
Abstract
The composition of eukaryotic membranes reflects a varied but precise amalgam of lipids. The genetic underpinning of how such diversity is achieved or maintained is surprisingly obscure, despite its clear metabolic and pathophysiological impact. The Arv1 protein is represented in all eukaryotes and was initially identified in the model eukaryote Sacccharomyces cerevisiae as a candidate transporter of lipids from the endoplasmic reticulum. Human Arv1 has been shown to directly bind cholesterol and fatty acid affinity probes. Murine in vivo studies point to a role for ARV1 in regulating obesity, glucose tolerance, insulin sensitivity and brain function. Multiple human ARV1 variants have been associated with epileptic encephalopathy, cerebellar ataxia, and severe intellectual deficits. We hypothesize that Arv1 acts as an energy independent, lipid scramblase at the endoplasmic reticulum thereby modulating membrane lipid asymmetry and thus the trafficking of sterols and the substituents of glycosyl-phosphatidylinositol and sphingolipid biosynthesis.
Introduction
The movement of diverse lipids between organelles is a major component of membrane homeostasis and establishes the long known heterogeneous distribution of lipids within eukaryotic cells. For example, the eukaryotic plasma membrane (PM) contains the vast majority of cellular sterols while the endoplasmic reticulum (ER) is sterol-poor, despite being the primary site of sterol biosynthesis as well as the sensing organelle for sterol homeostasis. Similarly the sphingolipid composition of organelle membranes varies but is precise; a case in point, the ganglioside GM1 is often used as a specific marker of the Golgi in many subcellular fractionation experiments. Lipid heterogeneity further persists within the membranes of organelles; sterols and sphingolipids readily form a complex that is unevenly distributed thereby creating membrane microdomains. This asymmetric dissemination of lipids between and within cellular membranes is consequential, facilitating a vast array of cellular processes such as protein folding and trafficking, osmotic integrity, and signal transduction (see review, (Gulati et al., 2010)).
In mammalian cells, lipids such as nascent cholesterol are rapidly transported by an ATP-dependent mechanism (Liscum and Underwood, 1995), a process only partially accounted for by the secretory pathway as treatment with Brefeldin A to collapse the Golgi does not abrogate lipid movement (Liscum and Underwood, 1995; Hao et al., 2002). Membrane juxtapositions or contact sites (MCS) may be sufficient for diffusion or protein-mediated lipid transport, however until recently, the overall molecular mechanisms in this process have remained obscure. Several candidate subcellular cholesterol or sphingolipid carriers, either as components of human pathophysiology or in model organisms such as Saccharomyces cerevisiae, have been identified. For example, the proteins defective in human Niemann Pick disease (NPC1 & 2), a fatal neurodegenerative lysosomal lipidosis bind cholesterol and other substrates. Deprivation of these conduits impacts the distribution of a host of lipids such that exogenous sterols and plasma-membrane derived sphingolipids accumulate in the lysosome. The process is an ancient one; the NPC channels are impressively well conserved throughout eukaryotic evolution (Higaki et al., 2004) to the extent that expression of the yeast NP-C-related gene 1 (NCR1), or yeast NPC2, restores lipid transport to Chinese Hamster Ovary mutant cells lacking the corresponding orthologous proteins (Malathi et al., 2004). In this short review we will describe the role of another pivotal lipid transfer protein encoded by the ARV1 gene. Initially identified in a yeast genetic screen, this protein is also functionally and structurally conserved throughout eukaryotic evolution to the extent that variation in human ARV1 is strongly associated with neuronal degeneration and encephalopathy.
ARV1, a Historical Perspective
The sterol pool of the plasma membrane is mobile, and continually recycles with intracellular reservoirs. This distribution is maintained by the orchestrated equilibration of sterol biosynthesis, uptake, utilization (as lipoproteins, steroid hormones or bile acids), intracellular transport, and storage as steryl ester in cytoplasmic neutral lipid droplets. This latter process is mediated by the acyl-CoA cholesterol acyltransferases (ACATs). In a search for novel lipid transporters, we reasoned that the loss of any one of these homeostatic pathways (e.g., sterol esterification), particularly in model organisms such as yeast, might be tolerated, however removal of multiple membrane “detoxifying” events would be lethal. In S. cerevisiae, the ACAT-related enzymes (Are1 and Are2) are 49 percent identical to each other and exhibit 23 percent identity to human ACAT1 (Yang et al., 1996; Yu et al., 1996). These enzymes represent the principal pathway for esterification and thus storage of sterols. Accordingly, deletion of ARE1 and ARE2 produces a viable yeast cell with no detectable esterification activity (Arthington-Skaggs et al., 1996; Yang et al., 1996). We created mutations that confer viability dependence upon sterol esterification to identify the ARV1 (ARE2 Required for Viability 1) gene with the concept that such pathways control the flow of sterol from the ER to the plasma membrane (Tinkelenberg et al., 2000; Swain et al., 2002). The deletion of ARV1 in the context of deficient sterol esterification, is inviable, possibly due to accumulation of unesterified sterols in ER membranes at the expense of plasma membrane sterol pools (Tinkelenberg et al., 2000). Mutations in ARV1 were also isolated independently in a genetic screen for mutants defective in sphingolipid biosynthesis (J. Nickels, unpublished results). Lower steady state levels of complex sphingolipids, with an accumulation of hydroxylated ceramides and many other defects in lipid metabolism were indicative of a generalized defect in lipid export from the ER in arv1Δ mutant cells (Swain et al., 2002). Subsequently, the ARV1 gene product was hypothesized to act as a glycosyl-phosphatidylinositol (GPI) flippase (Figure 1) (Kajiwara et al., 2008; Okai et al., 2020), although since it lacks demonstrable ATPase activity, it might be more accurately considered a scamblase. Interestingly, unlike many lipid homeostats, ARV1 is unique (with the exception of plants); it is not duplicated, nor does it share any domains with other proteins. Thus if alternate “bypass” pathways exist, they are truly Arv1 independent, although they likely intersect and interact.
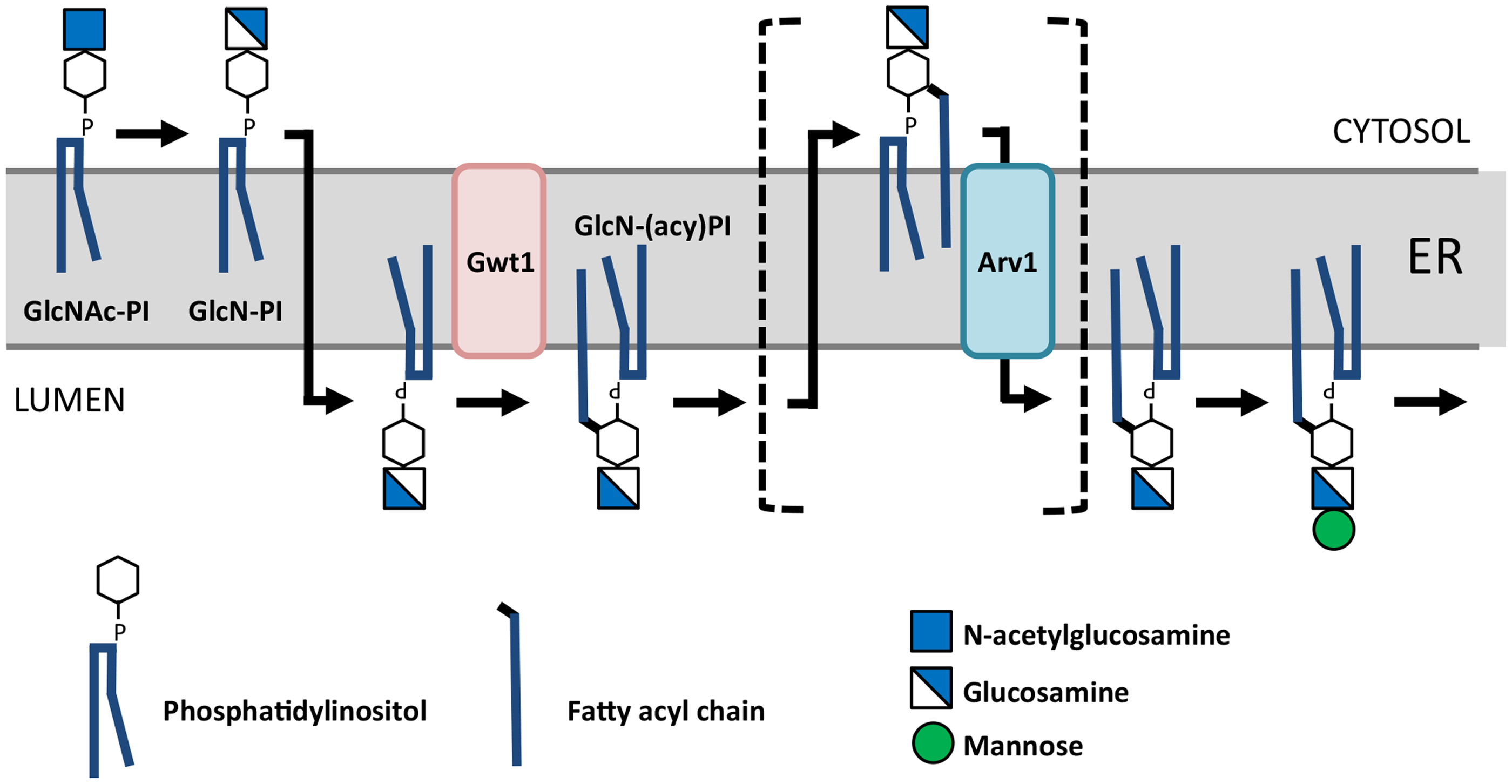
The Role of Arv1 in Glycosyl-Phosphatidylinositol (GPI) Synthesis. Reproduced with Permission (Okai et al., 2020). Arv1 is proposed to transfer (flip) glycosylamino-acyl-phosphatidylinositol (GlcN-PI) from the cytoplasmic side of the ER to the lumen side where it is used as a substrate for the further synthesis of GlcN-(acyl)PI and GPI anchors. It is plausible that as a consequence, sterols are rendered available to the membrane associated acylCoA cholesterol acyltransferases (ACATs) and intermediates in sphingolipid biosynthesis are further metabolized. Alternatively, ARV1 may encode a multi-substrate scramblase.
Evolutionary Conservation of ARV1
The yeast ARV1 gene predicts a 322 residue transmembrane protein that is conserved in every eukaryotic genome currently sequenced (Figure 2). Human ARV1 (hARV1) encodes a 271 amino acid protein, the expression of which complements all known phenotypes associated with loss of ARV1 in yeast (Scarv1Δ cells), including the GPI, sterol and sphingolipid transport deficiencies (Tinkelenberg et al., 2000; Swain et al., 2002; Ikeda et al., 2016). Human cell lines from individuals carrying an Arv1 Gly189Arg variant-associated with neurological deficiency, have reduced levels of GPI proteins. Interestingly, Scarv1Δ cells overexpressing the human Arv1 Gly189Arg variant showed only partial recovery of GPI synthesis and GPI-anchored protein maturation defects (Okai et al., 2020; Segel et al., 2020). A restoration in sphingolipid biosynthesis was suggested, but more detailed analyses need to be performed to resolve this issue. Nevertheless, the functional role of Arv1 seems likely to have been conserved throughout eukaryotic evolution.

Arv1 is maintained across eukaryotic evolution. The upper panel represents the evolutionary conservation of ARV across 17 vertebrates, 46 plants, 18 fungi and 12 protozoans. This conservation is entirely due to a ∼60 residue Arv1 homology domain (lower panel) which retains a cysteine rich motif that likely coordinates Zinc. No other domains other than trans membrane spanning α-helices are predicted.
Structural information can be highly informative and guide well-defined biochemical studies designed to elucidate function. However, Arv1, a transmembrane-associated protein, is recalcitrant to traditional biochemical structure/function analyses, in part due to limited homology at the prokaryotic species level, the lack of structural homology within the human genome, and absence of crystallographic or cryo-EM data. The primary sequence of Arv1 (Figure 2) from all eukaryotes indicates a cluster of cysteine residues, the spacing of which suggests a zinc ribbon, which may act to coordinate ligand complexing or protein-protein interactions. The predicted topology of Arv1p suggests this Arv1 homology domain (so termed because this 60 residue span displays the strongest conservation across organisms in the Arv1 molecule) faces the cytosol.
The AlphaFold Protein Structure Database is a repository of AI-generated 3D protein structures that contains the human proteome and is an invaluable source for uncovering protein function through identification and interpretation of predicted tertiary structure (Jumper et al., 2021; Varadi et al., 2022). AlphaFold predictions have human and yeast ARV1 structurally closely related although they share only 30% amino acid identity, making results gleaned from both species highly relevant for postulating the mechanistic function of human ARV1 based on structural predictions (Figure 3A).

Human ARV1 Topology. A, ribbon diagrams of human and yeast Arv1 proteins. B, transmembrane prediction profile. C, ribbon diagram showing human Arv1 amino acid substitutions found in individuals with epileptic encephalitis. The dashed faint horizontal lines define the approximate extent of the predicted bilayer.
Human ARV1 predicts a five transmembrane domain protein using Transmembrane DeepTMHMM (https://dtu.biolib.com/DeepTMHMM) (Hallgren et al., 2022) (Figure 3B). Amino acid substitutions of relevance to human disease, are shown in the ribbon diagram (Figure 3C). The neurological 101G > A nucleotide variant results in replacement of Cys34 with a tyrosine (Salian et al., 2021) within the zinc-binding domain motif of the AHD. Cys34 corresponds to Cys3 in S. cerevisiae and many pathogenic fungi that include C. albicans and C. glabrata, which are responsible for most fungal infections in individuals living with HIV (Perfect, 2017). Cys3 has been studied in detail in S. cerevisae and C. albicans. Studies have shown its requirement for fungal virulence (Villasmil et al., 2020), proper sterol distribution and localization (Gallo-Ebert et al., 2012), polarized cell growth during hyphal invasion (Gallo-Ebert et al., 2012), and normal sensitivity to sterol-targeting azole antifungal drugs (Villasmil et al., 2020). Similarly, the replacement of Gly189 with Arg within the third transmembrane domain and its placement based on ribbon diagrams may suggest a role in oligomerization of human Arv1 in cells (Figure 3B). Accordingly, fibroblasts from individuals expressing Arv1 (Gly189Arg) showed a significantly reduced level of the monomeric protein (Segel et al., 2020).
We have consistently found that E. coli expressed human Arv1-HIS recombinant protein purifies as a monomer, dimeric, and trimeric species, with enrichment in the dimeric and trimeric states even under denaturing conditions, suggesting Arv1 may exist as an oligomer whose subunit number is tied to its function. Dimeric and trimeric docking structures were predicted using the GalaxyWEB homomer predictive algorithm with the trimeric structure shown in Figure 4A and the hexameric shown in Figure 4B (Ko et al., 2012). The hexameric predictions show the presence of a tunnel within the Arv1 structure (Figure 4C) (Latterich and Schekman, 1994). Kajiwara et al., (Kajiwara et al., 2008) have suggested that ScArv1 is required to translocate glucosylamino-acyl-phosphatidylinositol (GlcN-acylPI) from the cytoplasmic side of the ER to the luminal side, possibly acting as a lipid scramblase (Figure 1), a process likely to be energy independent. It has been found that ScArv1 formed ER-associated, clustered foci in yeast cells that also contained Kar2 that is required for ER-membrane fusion (Latterich and Schekman, 1994; Kajiwara et al., 2008). These foci may represent an oligomeric form of ScArv1 or an oligomeric complex binding at specific membrane contact sites where it may shuttle lipids between organelles (K. Funato, personal communication). The proposed “Arv1 lipid tunnel” is made up of the transmembrane segments of the protein and could act as a conduit for lipid translocation by an Arv1 lipid scramblase activity (Hankins et al., 2015; Lyons et al., 2020; Giacometti et al., 2022). Determining whether Arv1 acts to either shuttle or flip lipids within membranes will require rigorous biochemical and biophysical analyses.

Human ARV1 May Oligomerize. A, ribbon diagram of a human Arv1 trimer. B, ribbon diagram of a human Arv1 hexamer. C, side view of a hexameric human Arv1 embedded in the ER membrane.
Purified human Arv1 protein has been shown to directly bind to cholesterol and fatty acid affinity probes using proteome-wide lipid binding methods. Along with severe defects in maintaining proper lipid distribution and localization (Swain et al., 2002; Villasmil et al., 2011; Gallo-Ebert et al., 2012; McCourt et al., 2016), Scarv1Δ cells show a fatty acid induced inviability phenotype (Ruggles et al., 2014). Therefore, the challenge remains how to discern the primary vs secondary defect in lipid metabolism caused by loss of ARV1.
Cellular Models of Arv1
The dual requirement of Arv1 and sterol esterification activity for viability in yeast was the basis of the initial identification of this putative sterol transporter. Subsequently, a more expansive role of the Arv1 protein in cell metabolism was proposed based on the vast array of phenotypes associated with loss of ARV1. The reduced viability of yeast ARV1 mutations under optimal growth conditions, was further exacerbated by elevated temperature, polyene antibiotics, or oxygen deprivation or fatty acid feeding. Biochemically, there is subcellular accumulation of endogenously or exogenously derived sterols at the expense of the plasma membrane, both by fluorometric assays and metabolic incorporation studies. Many of these phenotypes are clearly related to sterol metabolism, however Swain, Nickels and colleagues (Swain et al., 2002) also observed phenotypes associated with sphingolipid metabolism, consistent with a role for Arv1p in ceramide transport from the ER.
Many other Scarv1Δ cellular phenotypes (e.g., flocculence, calcofluor & caffeine sensitivity) are equally well explained by aberrant GPI-anchorage of cell wall proteins. Funato and colleagues (Kajiwara et al., 2008) were the first to postulate a role of Arv1p as a Glycosyl-phosphatidylinositol (GPI) scramblase, whereby an important GPI intermediate to the first mannosyltransferase of GPI synthesis fails to be flipped across the ER membrane into the lumen of in Scarv1Δ cells. Several lines of evidence strongly support the involvement of Arv1 in GPI biosynthesis. An Arv1-like protein in Trypaosoma brucei (TbArv1) was found to co-immunoprecipitate with a GPI-N-acetylglucoasmine transferase (GPI-GnT) involved in the first step of GPI anchor biosynthesis (Ji et al., 2021). Proteomic computational studies in S. cerevisae predicted that ScArv1 possibly interacts with Gpi1, the yeast ortholog of PIGQ (Humphreys et al., 2021). Liu et al. (2023) showed that human ARV1 associated with PIGQ, the human ortholog of GPI-GnT.
The pleiotropy of mutations in ARV1 led us to hypothesize that multiple homeostatic mechanisms must be disturbed by diminished transport of sterols to the plasma membrane. Microarray analysis of transcript profiles of ARV1 deficient cells compared to normal indicated multiple transcriptional responses (Shechtman et al., 2011; Ikeda et al., 2016). Some of the changes reflect the general response of any stressed cell. Strikingly, approximately 30% of the transcription responses reflect induction of the unfolded protein response (UPR) (Shechtman et al., 2011). Moreover, accumulation of CD55, a widely expressed GPI-anchored protein is correlated with activation of ER associated degradation (ERAD) and causes the upregulation of ARV1 expression and GPI synthesis in HEK293 cells (Li et al., 2023). We speculate that in the absence of sterol/lipid detoxification (by whatever mechanism), cell death is avoided or delayed by induction of stress responses. One such response is UPR activation, which serves to upregulate a series of ER associated folding chaperones and degradation pathways to deal with the accumulation of misfolded proteins before they aggregate and exacerbate the lipotoxicity (Kajiwara et al., 2012; Hong et al., 2024).
Murine Models of Arv1: A Role in Lipid Homeostasis, Obesity and Cognition
Murine in vivo studies point to a role for Arv1 in regulating obesity, glucose tolerance, and insulin sensitivity (Lagor et al., 2015; Gallo-Ebert et al., 2018) and mice deficient in neuronal ARV1 show multiple neurological defects (Palmer et al., 2016). Initial anti-sense oligonucleotide studies in mice and the use of mouse knockouts have built a fundamental understanding of the role of mammalian Arv1 in lipid biology, while uncovering an unexpected role in cognitive function and neurological development.
Early anti-sense oligonucleotides studies suggested that Arv1 may regulate FXR signaling and de novo cholesterol biosynthesis through altering changes in bile acid composition and SREBF1 gene expression (Tong et al., 2010). Knock down of ARV1 in the livers of mice caused hypercholesterolemia, elevated levels of serum bile acids, and increased gene expression of the FXR target gene, SHP. Expression levels of the ABCB11 efflux pump and taurocholate NTCP bile acid influx pump were increased and reduced, respectively, indicating increased bile acid excretion out of the liver. Expression of CYP7α1, which encodes the rate-limiting enzyme in bile acid synthesis, was reduced, possibly explaining the hypercholesterolemic phenotype.
In the same study, it was observed that ARV1 knockdown in HepG2 cells reduced CYP7α1 expression, consistent with in vivo findings, while also reducing SREBF1 and HMGCR expression. CYP7a1-deficient mice fed a western diet, like ARV1 knockdown mice, display altered bile acid composition and elevated FXR and SHP gene expression, Based on their data, the authors suggested that Arv1 acts as a central hub for directing cholesterol trafficking from the ER to various other organelles. When Arv1 function is ablated, cholesterol accumulation in the ER will result in increased bile acid synthesis and excretion, and decreased SREBP1c-dependent gene expression. However, eventually cholesterol accumulation would cause dysfunction within the ER.
Based on the metabolic phenotypes seen in Arv1−/Arv1− mice, ARV1 may act as a lipid “rheostat/sensor” that controls general/specific lipid transport in response to nutrient uptake and caloric overload. Arv1−/Arv1− knockout mice generated by Lagor and colleagues (Lagor et al., 2015) were tested in metabolic feeding studies. Fed a chow diet, these mice did not gain weight or increase their white adipose tissue mass when compared to matched wild type littermates. Blood cholesterol and high-density apolipoprotein levels were decreased, whereas energy expenditure rates were increased, and increased fatty acid oxidation was observed. Importantly, no loss of muscle mass was observed, so lack of weight gain due to ablation of ARV1 was related to altered nutrient utilization.
A second Arv1−/Arv1− mouse was later challenged with a high fat western diet and its metabolic response was characterized (Gallo-Ebert et al., 2018). These mice were resistant to high fat diet-induced obesity. Liver triglyceride levels were normal and there were no signs of hepatic steatosis. They remained insulin sensitive and glucose tolerant throughout the study. FXR signaling was activated, while de novo cholesterol and bile acid syntheses were reduced. Fibroblast growth factor 21 (FGF21) protein levels were elevated, suggesting activated peroxisome proliferator-activating receptor γ (PPARγ) signaling in the liver. Determining the effect of ablating ARV1 in genetically obese ob/ob and diabetic db/db mice on the phenotypes of these mice should help to see if inhibiting ARV1 is an avenue for drug discovery for treating weight loss and type 2 diabetes.
Cholesterol must be synthesized de novo in the brain representing ∼25% of the body's cholesterol, thus its proper handling is essential for maintaining brain function (Hussain et al., 2019). Neuronal function has been shown to be impaired due to either loss or aberrant accumulation of cholesterol in neurons (Hussain et al., 2019). At the subcellular level, aberrant distribution of cholesterol pools between the ER and mitochondria at membrane contact sites may lead to the neurodegeneration associated with Alzheimer's and Parkinson diseases (Arenas et al., 2017). Acccordingly, Palmer et al., (Palmer et al., 2016) showed that mice ablated for neuronal ARV1 (ARV1 NKO) displayed frequent circling behavior and were predisposed to severe seizures phenotypes as seen in humans with ARV1-variant epileptic encephalopathy (see below). Interestingly, ARV1 NKO mice fed a chow diet gained less weight and had reduced white adipose mass, like Arv1−/Arv1− mice (Lagor et al., 2015; Gallo-Ebert et al., 2018), indicating a function for brain ARV1 in regulating whole body lipid metabolism. Challenging these mice with a western diet and determining the metabolic responses could have the potential to differentiate what specific aspects of lipid metabolism are impacted by the activity of ARV1 in the brain.
Arv1 and Human Pathophysiology: A Role in Neuronal Function?
Given the marked hepatic and neuronal pathology associated with deprivation of Arv1p in murine models, we strongly anticipated a disease state to be associated with variation in this gene in humans. To date, this appears to predominantly manifest as neurological impairment; ten human ARV1 variants have been linked to epileptic encephalopathy, cerebellar ataxia, and severe intellectual deficits (Figure 5 and Table 1). A comprehensive review of each case report has been published elsewhere (Kamate and Basavanagowda, 2023). Below, we summarize data supporting a role for Arv1 in GPI-anchor biosynthesis that is consistent with the Arv1-variant phenotypes, particularly considering the pivotal role of neuronal GP I-anchored proteins in central nervous system development(Um and Ko, 2017).

Single Nucleotide Polymorphisms in Human ARV1. Note these variants are non-synonymous, 3 of which are in the ARV1 homology domain (AHD) and thus likely to impact activity of Arv1 (see table 1 for more details).
Human ARV1 Variants and Associated Consequences.

When overexpressed, human ARV1 suppresses the GPI biosynthesis defects of Scarv1Δ cells, indicating conservation in function (Ikeda et al., 2016). Subsequent complementation analyses suggest that mutations in human ARV1 linked to certain infantile seizure disorders are caused by defects in GPI biosynthesis. Further supporting evidence is the fact that cells from afflicted patients have reduced maturation of GPI-anchored proteins (Davids et al., 2020; Segel et al., 2020). Even with this mounting evidence, however, the question remains whether loss of human ARV1 causes a direct or indirect effect on GPI-anchor biosynthesis (Liu et al., 2023). It must be pointed out that in only one case has a human ARV1 mutant been tested for the ability to suppress the sphingolipid biosynthesis defects of Scarv1Δ cells (Segel et al., 2020). None were tested for the ability to restore normal sterol trafficking.
A homozygous ARV1 variant (565G > A:p.Gly189Arg), was first identified in children with neurogenetic disorders born of consanguineous parents (Alazami et al., 2015). Subsequently, two homozygous ARV1 mutant variants (294 + 1G > A:pLys59_Asn98del; 674-2A > T:p.Thr266_Phe271del) were linked to infantile seizure disorders including epileptic encephalopathy 38, which is an autosomal neurological disease linked to defects in GPI biosynthesis (Davids et al., 2020). While the Lys59_Asn98del was unable to complement the temperature sensitivity phenotype of Scarv1Δ cells when overexpressed, the Gly189Arg mutant could partially restore growth (Palmer et al., 2016; Segel et al., 2020). The Gly189Arg could not, however, suppress the GPI biosynthesis defects, as there remained the sustained decrease in the maturation of the GPI-anchored Gas1 protein (Segel et al., 2020). The Gly189Arg may have suppressed the sphingolipid biosynthesis defects observed in Scarv1Δ cells (Swain et al., 2002). Whether this allele restored normal sterol trafficking to mutant cells was not tested.
Cell culture experiments further tied ARV1 function to GPI-anchor protein trafficking, seen as reduced expression of GPI-anchor protein biomarkers at the plasma membrane. Neutrophils from patients harboring the ARV1 Thr266_Phe27del allele had reduced levels of the GPI-anchored proteins CD16, CD66b, CD55 & 59, and FLAER (Davids et al., 2020). Fibroblasts from a patient harboring the ARV1 Lys59_Asn98del allele had decreased expression of CD59 and CD87 (Palmer et al., 2016). Fibroblasts from a patient with a homozygous c.363_364del, p:Ser122Glnfs*7 frameshift allele had reduced levels of CD73 and CD109 (Salian et al., 2021). Lentiviral expression of human ARV1 in these cells fully rescued the reduced expression levels of GPI-anchor defects. Fibroblasts from patients carrying the p.Gly189Arg allele had decreased levels of FLAER, CD73, and CD59. Lentivirus expression of ARV1 partially rescued the decrease in FLAER expression while completely rescuing CD73 and CD59 cell surface levels. As with yeast complementation assays, cells were not tested for restoring normal cholesterol trafficking in HepG2 cells (Tong et al., 2010). Overall, results do suggest that ARV1 regulates some aspect of GPI-anchor biosynthesis. Only when the molecular function of ARV1 has been uncovered will we resolve how it does so at the molecular level and how this accounts for the striking metabolic consequences of variation in this lipid transporter.
Conclusions and Alternate Models: is ARV1 a Lipid Scramblase?
The endoplasmic reticulum represents a protected, sequestered environment for protein folding and lipid homeostasis. As such it is subjected to numerous levels of quality control that are pivotal to cell function. The discovery and characterization of Arv1 mediated pathways suggests a role for lipid metabolism, particularly lipid efflux, in the homeostasis of this key compartment of the cell. Arv1 appears to check all the boxes for a lipid transporter; it is clearly an ER resident, it binds lipids, it interacts with multiple lipid regulatory proteins and its deficiency results in numerous lipid related consequences. That said, an impartial reader might question the precise biological role of Arv1p, particularly in terms of primary vs. secondary roles. For example, what is the connection between Arv1's apparent role in lipid trafficking and GPI biosynthesis and regulation of membrane contact sites that links lipid homeostasis and cognitive function? The mouse and human phenotypic data implicating Arv1 in cognitive function has become well established, but cell culture studies are needed to uncover the causative mechanism. Very little is known about how Arv1 functions to regulate lipid metabolism and GPI synthesis, even though it was identified over twenty years ago.
Preliminary two-hybrid screening in our laboratory using the hAHD identified several proteins involved in endoplasmic reticulum/Golgi transport (Liu and Nickels, unpublished data). One such Arv1 ligand; the RING finger protein 139, also known as TRC8, is an E3 ubiquitin ligase known to interact with the sterol cleavage activating protein (SCAP)-SREBP-2 complex and hinder its translocation from the endoplasmic reticulum to the Golgi. A recent report has suggested that SCAP regulates an oxysterol binding protein complex involved in cholesterol transport at ER-Golgi membrane contact sites (Apodaca and Brown, 2014; Wakana et al., 2021). Fitting in with this result is the fact that cholesterol has been shown to regulate multiple different organelle membrane contact sites (Ridgway and Zhao, 2018). Perhaps TRC8, by causing the rapid degradation of the SCAP-SREBP-2 complex, decreases cholesterol biosynthesis. If ARV1 is involved in human sterol transport, as in both yeast (ergosterol) (Villasmil et al., 2011; Gallo-Ebert et al., 2012; McCourt et al., 2016) and mice (Tong et al., 2010), there may exist an ARV1-TRC8-SREBP2 axis regulating sterol levels to sustain proper membrane contact site fusion. Arv1's role would be cholesterol transport, and possibly GPI synthesis that would be required for modification of proteins necessary for contact site biogenesis/function.
Is Arv1a lipid scamblase whose function is essential for maintaining metabolic and neurologic health? It is tempting to speculate that by being localized within plasma membrane-associated tubules of the cortical ER and Golgi-associated ER (Swain et al., 2002; Tong et al., 2010), Arv1 acts as a lipid “sensor” and/or lipid scamblase necessary for maintaining proper membrane contact site interactions. The lipid-specific Arv1-dependent activity would depend on the lipid to which it is bound (“sensing”), which would then trigger its lipid transport activity that is not only involved in ensuring proper lipid symmetry between organelles but lipid shuttling through specific pathways for sphingolipid and/or GPI synthesis (Devaux et al., 2008; Ristovski et al., 2021). Saccharomyces cerevisae cells lacking ScARV1 have abnormal ER and PM morphologies (Georgiev et al., 2013). It is known that ER-mitochondrial membrane contact sites are enriched in ceramides and sterol (Scorrano et al., 2019). Loss of ARV1 in yeast results in accumulation of ceramides and defects in sterol trafficking (Swain et al., 2002; Villasmil et al., 2011), suggesting the possibility that the putative transport activity of Arv1 may include sterols and sphingolipids. Human Arv1 contains several cholesterol binding motifs (CRAC/CARC) (Fantini and Barrantes, 2013), some within the AHD, consistent with an interaction with cholesterol or other sterol intermediates at membrane contact sites (Muallem et al., 2017).
The list of lipid transporters within membrane contact sites is ever increasing (Scorrano et al., 2019; Castro et al., 2022). Yeast Csf1 has been proposed to act at membrane contact sites and shuttle phosphatidylethanolamine to Mcd4 to be used as a substrate for GPI synthesis (Toulmay et al., 2022). An ortholog of Csf1 is found in humans and is required for synaptic vesicle recycling (Verstreken et al., 2009). The yeast Lam6/Ltc1 protein (Gatta et al., 2015) that is an ortholog of the StART family of cholesterol binding proteins (Epand, 2006) transports sterol across ER-mitochondrial membranes (Elbaz-Alon et al., 2015) and acts as a tether for stabilizing ER-mitochondrial contact sites (Kornmann et al., 2009; Murley et al., 2015).
If Arv1 is a bona fide lipid scramblase, its function must be critical for the exchange of lipids back and forth between adjacent membranes to maintain proper MCS lipid composition under different physiological conditions (Montigny et al., 2016; Hammond and Burke, 2020; Lopez-Marques, 2021). The activity of Arv1 may translocate lipids with diverse structures based on the pleiotropic defects seen with its loss in yeast and possible propensity for binding several lipid species in human cells. The profound consequences of loss of Arv1to any cell, from the unicellular context of a yeast to the multicellular complex environment of the human brain strongly suggests a key role of the MCS in pathophysiology and merits further pursuit.
Footnotes
Declaration of Conflicting Interests
The authors declared no potential conflicts of interest with respect to the research, authorship, and/or publication of this article.
Funding
The authors disclosed receipt of the following financial support for the research, authorship, and/or publication of this article: Supported in part by awards from the Ara Parseghian Medical Research Foundation and Dana's Angels Research Trust, NIH, (grant number DK54320).