high affinity (Ki , 0.07 nM), 21 and clinical data indicate that treatment with ubrogepant provides substantial pain relief and meaningful improvement in functional disability for people with migraine. 22,23 Here we report results from two phase 1 studies conducted to assess the potential for pharmacokinetic (PK) DDIs between ubrogepant and acetaminophen or naproxen in healthy participants. Additionally, pooled data from two phase 3 trials (ACHIEVE I (NCT02828020) and II (NCT02867709)) are analyzed to evaluate the safety and tolerability of ubrogepant administered alone and in combination with acetaminophen or an NSAID when used as rescue medication in participants with migraine.
Methods
Phase 1 studies
Two phase 1, single-center, single-dose, open-label, three-way, crossover PK interaction studies were conducted in healthy participants. Study A evaluated the effects of 1000 mg acetaminophen on the PK of 100 mg ubrogepant as well as the effects of ubrogepant on the PK of acetaminophen. Similarly, study B evaluated the effects of 500 mg naproxen on the PK of 100 mg ubrogepant and the effects of ubrogepant on the PK of naproxen. Both study protocols were approved by properly constituted Institutional Review Boards (IRBs) before study initiation, and all participants provided written informed consent. The IRB for study A was Chesapeake IRB, Columbia, Maryland (approval: June 29, 2016). The IRB for study B was IntegReview IRB, Austin, Texas (approval: March 16, 2018). Studies were conducted in conformance with the International Council for Harmonisation E6 Guidance for Good Clinical Practice and the principles of the Declaration of Helsinki.
Participants
Both phase 1 studies included healthy men and women between 18 years and 45 years of age with a body mass index ≥18 and ≤30 kg/m2 and sitting pulse rate ≥50 and ≤100 bpm at screening. Participants were required to be nonsmokers, defined as having never smoked or not having smoked within the previous 2 years. Additionally, men and women of childbearing potential agreed to use an effective method of contraception, and women were required to have a negative serum pregnancy test at screening and a negative serum or urine pregnancy test on day −1.
Participants were excluded from both phase 1 studies if they had known hypersensitivity to ubrogepant or other CGRP receptor antagonists, or to acetaminophen (study A) or naproxen (study B). Other exclusion criteria were as follows: sitting systolic blood pressure ≥140 or ≤90 mm Hg or diastolic blood pressure ≥90 or ≤50 mm Hg at screening; potentially clinically significant abnormal electrocardiogram (ECG) results or QT prolongation (QTcF ≥450 ms for men; QTcF ≥470 ms for women, or, in study B only, uncorrected QT ≥500 ms) at screening; breastfeeding; positive urine test for benzoylecgonine (cocaine), methadone, barbiturates, amphetamines, benzodiazepines, alcohol, cannabinoids, opiates, phencyclidine, or cotinine at screening or day −1; any clinically significant disease (study B); or any clinical condition or previous surgery that could have affected the absorption, distribution, biotransformation, or excretion of ubrogepant and/or acetaminophen (study A) and/or naproxen (study B). Participants were also prohibited from consuming caffeine or xanthine-containing compounds within 48 h; alcohol within 72 h; grapefruit-containing products, vegetables from the mustard green family, or charbroiled meats within 14 days (2 weeks); or St. John’s Wort within 4 weeks before administration of the first dose of study treatment and during the study.
Study designs
Both phase 1 studies consisted of a 14-day screening period (day −14 to day −1), a randomized treatment period, and a follow-up period of 30 days after the last dose of study treatment for liver function testing. The treatment periods of both studies had open-label, six-sequence, three-way crossover designs in which eligible participants were randomized in a 1:1:1:1:1:1 ratio to receive treatments A, B, or C in one of six sequences under fasted conditions (Online Supplemental Material 1). Participants were assigned consecutive numbers by the investigators at the time of recruitment or at the time of consent. Randomization codes were generated by statistical programming.
In study A, treatment A was a single oral dose of 1000 mg acetaminophen (2 × 500 mg tablets; Geri-Care Pharmaceutical Corp, Brooklyn, New York, USA 24 ) alone; treatment B was a single oral therapeutic dose of 100 mg ubrogepant (2 × 50 mg hot-melt extrusion (HME) tablets used in phase 3 clinical trials) alone; and treatment C was a single oral dose of 100 mg ubrogepant plus 1000 mg acetaminophen. In study B, treatment A was a single oral therapeutic dose of 100 mg ubrogepant (2 × 50 mg HME tablets) alone; treatment B was a single oral dose of 500 mg naproxen (1 × 550 mg naproxen sodium tablet containing 500 mg of naproxen; Amneal Pharmaceuticals of New York LLC 25 ) alone; and treatment C was a single oral dose of 100 mg ubrogepant plus 500 mg naproxen. In both studies, treatments were separated by 7-day washout periods.
Participants were admitted to the study center on days −1, 7, and 14 and were required to undergo a 10-h overnight fast prior to dosing. Study treatment was administered with 240 mL of water at approximately 0800 h on days 1, 8, and 15, after which time participants were required to maintain a fasted condition and remain seated upright for an additional 4 h. Participants remained in the study center until 24 h after dose administration.
Venous blood samples to determine ubrogepant and acetaminophen (study A) or ubrogepant and naproxen (study B) concentrations were collected on days 1, 8, and 15 at time 0 (before dose) and at 0.5, 1, 1.5, 2, 3, 4, 5, 6, 8, 12, 14, and 24 h post dose. In study B, additional blood samples were collected at 36, 48, and 72 h after administration of treatments B and C (500 mg naproxen and 100 mg ubrogepant plus 500 mg naproxen, respectively) to determine plasma naproxen concentrations only.
Bioanalytical assessments
Plasma PK samples from phase 1 studies were analyzed using separate validated liquid chromatography with tandem mass spectrometry (LC-MS/MS) assays for the quantitation of ubrogepant (studies A and B), acetaminophen (study A), and naproxen (study B). In study A, validated LC-MS/MS assay calibration ranges were 0.1–150 ng/mL for ubrogepant and 0.1–30 µg/mL for acetaminophen for plasma samples. The same ubrogepant LC-MS/MS bioanalytical method was used in study B, with a higher calibration range of 1–1000 ng/mL. The LC-MS/MS bioanalytical method for analysis of naproxen had a calibration range of 0.5–100 µg/mL.
Safety assessments
In both phase 1 studies, safety was assessed by monitoring AEs, clinical laboratory tests, vital sign parameters, 12-lead ECGs, and physical examinations at specified time points. An AE was defined as any unfavorable and unintended sign, symptom, or disease temporarily associated with the use of a medicinal product. AEs were classified as treatment-emergent AEs (TEAEs) if the events started or worsened in severity during a treatment period or the subsequent washout period. Blood samples for liver function tests were obtained 30 days after the last dose of study treatment.
Pooled phase 3 ACHIEVE trials safety assessment
Full methods for these studies have been previously described. 22,23 Briefly, the ACHIEVE trials were two phase 3, multicenter, single-attack, placebo-controlled, randomized trials in adults with a history of migraine with or without aura. Data from the ubrogepant 50 mg and placebo treatment groups of ACHIEVE I and ACHIEVE II were pooled for this subanalysis. Data from the 25 and 100 mg groups were not pooled and represent data from the individual studies. An optional second dose or rescue medication was allowed for the treatment of moderate or severe headache starting from 2 to 48 h after the initial dose of study medication. Rescue medication options included acetaminophen, NSAIDs, opioids, antiemetics, and triptans. In this subanalysis, we compared the prevalence of TEAEs in participants who took ubrogepant without an optional rescue medication (ubrogepant alone) and those who took ubrogepant and acetaminophen or an NSAID as a rescue medication. Eligible NSAIDs included aspirin, diclofenac, ibuprofen, ketoprofen, and naproxen. No fixed-dose combination medications were included.
Statistical analyses
Based on a within-participant coefficient of variation observed in previous studies of 25% for ubrogepant and a ratio of 1.0 for ubrogepant in combination with acetaminophen/naproxen versus ubrogepant alone, a sample size of 30 participants with at least 24 participants completing the study was required to ensure a probability of at least 90% that the 90% confidence interval (CI) for the ratio of treatment administered in combination (test) versus treatment administered alone (reference) was within the limits of 0.8–1.25.
PK parameters derived from plasma concentrations of ubrogepant (studies A and B), acetaminophen (study A), and naproxen (study B) included area under the plasma drug concentration-versus-time (AUC) from time 0 to t (AUC0–t ) and from time 0 to infinity (AUC0–∞), maximum plasma concentration (C max), time to C max (t max), terminal elimination half-life (t 1/2), terminal elimination rate constant (λz ), apparent total body clearance of medication from plasma after extravascular administration (CL/F), and apparent volume of distribution during the terminal phase after extravascular administration (V z/F). PK parameters were derived from plasma concentrations using noncompartmental analysis with Phoenix WinNonlin software (version 6.2 for study A and version 8.0 for study B). Plasma concentrations below the limit of quantification were treated as 0.
Log-transformed C max, AUC0–t , and AUC0–∞ values for treatment in combination versus treatment alone were compared using a linear mixed-effects model with sequence, treatment, and period as fixed effects and participant within sequence as a random effect. Two-sided 90% CIs were constructed for the least squares geometric mean ratio (GMR) of C max and AUC of ubrogepant in combination with acetaminophen (study A) or naproxen (study B) versus ubrogepant alone; 90% CIs were also constructed for acetaminophen (study A) or naproxen (study B) in combination with ubrogepant versus acetaminophen or naproxen alone, respectively. No DDI was to be concluded if the 90% CIs for the ratios of C max and AUC parameters of treatment in combination versus treatment alone were within the limits of 0.8 and 1.25.
Results
PK—Phase 1 studies
Participants
Thirty participants were enrolled in each phase 1 study. Participant demographic and baseline characteristics are summarized in Table 1. In study A (ubrogepant–acetaminophen DDI), all 30 participants received all designated treatments and completed the study (Online Supplemental Material 2). In study B (ubrogepant–naproxen DDI), 29 of 30 participants received all designated treatments and completed the study. One participant assigned to treatment sequence B-A-C withdrew consent after administration of treatment B (i.e. naproxen alone) in study B (Online Supplemental Material 3).
Participant demographic and baseline characteristics.

BMI: body mass index; DDI: drug–drug interaction; SD: standard deviation.
Study A (ubrogepant–acetaminophen DDI)
The plasma concentration–time profile of ubrogepant following its administration alone and when coadministered with acetaminophen in study A is shown in Figure 1. A summary of the mean PK parameters for ubrogepant when administered alone or in combination with acetaminophen is presented in Table 2. Notably, C max and AUC of ubrogepant were increased following coadministration with acetaminophen. However, median t max (approximately 2 h) and mean apparent terminal t 1/2 were similar for ubrogepant when administered alone and in combination with acetaminophen. These similarities in the terminal elimination phase imply that there was no impact of acetaminophen on the elimination of ubrogepant.

Mean (SD) plasma ubrogepant concentrations when administered alone and in combination with acetaminophen. SD: standard deviation.
Mean (SD) 100 mg ubrogepant PK parameters when administered alone or in combination with 1000 mg acetaminophen.

AUC0–t : area under the plasma drug concentration–time curve from time 0 to t; AUC0–∞: area under the plasma drug concentration–time curve from time 0 to infinity; CL/F: apparent total body clearance of drug from plasma after extravascular administration; C max: maximum plasma drug concentration; PK: pharmacokinetics; SD: standard deviation; t 1/2: terminal elimination half-life; t max: time to C max; V z/F: apparent volume of distribution during the terminal phase after extravascular administration.
a Median (range).
The plasma concentration–time profile of acetaminophen following its administration alone and in combination with ubrogepant is shown in Figure 2. The median acetaminophen t max value (approximately 1 h) and mean apparent terminal t 1/2 were similar following acetaminophen administration alone and in combination with ubrogepant. Mean C max was decreased and AUC was similar following coadministration of acetaminophen and ubrogepant compared with acetaminophen alone (Table 3).

Mean (SD) plasma acetaminophen concentrations when administered alone and in combination with ubrogepant. SD: standard deviation.
Mean (SD) 1000 mg acetaminophen PK parameters when administered alone or in combination with 100 mg ubrogepant.

AUC0–t : area under the plasma drug concentration–time curve from time 0 to t; AUC0–∞: AUC from time 0 to infinity; CL/F: apparent total body clearance of drug from plasma after extravascular administration; C max: maximum plasma drug concentration; PK: pharmacokinetics; SD: standard deviation; t 1/2: terminal elimination half-life; t max: time to C max; V z/F: apparent volume of distribution during the terminal phase after extravascular administration.
a Median (range).
Based on statistical comparisons using a linear mixed-effects model, the GMRs and 90% CIs for ubrogepant and acetaminophen PK parameters when administered in combination versus when administered alone are summarized in Table 4. The GMRs for ubrogepant C max, AUC0–t , and AUC0–∞ when coadministered with acetaminophen versus ubrogepant administered alone were 1.42, 1.44, and 1.43, respectively. These results indicate that ubrogepant C max, AUC0–t , and AUC0–∞ were approximately 40% higher when coadministered with acetaminophen than when administered alone. The GMR of the acetaminophen C max when coadministered with ubrogepant versus acetaminophen alone was 0.76, corresponding to a 24% decrease in acetaminophen C max when coadministered with ubrogepant. The GMRs and 90% CIs for acetaminophen AUC were within the range of 0.8–1.25, suggesting no change in total systemic exposure to acetaminophen.
Results of study A statistical analysis of PK parameters (N = 30).
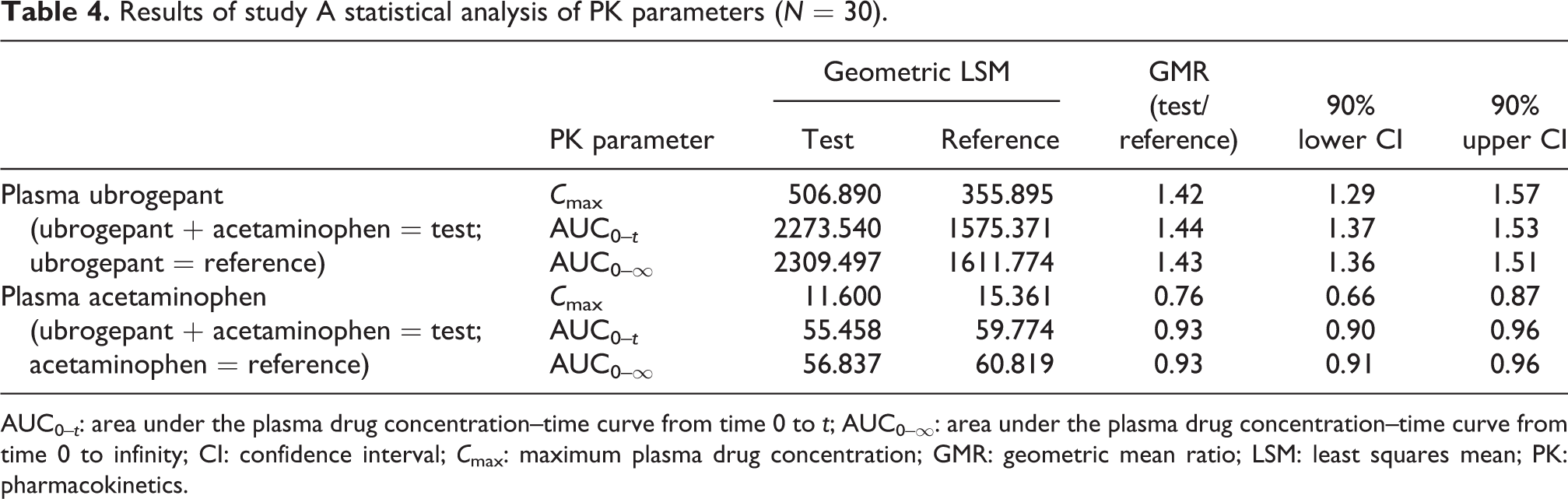
AUC0–t : area under the plasma drug concentration–time curve from time 0 to t; AUC0–∞: area under the plasma drug concentration–time curve from time 0 to infinity; CI: confidence interval; C max: maximum plasma drug concentration; GMR: geometric mean ratio; LSM: least squares mean; PK: pharmacokinetics.
Study B (ubrogepant–naproxen DDI)
The plasma concentration–time profile of ubrogepant following administration alone and when coadministered with naproxen during study B is shown in Figure 3, and mean PK parameters are summarized in Table 5. The median t max and mean apparent terminal t 1/2 for ubrogepant were generally similar when ubrogepant was administered alone and in combination with naproxen. The overall systemic exposure (AUC) and C max of ubrogepant were also unchanged when coadministered with naproxen.

Mean (SD) plasma ubrogepant concentrations when administered alone and in combination with naproxen. SD: standard deviation.
Mean (SD) 100 mg ubrogepant PK parameters when administered alone or in combination with 500 mg naproxen.

AUC0–t : area under the plasma drug concentration–time curve from time 0 to t; AUC0–∞: AUC time 0 to infinity; CL/F: apparent total body clearance of drug from plasma after extravascular administration; C max: maximum plasma drug concentration; PK: pharmacokinetics; SD: standard deviation; t 1/2: terminal elimination half-life; t max: time to C max; V z/F: apparent volume of distribution during the terminal phase after extravascular administration.
a Median (range).
The plasma concentration–time profile of naproxen following its administration alone and in combination with ubrogepant is shown in Figure 4. Median naproxen t max and mean apparent terminal t 1/2 values were similar when naproxen was administered alone and in combination with ubrogepant. There were no changes in overall systemic exposure (AUC) or mean C max following coadministration of naproxen and ubrogepant compared with naproxen alone (Table 6).

Mean (SD) plasma naproxen concentrations when administered alone and in combination with ubrogepant. SD: standard deviation.
Mean (SD) 500 mg naproxen PK parameters when administered alone or in combination with 100 mg ubrogepant.
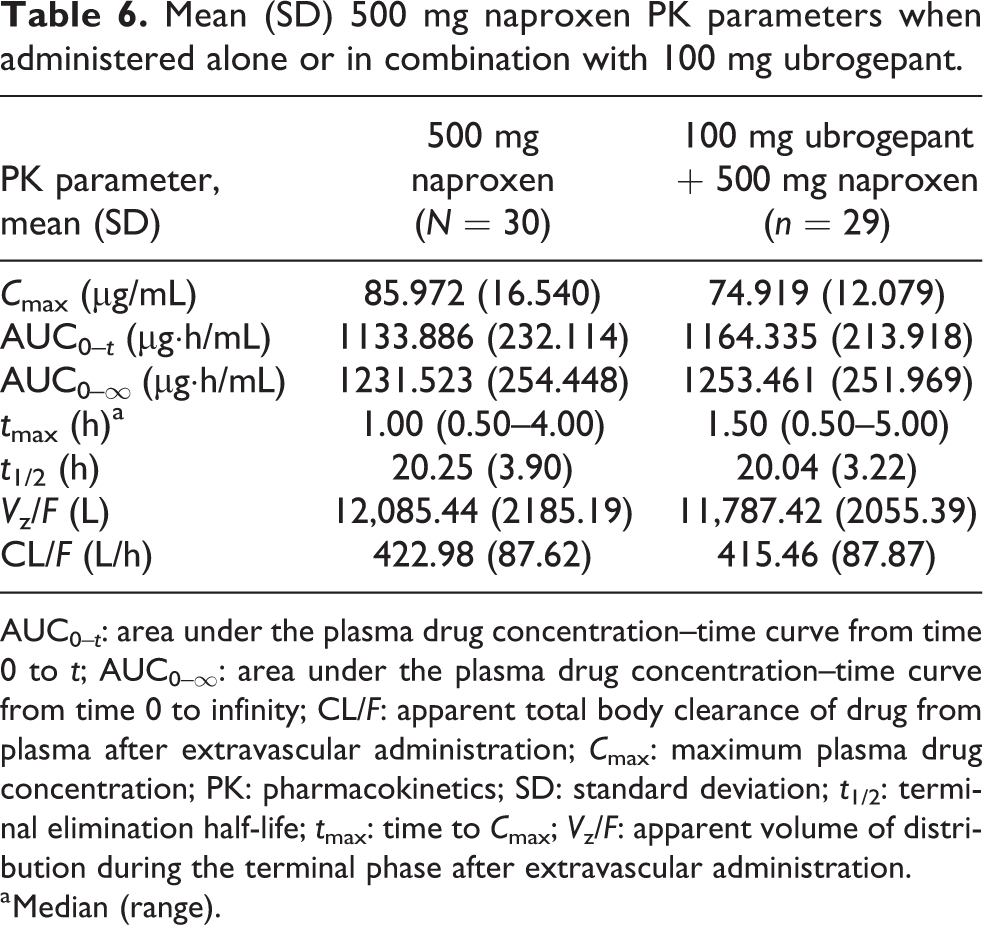
AUC0–t : area under the plasma drug concentration–time curve from time 0 to t; AUC0–∞: area under the plasma drug concentration–time curve from time 0 to infinity; CL/F: apparent total body clearance of drug from plasma after extravascular administration; C max: maximum plasma drug concentration; PK: pharmacokinetics; SD: standard deviation; t 1/2: terminal elimination half-life; t max: time to C max; V z/F: apparent volume of distribution during the terminal phase after extravascular administration.
a Median (range).
Based on statistical comparisons, the GMRs and 90% CIs for ubrogepant and naproxen PK parameters when administered in combination versus their administration alone are summarized in Table 7. As the GMRs and 90% CIs for all study B comparisons were within the range of 0.8–1.25, ubrogepant and naproxen PK were considered to be unchanged when these treatments were coadministered.
Results of study B statistical analysis of PK parameters (n = 29).

AUC0–t : area under the plasma drug concentration–time curve from time 0 to t; AUC0–∞: area under the plasma drug concentration–time curve from time 0 to infinity; CI: confidence interval; C max: maximum plasma drug concentration; GMR: geometric mean ratio; LSM: least squares mean; PK: pharmacokinetics.
Safety and tolerability
Phase 1 studies
Safety and tolerability results were similar in studies A and B. In both studies, there were no serious AEs, deaths, or premature discontinuations due to AEs. The incidence of TEAEs was low and comparable across treatments. In study A, four participants (13.3%) experienced at least one TEAE, all of which were mild in severity. One participant experienced treatment-related TEAEs of dizziness and ocular hyperemia during the acetaminophen plus ubrogepant treatment period. In study B, four participants (13.3%) experienced at least one TEAE, all of which were mild in severity. Three participants each had one TEAE of headache, and one participant had a TEAE of flatulence. Two of the TEAEs of headache were considered treatment-related.
For all clinical laboratory parameters and vital signs, mean changes from baseline were small and not considered clinically meaningful. Few abnormalities were noted and no clinically relevant trends or patterns were observed. All ECG results were similar at baseline and end-of-study visits, and no clinically significant abnormal ECG results were observed in either study.
Phase 3 trials
A pooled analysis of ACHIEVE trial data was conducted to evaluate safety in participants randomized to ubrogepant who took acetaminophen (n = 32) or an NSAID, including naproxen (n = 176), as an optional rescue medication. For comparison, this analysis included 1344 participants who did not take any rescue medication after receiving ubrogepant to treat their migraine attack. AE data for these participants are summarized in Table 8. Overall, the frequency of TEAEs was similar among participants who took ubrogepant alone and among those who took an NSAID as a rescue medication. Very few participants in any ubrogepant dose group took acetaminophen as their rescue medication (n ≤ 14); however, comparison of TEAEs between participants who took ubrogepant alone and those who took ubrogepant with acetaminophen showed no evidence of any differences in the safety profile. Specific TEAEs occurring in ≥2 participants in the ubrogepant-NSAID group included nausea, diarrhea, upper respiratory tract infection, nasopharyngitis, and urinary tract infection. No specific AE occurred in ≥2 participants in any single-dose group for the ubrogepant–acetaminophen subgroup. A complete list of specific AEs, regardless of relation to treatment, is included in Online Supplemental Material 4.
Overall summary of adverse events from ACHIEVE trials for ubrogepant alone or in combination with acetaminophen or NSAIDs as rescue medication within 30 days.
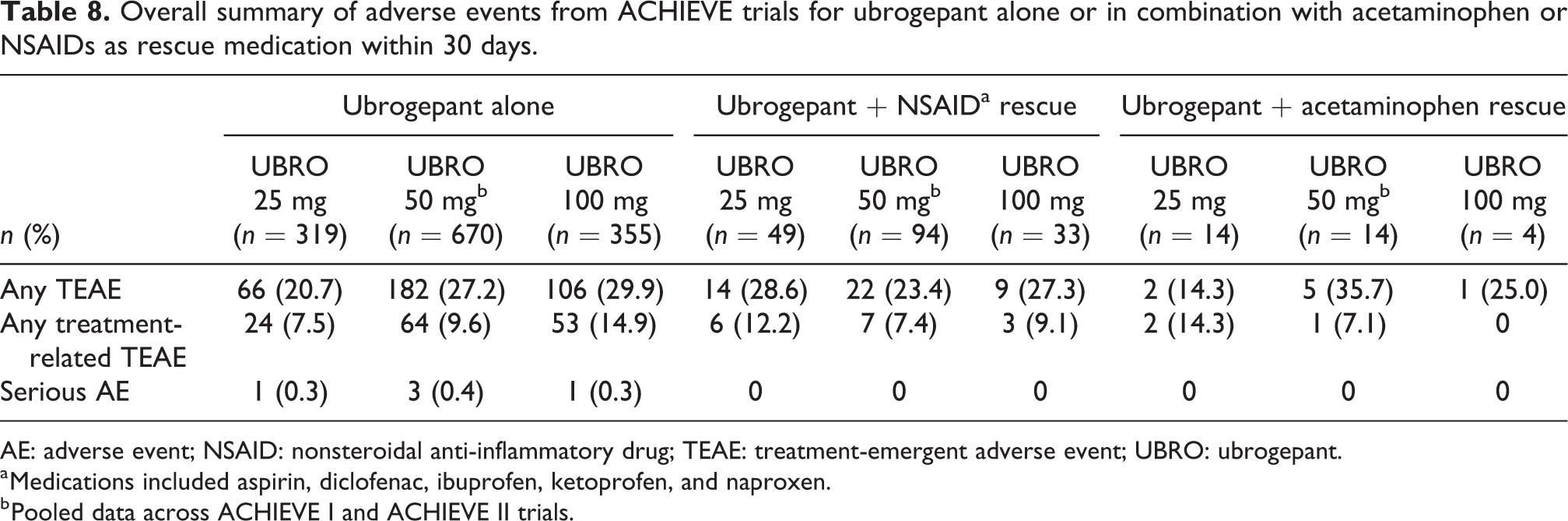
AE: adverse event; NSAID: nonsteroidal anti-inflammatory drug; TEAE: treatment-emergent adverse event; UBRO: ubrogepant.
a Medications included aspirin, diclofenac, ibuprofen, ketoprofen, and naproxen.
b Pooled data across ACHIEVE I and ACHIEVE II trials.
Discussion
Results of the two phase 1 PK DDI studies reported here provide important information on the safety, tolerability, and PK effects of coadministration of 100 mg ubrogepant with 1000 mg acetaminophen or 500 mg naproxen. Coadministration of ubrogepant with acetaminophen increased exposure to ubrogepant by approximately 40% (C max and AUC) and reduced mean acetaminophen C max by approximately 24% while having only minimal impact on AUC (7% reduction) and no impact on median tmax; these interactions are not considered to be clinically relevant, both in terms of efficacy or safety. Given the confirmed safety and tolerability of ubrogepant in combination with other rescue medications in phase 3 trials, the increase in exposure to ubrogepant observed with acetaminophen is unlikely to impact patients at the anticipated clinical doses. 22,23 Furthermore, single ubrogepant doses of 400 mg were found to be safe and generally well tolerated in a thorough QT trial in healthy adults. 26 The observed decrease in acetaminophen C max when coadministered with ubrogepant and the minimal impact on AUC was not considered clinically relevant, as safety concerns with acetaminophen-associated toxicity are due to increased acetaminophen dose and exposure. 27 Coadministration of ubrogepant with naproxen did not alter the maximum plasma concentrations or overall exposures to either treatment.
Results of these studies are important because combining analgesic treatments with different mechanisms of action is a common strategy in the acute treatment of migraine attacks. 28 First-line treatment for migraine attacks often includes acetaminophen or an NSAID, with patients adding a prescription treatment if symptoms persist or intensify. 3,14,17,28 As treatments that target CGRP or its receptor emerge as novel treatment options for migraine, 29 it is important for clinicians to understand how these treatments are best utilized and any potential interactions with other medications, particularly those used to treat migraine.
Our findings that PK interactions between ubrogepant and acetaminophen or naproxen are not clinically relevant are consistent with the knowledge that these treatments have largely different primary metabolic pathways and are not inducers or inhibitors of the same cytochrome P450 (CYP) isozymes at clinically relevant concentrations. Ubrogepant is metabolized almost exclusively by CYP3A4, 30 whereas acetaminophen is metabolized primarily via hepatic glucuronidation and sulfation 31 and naproxen is metabolized by hepatic CYP2C9. 32 However, because glucuronidation is a common elimination pathway for all three treatments, 30,31,33 minimal PK interactions were likely. Additionally, ubrogepant is a substrate of the P-glycoprotein (P-gp) efflux transporter 18 and acetaminophen is an inhibitor of P-gp. 34 It is possible that inhibition of P-gp by acetaminophen might have decreased the efflux of ubrogepant across intestinal membranes, resulting in increased oral bioavailability (40% increase in C max and AUC) of ubrogepant. Given the safety margin of ubrogepant (a 400-mg once-daily dose for 10 days was well tolerated in healthy subjects), 35 and that the increase in ubrogepant exposure in the presence of P-gp inhibitors is not expected to be more than twofold, 18 such an interaction is likely to be of low clinical significance even if P-gp is completely inhibited due to overuse of acetaminophen.
The incidence of TEAEs was low and comparable across treatments. There were no deaths or study discontinuations due to TEAEs in the phase 1 studies. Treatment with ubrogepant alone or in combination with acetaminophen or an NSAID was safe and well tolerated in both phase 1 studies and in the phase 3 trials.
There are a number of differences between the phase 1 and phase 3 studies that are relevant to the interpretation of the results. The small sample size (n = 14) in the ubrogepant–acetaminophen subgroup in the phase 3 trials limits the comparison between treatments; however, even with this small number of participants, there was no evidence of a pronounced difference in the safety profile between ubrogepant alone and ubrogepant with acetaminophen in the phase 3 trials.
The average incidence of TEAEs reported in the ACHIEVE pooled analysis (25.9% (ubrogepant alone), 26.4% (ubrogepant-NSAIDs), and 25.0% (ubrogepant–acetaminophen)) was approximately double that of both phase 1 studies (13.3%; 4/30 in each phase 1 study). The ACHIEVE trials included a larger number of overall participants, were outpatient, placebo-controlled trials, and enrolled participants with migraine. Phase 1 studies enrolled a smaller number of healthy adults and were conducted as inpatient studies. These differences may have contributed to the between-study differences in overall rates of TEAEs; however, results for the within-study comparison of ubrogepant 100 mg alone (29.9%, 106/355) versus ubrogepant 100 mg with acetaminophen (25%, 1/4) or an NSAID (27.3%, 9/33) showed similar rates of TEAEs between subgroups.
The timing of acetaminophen or naproxen administration relative to ubrogepant differed in the phase 1 and phase 3 studies. As evaluation of any potential PK interaction was a primary objective in the phase 1 studies, both medications were administered together. However, in the phase 3 studies, participants were allowed to administer rescue medication no earlier than 2 h post ubrogepant dose if headache pain was not adequately addressed, similar to expected real-world use. Nevertheless, the PK properties of ubrogepant were similar in the phase 1 and phase 3 studies (data on file, Allergan plc) consistent with the results of a population PK analysis that indicated no significant effect of age, sex, race, and body weight on the PK (C max and AUC) of ubrogepant. 18
Finally, the phase 1 studies in healthy participants included more men than women (70% men in study A and 57% men in study B). 2,36 In contrast, the population in the phase 3 studies included approximately 90% women. 22,23 Individuals with migraine are disproportionately female (migraine prevalence is approximately threefold higher in women than in men); therefore, the phase 3 studies were more representative of the target migraine population. 2,36 However, the population PK study found no clinically relevant impact of sex on the PK profile of ubrogepant. 18 Furthermore, noncompartmental PK analyses of data from a subset of patients enrolled in the phase 3 studies and who had serial PK sampling showed PK profiles similar to those observed in phase 1 studies (data on file, Allergan plc). Therefore, it is expected that the findings of the phase 1 studies apply to the migraine population overall.
Conclusions
Although coadministration of 100 mg ubrogepant and 1000 mg acetaminophen resulted in a statistically significant increase in exposure to ubrogepant of approximately 40% and a 24% decrease in C max of acetaminophen, no dose adjustment of either treatment is needed. Coadministration of 100 mg ubrogepant and 500 mg naproxen had no statistically significant or clinically relevant effects on C max or overall exposure to either treatment. Coadministration of ubrogepant with either acetaminophen or an NSAID is safe and well tolerated in healthy participants and patients with migraine.
Clinical implications
Two phase 1, open-label, randomized, crossover studies in healthy adults evaluated the PK interaction and safety of 100 mg ubrogepant with 1000 mg acetaminophen or 500 mg naproxen. TEAEs for ubrogepant alone or in combination with acetaminophen or naproxen were evaluated in two phase 3 randomized trials of adults with migraine.
Exposure to ubrogepant increased by approximately 40% when coadministered with acetaminophen; however, this change is not considered clinically relevant for anticipated dosing.
The observed C max of acetaminophen decreased by 24% when coadministered with ubrogepant.
When ubrogepant and naproxen were coadministered, no statistically significant or clinically relevant changes in PK were observed.
Coadministration of ubrogepant with either acetaminophen or NSAIDs (including naproxen) was safe and well tolerated in healthy adults and people with migraine.
Supplemental material
Supplemental Material, Jakate-et-al_SUPPLEMENTAL-MATERIAL-FOR-SUBMISSION - Evaluation of the pharmacokinetic interaction and safety of ubrogepant coadministered with acetaminophen or nonsteroidal anti-inflammatory drugs: A randomized trial
Supplemental Material, Jakate-et-al_SUPPLEMENTAL-MATERIAL-FOR-SUBMISSION for Evaluation of the pharmacokinetic interaction and safety of ubrogepant coadministered with acetaminophen or nonsteroidal anti-inflammatory drugs: A randomized trial by Abhijeet Jakate, Ramesh Boinpally, Matthew Butler, Kaifeng Lu, Kristi Womack, Danielle McGeeney and Antonia Periclou in Cephalalgia Reports
Footnotes
Authors’ note
Writing and editorial assistance was provided to the authors by Peloton Advantage, LLC, an OPEN Health company, Parsippany, NJ, USA, and was funded by Allergan plc. Results of these studies were presented, in part, at the 61st Annual Scientific Meeting of the American Headache Society, July 11–14, 2019, Philadelphia, PA, USA.
Acknowledgements
The authors would like to acknowledge the participants in these studies.
Declaration of conflicting interests
The author(s) declared the following potential conflicts of interest with respect to the research, authorship, and/or publication of this article: Abhijeet Jakate, Ramesh Boinpally, Matthew Butler, Kaifeng Lu, Kristi Womack, Danielle McGeeney, and Antonia Periclou are employees and stockholders of Allergan plc.
Funding
The author(s) disclosed receipt of the following financial support for the research, authorship, and/or publication of this article: These studies were sponsored by Allergan plc, Dublin, Ireland.
Data accessibility statement
Data reported in this manuscript are available within the article and its supplemental material. Allergan may share de-identified patient-level data and/or study-level data, including protocols and clinical study reports, for phase 1 trials completed after 2008 that are registered on ClinicalTrials.gov or EudraCT. The indication studied in the trial must have regulatory approval in the United States and/or the European Union and the primary manuscript from the trial must be published prior to data sharing. To request access to the data, the researcher must sign a data use agreement. All shared data are to be used for noncommercial purposes only. More information can be found on ![]() .
.
Supplemental material
Supplemental material for this article is available online.
References
Supplementary Material
Please find the following supplemental material available below.
For Open Access articles published under a Creative Commons License, all supplemental material carries the same license as the article it is associated with.
For non-Open Access articles published, all supplemental material carries a non-exclusive license, and permission requests for re-use of supplemental material or any part of supplemental material shall be sent directly to the copyright owner as specified in the copyright notice associated with the article.