
Abstract
Membrane contact sites (MCSs) are specialized regions where two or more organelle membranes come into close apposition, typically separated by only 10–30 nm, while remaining distinct and unfused. These sites play crucial roles in cellular homeostasis, signaling, and metabolism. This review focuses on ion channels, transporters, and receptors localized to MCSs, with particular emphasis on those associated with the plasma membrane and endoplasmic reticulum (ER). We discuss the molecular composition and functional significance of these proteins in shaping both organelle and cellular functions, highlighting their importance in excitable cells and their influence on intracellular calcium signaling. Key MCSs examined include ER–plasma membrane, ER–mitochondria, and ER–lysosome contacts. This review addresses our current knowledge of the ion channels found within these contacts, the dynamic regulation of MCSs, their importance in various physiological processes, and their potential implications in pathological conditions.
Keywords
Introduction
Eukaryotic cells contain a variety of membrane-bound organelles, each with specialized functions and distinct spatial organizations. The nucleus, housing genetic material, typically occupies a central position, while the endoplasmic reticulum (ER) extends throughout the cytoplasm, forming an intricate network. Mitochondria, Golgi apparatus, endo/lysosomes, lipid droplets, and peroxisomes are generally distributed throughout the cell, with their positioning often dynamic and responsive to cellular needs (Valm et al., 2017; Guo et al., 2018). These morphologies and spatial organizations facilitate inter-organelle communication at membrane contact sites (MCS). Historically, intracellular organelle MCS were broadly defined as regions of close apposition between the membranes of two organelles. However, recent advances in our understanding of the molecular mechanisms underlying MCS formation and function necessitate a more precise and evolving definition. Here, we define MCS as specialized cellular domains where two distinct cellular membranes are tethered in close proximity (typically within 10–30 nanometers) without fusing, allowing for communication and exchange of molecules between different cellular compartments (Figure 1A) (Eden, 2016; Phillips and Voeltz, 2016; Scorrano et al., 2019; Prinz et al., 2020; Guillén-Samander and De Camilli, 2022). These MCSs play crucial roles in processes such as lipid transfer, calcium (Ca2+) signaling, and metabolic regulation. This review concentrates on Ca2+ transfer at MCS in excitable cells by examining the molecular tethering mechanisms, regulatory processes, maintenance of MCS architecture, and their functional roles in Ca2+ homeostasis and signaling.
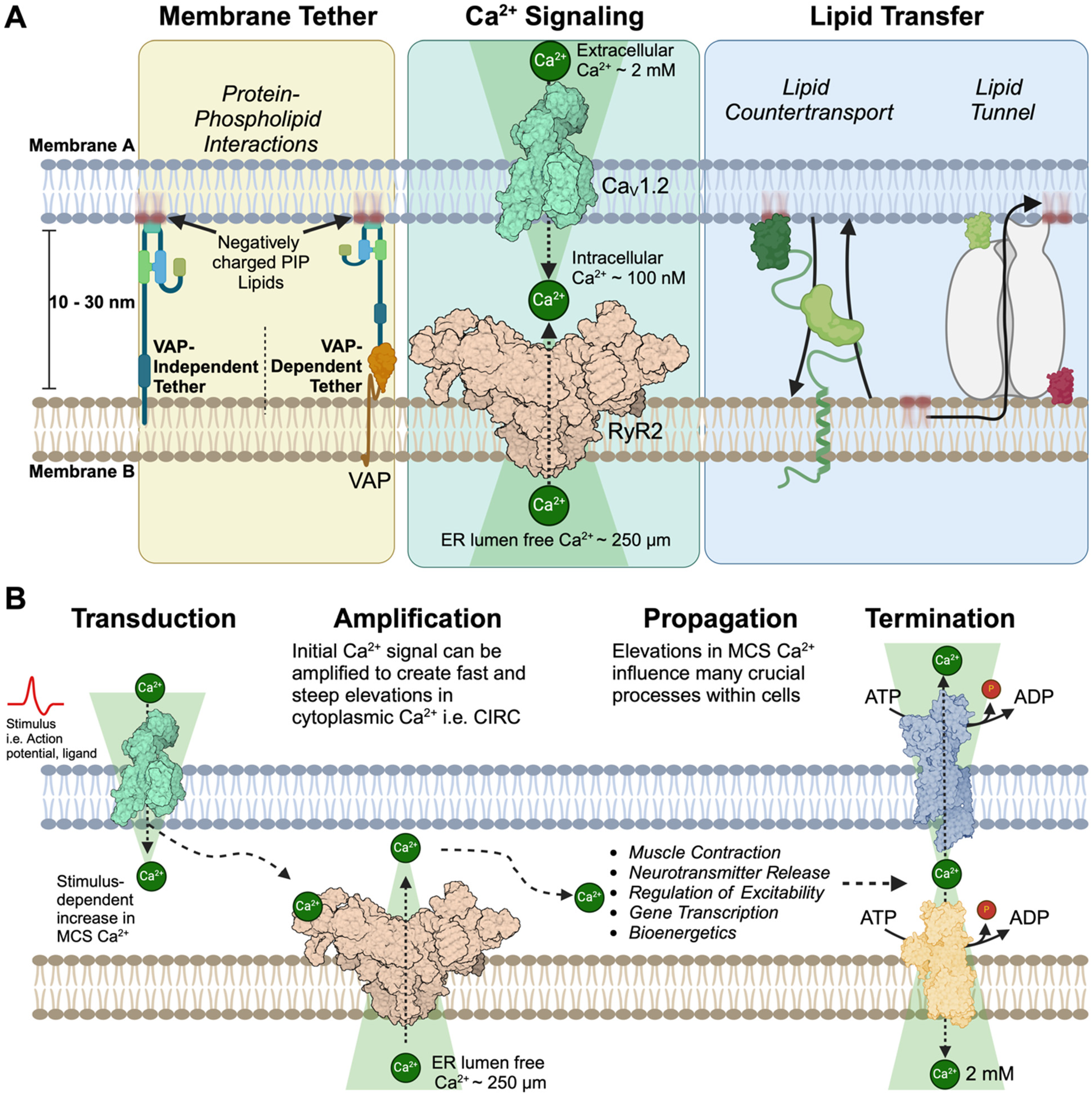
Molecular composition of a membrane contact site.
The center of MCS formation and function is the ER, a large, multifaceted, and dynamic organelle comprised of interconnected tubules and cisternae that extend throughout the cytoplasm. It's extensive surface area, motility, and close association with other organelles make the ER an ideal platform for establishing contacts with other organelles. MCS between the ER and other organelles are not the result of stochastic interactions between adjacent lipid bilayers. Rather, they are highly regulated structures formed through the selective enrichment of specialized proteins that mediate the tethering of membranes and functionality of inter-organelle communication (Voeltz et al., 2024) (Figure 1A). This review does not focus on the numerous proteins that have been identified as bona fide MCS tethering proteins i.e., extended-synaptotagmins (E-Syts) (Giordano et al., 2013), oxysterol-binding protein-related proteins (ORPs) (Chung et al., 2015), or, proteins essential for membrane contact site formation i.e., vesicle-associated membrane protein (VAMP)-associated proteins (VAPs) (Murphy and Levine, 2016). Instead, we focus on specific ion channels that have been identified and characterized as being present and functionally important at MCS.
At MCS, various membrane proteins—including ion channels, transporters, receptors, and pumps play pivotal roles in orchestrating both electrical and chemical signaling cascades. These proteins function as initiators, transducers, amplifiers, and terminators of signals within specialized nanodomains (Figure 1B). Among the key signals regulated by ion channels at MCS is cytoplasmic Ca2+. Under resting conditions, cytoplasmic Ca2+ concentrations are maintained at low levels (∼100–200 nM) in most cell types. Localized elevations in intracellular Ca2+ at MCS serve as instructive signals, modulating a diverse array of cellular processes, including signal transduction, muscle contraction, energy production, synaptic plasticity, cell survival, and stress responses (for review please see Wei et al., 2012; Clapham, 2007; Bers, 2002; Berridge, 1998; Ghosh and Greenberg, 1995). The need for precise spatial and temporal control of Ca2+ signaling at MCS is underscored by the wide range of pathological conditions that result following dysregulation of Ca2+ signaling at these MCS nanodomains (Gomez et al., 2001; Bers and Guo, 2005; Bezprozvanny, 2010; Navedo et al., 2010; Kawamoto et al., 2012; Brini et al., 2014; Alzheimer's Association Calcium Hypothesis Workgroup and Khachaturian, Z.S, 2017; Pchitskaya et al., 2017; Zaichick et al., 2017).
This review focuses on ion channels that are found in the ER and apposing MCS membranes, we discuss the molecular composition and functional significance of these ion channels in shaping both organelle and cellular functions with a particular emphasis on MCS in excitable cells and how they influence a broad range of physiological outputs through elevations in intracellular Ca2+. The importance of reviewing and understanding the molecular mechanisms governing MCSs provides insights into their roles in health and disease, potentially unveiling new therapineutic targets for conditions associated with dysfunctional organelle interactions.
Endoplasmic Reticulum—Plasma Membrane MCS
Endoplasmic reticulum–plasma membrane (ER–PM) MCS, often referred to as ER–PM junctions, have been the subject of intense study in excitable cells for more than six decades. This sustained interest stems from several key factors that have shaped our understanding of these crucial cellular structures. The initial spark in interest came from early electron microscopy studies on the sarcoplasmic reticulum (SR) (a specialized form of ER in muscle cells) in striated muscle, which revealed the close apposition between the SR and the plasma membrane (SR–PM) (Porter and Palade, 1957). This discovery laid the groundwork for further investigations into the nature and function of these junctions. Following this initial observation, researchers discovered in skeletal muscle that Ca2+ influx through dihydropyridine receptors (DHPRs) on the plasma membrane—also known as voltage-gated Ca2+ channels or CaV—is not necessary to activate ryanodine receptors (RyRs) in SR membranes. Instead, a direct mechanical coupling mechanism occurs across the MCS junction between CaV and RyRs, efficiently translating action potentials into muscle contraction (Figure 2A) (Schneider and Chandler, 1973; Rios and Brum, 1987; Block et al., 1988). This finding highlighted the crucial role of SR-PM MCS in excitation-contraction (EC) coupling, the fundamental process that facilitates contraction of muscle myocytes and enables us to breath, stand, and walk. Further facilitating investigations into SR–-PM MCS is their peripheral location within cells. This accessibility has enabled researchers to employ techniques such as patch-clamp electrophysiology and total internal reflection fluorescence (TIRF) microscopy, allowing MCS, the ion channels localized to MCS, and their downstream processes to be resolved at high temporal and spatial resolution. Another factor contributing to the extensive research in this field is the evolutionary conservation of key proteins present in ER–PM contacts. Proteins such as STIM1, Orai1, and junctophilins are found across diverse species, enabling comparative studies that have provided valuable insights into the fundamental nature and importance of these structures in cellular physiology. In the sections that follow, we will detail our current understanding of ion channels at ER–PM MCS, focusing on muscle cells (skeletal, cardiac, and smooth), and neurons. We will highlight the adaptations, molecular elements and functional significance of these structures in each cell type, providing a comprehensive overview of their role in cellular physiology and underscoring their importance in health and disease.

Ion channels in SR-PM MCS in muscle cells.
Muscle
As introduced above, EC coupling in muscle cells is the process that translates electrical stimuli into mechanical contraction. This mechanism critically depends on the intimate spatial relationship between ion channels in the plasma membrane (termed sarcolemma in muscle tissue) and ligand-gated receptors in the SR (referred in this section as SR–PM MCS). In both skeletal muscle fibers and cardiac myocytes, EC coupling is initiated by the depolarization of the sarcolemma which rapidly propagates into specialized membrane invaginations called transverse tubules (T-tubules; Figure 2A). The depolarization activates voltage-gated L-type calcium channels located within these T-tubules, specifically CaV1.1 in skeletal muscle and CaV1.2 in cardiac muscle. These activated CaV channels then interact with RyRs situated on the adjacent SR membrane, which serves as the primary intracellular Ca2+ reservoir in muscle cells. The interaction between L-type calcium channels and RyRs, which occurs through either mechanical (CaV1.1) or chemical coupling (CaV1.2; to be discussed in detail in subsequent sections), triggers Ca2+ release from the SR into the cytoplasm. This localized elevation in cytoplasmic Ca2+ concentration is crucial for initiating the molecular events that ultimately lead to muscle contraction. Therefore, the process of EC coupling in different muscle types relies on precise localization and communication between specific ion channels and receptors at SR–PM MCS. In the following sections, we will detail the molecular components across various muscle tissues. These sections will highlight the specialized adaptations of EC coupling machinery, underscoring the importance of SR–PM junctions in muscle physiology and their role in facilitating the precise and rapid signal transduction necessary for muscle contractions.
Skeletal Muscle
Skeletal muscle triads are specialized structures crucial for EC coupling. These triads consist of three distinct elements: a central T-tubule flanked by two terminal cisternae of the SR (1:2 ratio PM:SR; Figure 2A). This unique arrangement forms two closely apposed SR–PM MCSs within each triad, typically spanning 10–15 nm.
Membrane Tethers
At skeletal muscle triads Junctophilin 1 and 2 (JPH1 and JPH2) play key roles as membrane tethers (Ito et al., 2001; Komazaki et al., 2002). Evidence supporting a role for JPH1 and JPH2 as MCS tethers in skeletal muscle myocytes include their localization and enrichment at Triads (Rossi et al., 2019), electron microscopy analysis of JPH1 and JPH2 knockout mice showing reduced formation of triad junctions between T-tubules and SR (Ito et al., 2001; Komazaki et al., 2002), and functional studies correlating changes in MCS formation with impaired EC coupling, leading to reduced contractile force at low stimulation frequencies and abnormal sensitivity to extracellular calcium levels (Ito et al., 2001; Komazaki et al., 2002). In addition to JPH1 and JPH2, triad morphology in skeletal muscle is supported by mitsugumin29 (MG29) (Komazaki et al., 1999; Nishi et al., 1999) and bridging integrator 1 protein (BIN1) (Lee et al., 2002). A role for MG29 in supporting triad morphology comes from electron microscopy studies characterizing its localization to the SR during muscle development and MG29 knockout mice showing abnormal ultrastructure of skeletal muscle, including swollen T-tubules, irregular SR structures, and partial alteration of triad junctions (Komazaki et al., 1999; Nishi et al., 1999). Functionally, MG29 knockout mice models also have impaired EC coupling (Nishi et al., 1999). A final component implicated in MCS formation/maintenance in skeletal muscle is BIN1 where a muscle-specific isoform of BIN1 has been shown to localize to T-tubules in skeletal muscle (Lee et al., 2002), is necessary for triad formation, and its constitutive deficiency leads to death at birth due to a skeletal muscle feeding defect (Prokic et al., 2020). While these data support a role for BIN1 in early muscle organization and position, inducible knockout of BIN1 in adult murine models had little impact (Prokic et al., 2020) on triad organization or function, presenting a model where BIN1 is necessary for triad development and function whereas other tethers contribute to the stability of triads with aging.
Molecular Organization of Skeletal Muscle ion Channels, Receptors, and Transporters
As noted above, the molecular elements responsible for EC at SR-PM MCS begins with DHPR on transverse (T) tubule membranes. DHPR are voltage-gated Ca2+ channels consisting of α1, β, γ, and α2δ subunits, with the α1 subunit forming the ion-conducting pore and voltage sensor (Wu et al., 2015) (Figure 2A). In skeletal muscle the predominant voltage-gated Ca2+ channel isoform is CaV1.1 (Catterall, 2000). Apposing DHPR channels are Ryanodine receptor 1 (RyR1) channels on SR membranes (Figure 2A). Upon membrane depolarization, CaV1.1 undergoes conformational changes that are physically communicated to RyR1, triggering the opening of RyR1 channels on the SR membrane (Schneider and Chandler, 1973; Gonzalez-Serratos et al., 1982; Rios and Brum, 1987; Dirksen and Beam, 1999). This allows Ca2+ ions to move down their concentration gradient and flow from the SR into the cytosol (concentration gradient ∼ 5,000 : 1, SR : cytosol), initiating the cascade of events leading to muscle contraction. In this way, CaV1.1 functions as a voltage sensor, translating membrane potential changes in the plasma membrane into structural modifications of RyR1 in the SR membrane enabling rapid Ca2+ release into the cytoplasm. In skeletal muscle, Ca2+ released from the SR through RyR1 channels binds to troponin C, causing a conformational change that exposes myosin-binding sites on actin filaments. This allows myosin heads to interact with actin, initiating cross-bridge cycling and muscle contraction, with the force and speed of contraction directly related to the concentration of free cytosolic Ca2+ (Reviewed in Geeves and Holmes, 1999; Gordon et al., 2000; Rebbeck et al., 2014; Sweeney and Houdusse, 2010). Collectively, these tightly orchestrated elements ensure efficient EC coupling in skeletal muscle. At junctional SR–PM MCS RyR1 can be regulated by various factors to tune Ca2+ release, including Ca2+ itself, ATP, Mg2+, phosphorylation, and interacting proteins such as FKBP12 and calmodulin (Rodney et al., 2000; Danila and Hamilton, 2004; Lanner et al., 2010; Rebbeck et al., 2014). Skeletal muscle also contains a high concentration of sarco/endoplasmic reticulum Ca2+-ATPase type 1 (SERCA1). These pumps are primarily localized to the longitudinal SR tubules rather than at junctional SR–PM contact sites (Rossi et al., 2022). SERCA1 activity in the longitudinal tubules facilitates rapid Ca2+ reuptake, promoting muscle relaxation and enabling subsequent contraction cycles. For more detailed information on the organization and molecular elements responsible for EC coupling in skeletal muscle please see following excellent reviews (Franzini-Armstrong, 1970; Block et al., 1988; Franzini-Armstrong and Protasi, 1997; Franzini-Armstrong et al., 1999; Protasi et al., 2002; Al-Qusairi and Laporte, 2011)
Cardiac Myocytes
In cardiac muscle cells, EC coupling is a complex process that also links electrical excitation of the sarcolemma to contraction of the myofilaments. Central to this process is the dyad, which consists of a junctional complex formed by T-tubules of the sarcolemma and a single SR membrane (1:1 ratio; Figure 2B). This arrangement ensures efficient coupling between the depolarization of the sarcolemma and the release of Ca2+ from the SR, essential for initiating cardiac myocyte contraction.
Membrane Tethers
in cardiac myocytes, the formation and maintenance of SR-PM MCS are primarily mediated by three key proteins: junctophilin-2 (JPH2), amphiphysin II/bridging integrator 1 protein (BIN1), and nexilin (NEXN).
Molecular Organization of Cardiac Ion Channels, Receptors, and Transporters
On the sarcolemma side of the dyad, CaV1.2 voltage-gated L-type Ca2+ channels are the most prominent isoform that sense T-tubule membrane depolarization and undergo conformational changes allowing the pore forming alpha subunit (α1) to open and permit Ca2+ to flow down its concentration gradient into the cytosol (Figure 2B). In cardiac dyads, CaV1.2 channels form distinct clusters of approximately 5–8 channels that profoundly influence their functional properties (Navedo et al., 2005; Dixon et al., 2012; Dixon et al., 2015). This clustering phenomenon facilitates cooperative gating, a mechanism that enhances channel activity. Substantial evidence demonstrates that the spatial proximity of CaV1.2 channels within these clusters promotes Ca2+/calmodulin-dependent C-terminal interactions between adjacent channels, leading to an increased open channel probability (Dixon et al., 2012; Dixon et al., 2015). This cooperative behavior effectively amplifies the Ca2+ influx, which in turn amplifies CaV1.2 clustering to help tune EC coupling. As noted, the size and composition of CaV1.2 clusters are not static but dynamically regulated by various physiological and pathological factors. Adrenergic receptor signaling, a key modulator of cardiac function, can increase cluster size and arrangement, thereby fine-tuning Ca2+ influx in response to sympathetic stimulation to meet hemodynamic demands (Ito et al., 2019). Age-related increases in CaV1.2 clustering (to form “super-clusters” Westhoff et al., 2024) also impacts cardiac physiology and may contribute to altered Ca2+ handling and lack of sympathetic nervous system responsivity in the aging heart (Westhoff et al., 2024). Moreover, certain disease-associated mutations can disrupt normal clustering patterns, leading to aberrant channel function and potentially contributing to cardiac pathologies (Navedo et al., 2010).
Unlike skeletal muscle, opening of CaV1.2 channels on the sarcolemma in cardiac myocytes facilitates rising Ca2+ concentrations in the dyadic cleft which bind and activate nearby RyR2 to facilitate a process called Ca2+-induce Ca2+-release (CICR). Here, Ca2+-binding to RyR2 sensitizes the receptor and allows Ca2+ release from the SR to amplify the dyad Ca2+ signal, resulting in a large and rapid elevation in cytosolic Ca2+ (Figure 2B). Like CaV1.2, RyR2 exhibit the ability to form clusters (Sun et al., 1995; Baddeley et al., 2009). The spatial arrangement of RyR2 channels within these clusters facilitates inter-channel interactions, leading to coordinated gating behavior (Dixon et al., 2022). This cooperative activity can amplify Ca2+ release events, thereby fine-tuning CICR for cardiac EC coupling (Cheng et al., 1993). As with C aV1.2 channels, RyR2 clusters increase with aging to increase Ca2+ release (Westhoff et al., 2024).
In cardiac myocytes, elevated cytosolic Ca2+ binds to troponin C on the actin filaments, initiating a series of events that lead to muscle contraction (Gomez et al., 2001; Wang et al., 2001; Bers, 2002). While junctional SR-PM MCS are responsible for Ca2+ entry into the cytoplasm, the longitudinal SR is dedicated to the removal of Ca2+ from the cytosol. Following myocyte contraction, Ca2+ ions are rapidly removed from the cytosol by the SR Ca2+-ATPase (SERCA2a), located on the SR membrane and exchanged via the Na+/Ca2+ exchanger (NCX) on the sarcolemma. Therefore, SERCA2a and NCX maintain low cytosol Ca2+ levels and prepare myocytes for the next contraction cycle. CICR is tightly regulated by various factors, including membrane potential, Ca2+ concentration gradients, and the activity of regulatory proteins such as calmodulin, calsequestrin, and FKBP12 (Bers, 2002; Bers and Guo, 2005).
Smooth Muscle
Smooth muscle tissue, found in the walls of hollow organs such as blood vessels, the gastrointestinal tract, bladder, and uterus, plays a crucial role in various physiological processes including vasoconstriction, peristalsis, and the control of blood flow and pressure (Berridge, 2008). Unlike skeletal and cardiac myocytes, smooth muscle cells rely on multiple calcium (Ca2+) signaling mechanisms for both contraction and relaxation. There are two primary mechanisms through which intracellular Ca2+ can be increased leading to a contraction: (i) electromechanical coupling, and (ii) pharmacomechanical coupling (Somlyo and Somlyo, 1968). Electromechanical coupling refers to depolarization-induced opening of voltage-gated Ca2+ channels (CaV1.2) leading to a contraction (Figure 2C). Whereas, pharmacomechanical coupling refers to the process by which chemical signals (typically drugs or hormones) directly cause muscle contraction or relaxation without requiring changes in membrane potential.
Molecular Organization of Smooth Muscle ion Channels, Receptors, and Transporters
In smooth muscle, CaV1.2 channels are the primary mediators of contraction, but Ca2+ influx across the smooth muscle sarcolemma can come from a variety of sources including T-type calcium channels (Kuo et al., 2010; Abd El-Rahman et al., 2013), TRP channels (Gonzales et al., 2010; Mercado et al., 2014; Thakore et al., 2020), and Orai channels (Potier et al., 2009; Trebak, 2012). Like in Cardiac muscle, CaV1.2 channels in smooth muscle demonstrate the ability to cluster and cooperatively interact to increase the open probability of channels (Navedo et al., 2005; Navedo et al., 2010; Nystoriak et al., 2017). Pathologically, aberrant increases in CaV1.2 clustering in smooth muscle is observed in several conditions including diabetes (Nystoriak et al., 2017), hyperglycemia (Prada et al., 2019), hypertension (Navedo et al., 2010), and timothy syndrome (Navedo et al., 2010). Ca2+ entry through CaV1.2 channels elevates intracellular Ca2+ which increase the probability of cytosolic Ca2+-binding to calmodulin (CaM). Formation of Ca2+-CaM complexes activate myosin light chain kinase (MLCK). MLCK phosphorylates myosin I regulatory light chains, enabling myosin-actin interactions. This leads to cross-bridge formation, filament sliding, and muscle contraction (Somlyo and Somlyo, 2003). The critical role of CaV1.2 channels in the development and maintenance of vascular tone and reactivity is underscored by the efficacy of L-type calcium channel blockers as potent antihypertensive drugs (Brozovich et al., 2016).
In smooth muscle, Ca2+ signaling exhibits a dual role, mediating both contraction and relaxation. This seemingly paradoxical function can be explained by the functional coupling between ion channels at SR–PM MCS. At SR–PM MCS Ca2+ release from RyRs on the SR, known as Ca2+ sparks (Cheng et al., 1993), occurs in close proximity to large-conductance Ca2+-activated K+ channels (BKCa) on the plasma membrane (Figure 2C, left). These BKCa channels are uniquely positioned to respond to both membrane depolarization and localized elevations in cytoplasmic Ca2+ (Berkefeld et al., 2006; Fakler and Adelman, 2008; Muller et al., 2010; Irie and Trussell, 2017). Further their low affinity Ca2+-binding site means that a large Ca2+ release is required for their activation. When activated, they facilitate K+ efflux, resulting in membrane hyperpolarization. This hyperpolarization decreases the open probability of voltage-gated L-type Ca2+ channels (CaV1.2), thereby reducing Ca2+ entry and promoting smooth muscle relaxation (Figure 2C, left). Unlike in cardiac and skeletal muscle, where there is tight coupling between CaV1.2 channels and RyRs, smooth muscle CaV1.2 channels exhibit only loose coupling to RyR2 on SR membranes. The primary influence of CaV1.2 on RyR2 activity is thought to be indirect, primarily through the regulation of SR Ca2+ content (Essin et al., 2007; Takeda et al., 2011). Recent studies that optically quantified Ca2+ release from CaV1.2 channel clusters, termed Ca2+ sparklets (Navedo et al., 2005), have revealed that these events occur in proximity to both the SR and sarco/endoplasmic reticulum Ca2+-ATPase (SERCA) pumps (Takeda et al., 2011). These Ca2+ sparklets can coexist within the same microenvironment as Ca2+ sparks, allowing for complex local Ca2+ signaling. In this arrangement, Ca2+ sparklets can indirectly modulate RyR activity through the regulation of SR Ca2+ concentration ([Ca2+]SR), creating a sophisticated feedback mechanism for fine-tuning smooth muscle contractility. This intricate system of Ca2+ signaling at SR–PM MCS allows smooth muscle to precisely regulate its contractile state in response to various physiological stimuli, highlighting the importance of spatial organization in cellular signaling processes
Pharmacomechanical coupling is a crucial mechanism in smooth muscle contraction, whereby pharmacological ligands bind to and activate specific receptors, leading to increased intracellular Ca2+ and subsequent muscle contraction. This process is highlighted during activation of Gq-coupled receptors by the ligand angiotensin II (AngII). In smooth muscle cells, Gq-receptor activation by AngII initiates a signaling cascade that activates phospholipase C (PLC). PLC then catalyzes the hydrolysis of phosphatidylinositol 4,5-bisphosphate (PI(4,5)P2) into two second messengers: soluble inositol 1,4,5-trisphosphate (IP3) and membrane-bound diacylglycerol (DAG). IP3 rapidly diffuses through the cytoplasm and binds to IP3 receptors (IP3Rs) located on the SR membrane. This binding increases the open probability of IP3Rs (Prole and Taylor, 2019), allowing Ca2+ to flow down its concentration gradient from the SR into the cytoplasm. The resultant localized increase in cytoplasmic Ca2+ near the plasma membrane has significant downstream effects. One key consequence of this IP3R-mediated Ca2+ release is the activation of transient receptor potential vanilloid 4 (TRPV4) (Mercado et al., 2014) or transient receptor potential cation channel subfamily M member 4 (TRPM4) (Gonzales et al., 2010) channels located on the plasma membrane. The proximity of IP3Rs to TRP channels creates a functional microdomain where localized Ca2+ increases can efficiently modulate TRP activity (Bandell et al., 2006; Gonzales et al., 2010; Mercado et al., 2014). Activation of TRP channels leads to membrane depolarization, which in turn opens voltage-gated L-type Ca2+ channels (CaV1.2). This further increases intracellular Ca2+, ultimately causing smooth muscle contraction (Figure 2C, right). Recent research has revealed that TRPV4 channels are anchored by A-kinase anchoring protein 150 (AKAP150), which helps create a localized Ca2+ signaling microdomain within the plasma membrane (Mercado et al., 2014). However, it remains unclear whether AKAP150 also influences the positioning of IP3Rs or if other anchoring proteins are involved in organizing this signaling complex. Interestingly, in addition to the Gq-mediated activation of TRP–IP3R signaling at smooth muscle SR-PM MCS, studies on freshly isolated cerebral artery smooth muscle cells have suggested the presence of a basal level of PLC-dependent IP3 generation and IP3R-mediated Ca2+ release. This basal activity could potentially support TRP-mediated depolarizations even in the absence of explicit Gq-receptor activation.
Membrane Tethers
While substantial research has elucidated the functional coupling of ion channels at sarcoplasmic reticulum-plasma membrane (SR-PM) MCS, there has been an absence of research attention on the tethering proteins that generate and maintain these crucial junctionsr in smooth muscle. This gap in knowledge may be attributed to various factors, including historical perspectives, perceived functional importance, disease relevance, structural complexity, and the inherent diversity of smooth muscle types. In smooth muscle cells, several proteins have been identified as bona fide SR-PM MCS tethers, including JPH2 (Pritchard et al., 2019; Saeki et al., 2019), STIM1-Orai1 (Potier et al., 2009), and voltage-gated potassium channel 2.1 (KV2.1) (O'Dwyer et al., 2020; Matsumoto et al., 2023). Each of these proteins plays a unique role in maintaining the structural and functional integrity of SR-PM MCS. JPH2, well-known for its role in striated muscle, has been shown to be equally important in smooth muscle. Knockout studies of JPH2 in smooth muscle demonstrate a reduction in the proximity between the SR and PM (Pritchard et al., 2019). This loss of tethering significantly alters the functional coupling between ryanodine receptors (RyR2) and large-conductance Ca2+-activated K+ channels (BKCa) (Gonzales et al., 2010; Mercado et al., 2014; Mercado et al., 2014). Physiologically, the disruption of RyR2–BKCa coupling decreases the local Ca2+ available to activate BKCa channels, reducing their conductance and resulting in vascular hypercontractility (Pritchard et al., 2019). These findings underscore the critical role of JPH2 in facilitating SR–PM contacts and localizing ion channels at these sites. KV2.1 has emerged as another important SR–PM tethering protein in smooth muscle (O'Dwyer et al., 2020). In neurons, somatic and dendritic KV2.1 serves dual roles: controlling membrane potential and generating ER-PM MCS in a phosphorylation-dependent manner (see neuron section for more details). In smooth muscle, KV2.1 exhibits a similar multifaceted role, functioning both as a regulator of membrane potential and a structural organizer of CaV1.2 complexes (O'Dwyer et al., 2020; Matsumoto et al., 2023). In smooth muscle, KV2.1 clusters CaV1.2 channels to enhance their function and increase contraction in a sex-specific manner (O'Dwyer et al., 2020; Matsumoto et al., 2023). In male myocytes KV2.1 channels primarily function as regulators of membrane potential, whereas knockout of KV2.1 or KV2.1S590A mutations (non-phosphorylated mutation leading to reduced SR–PM MCS) in female myocytes reduce the clustering of CaV1.2 and Ca2+ influx. Thus, Kv2.1 could control both relaxation (by controlling the membrane potential) and contraction (by influencing CaV1.2 channel clustering) in smooth muscle, depending on sex. The potential integration of KV2.1–CaV1.2 complexes with RyR–BKCa signaling pathways at SR–PM MCS in smooth muscle remains an open question. Evidence from neuronal studies demonstrating KV2.1's capacity to interact with and cluster CaV1.2, RyR, SERCA, and PKA at ER-PM junctions (Vierra et al., 2019; Vierra et al., 2021; Vierra et al., 2023) (see below) suggests the possibility of a similar integrative signaling hub in smooth muscle cells. This hypothesis warrants further investigation, as it could reveal novel mechanisms of Ca2+ signaling coordination and provide insights into the fine-tuning of smooth muscle contractility.
Neurons
A typical neuron has a complex structure that includes several specialized regions, each with distinct functions. The soma, or cell body, contains the nucleus and most of the cell's organelles, serving as the metabolic center. Extending from the soma are dendrites, which are branching projections that receive signals from other neurons and convey them towards the soma. The synaptic region, located at the end of the axon, is where neurons communicate with each other through synapses. Together, these regions enable neurons to process and transmit information efficiently within the nervous system. Given this distinct and specialized architecture, it is perhaps not surprising that ER–PM MCS, or junctions as they are often referred to in neurons, have been identified across each of these areas. Despite these findings, we still lack specific tools to clearly quantify the distribution of ER–PM MCS proteins and ion channels in vitro and in vivo across each distinct area of a neuron. Additionally, generalized characterizations of MCS in neurons should be avoided, as each brain region is unique in terms of its architecture, innervation, complement of neurons, and function. Quantitative ultrastructural analyses of distinct regions have revealed that significant portions of the plasma membrane are involved in ER–PM MCS signaling. Specifically, around 12% of the somatic PM is engaged with the cisternae of the ER, while dendritic and synaptic regions show approximately 3% engagement (Wu et al., 2017). Despite the extensive presence of ER–PM MCS in neurons, the characterization of proteins responsible for forming, regulating, and signaling at these MCS has been limited. Understanding the molecular underpinnings of these contact sites will enhance our knowledge of neuronal physiology and pathophysiology. Below, we broadly define the molecular elements of MCS in neurons, noting that each protein complex may be heterogeneously distributed within neurons (i.e., somatic, dendritic, etc.) and across regions.
KV2.1-Mediated MCS
At somatodendritic regions of neurons, the voltage-gated potassium channel KV2.1 is the best-characterized ion channel/MCS tether (Figure 3A). Historically, research has focused on KV2.1's non-clustering role in regulating the resting membrane potential (Figure 3A, left). Here, following membrane depolarization, KV2.1 opens, allowing K+ ion efflux to repolarize the neuronal membrane potential (Du et al., 2000). Beyond this essential role, KV2.1 is regulated by protein kinases such as cyclin-dependent kinase-5 (CDK5) and Ca2+/calmodulin-dependent protein kinase II (CAMK2) (Misonou et al., 2006; Cerda and Trimmer, 2011). Channel phosphorylation acts as a molecular switch, converting KV2.1 from a non-clustering (Figure 3A, left), conducting state (electrical dampener) to a clustering, non-conducting state (ER–PM MCS tether; see Figure 3A, right). The dynamic equilibrium between clustered and non-clustered KV2.1 channels is further regulated by the Ca2+-dependent protein phosphatase calcineurin (Figure 3) (Misonou et al., 2004). Phosphorylation of KV2.1's proximal restriction and clustering (PRC) domain creates a noncanonical FFAT motif enabling the cytoplasmic MSP domain of ER–localized VAP proteins to bind KV2.1, forming ER-PM MCSs (Johnson et al., 2018; Kirmiz et al., 2018). Thus, the net phosphorylation status of the PRC domain determines the proportion of KV2.1 available for electrical (K+ efflux) versus chemical signaling (Ca2+ nanodomain formation, see below). This dynamic regulation underscores KV2.1's critical role in modulating neuronal signaling and maintaining cellular homeostasis. Kv2.1–mediated ER–PM MCSs have been characterized in hippocampal, cortical, substania nigra, and Purkinje neurons of the cerebellum (Lim et al., 2000; Kirmiz et al., 2018; Lebowitz et al., 2019; Casas et al., 2023). Given KV2.1's extensive distribution throughout the brain, it is likely that KV2.1 MCSs are present in most neurons and brain regions. At MCS, Kv2.1 serves dual functions: it acts as an MCS tether and recruits other ion channels to generate MCS Ca2+ nanodomains (Figure 3A). KV2.1 physically and functionally interacts with several Ca2+-signaling proteins including CaV1.2 and CaV1.3 on the PM, and RyRs, and SERCA on ER membranes (Figure 3A, right) (Vierra et al., 2019) to create Ca2+ nanodomains that are essential for depolarization-induced activation of transcription factors CREB and c-Fos, and protein kinase A (PKA) signaling (Vierra et al., 2021; Vierra et al., 2023). The molecular motif that nucleates KV2.1-mediated Ca2+-signaling complexes is called the Ca2+ channel association domain (CAD) located at KV2.1's C-terminus. This domain is necessary for the recruitment of CaV1.2 to Kv2.1-containing ER–PM MCSs. Mutations or loss of function in KV2.1 reduce channel clustering, MCS formation, Ca2+-signaling, PKA signaling, and transcription factor activation, and lead to hyperexcitability (Speca et al., 2014; Thiffault et al., 2015). Increases in KV2.1 MCS formation have also been observed in neurodegenerative diseases, including Niemann-Pick type C1 (NPC1) disease (Casas et al., 2023). In NPC1 disease, the loss of the lysosomal cholesterol transporter NPC1 leads to CDK5-dependent increases in KV2.1 MCS, which increases Ca2+ influx to drives Ca2+ entry into mitochondria and results in neuronal toxicity. Collectively, these data suggest that KV2.1–containing ER-PM MCS play an important role in healthy neurons by facilitating compartmentalized Ca2+-dependent excitation-transcription coupling and PKA signaling. While excessive KV2.1 MCS formation could be a trigger for neuronal degeneration.

Ion channels in SR-PM MCS in neurons.
CaV-RyR-KCa Crosstalk
The interactions between voltage-gated Ca2+ channels (CaV1.2, 1.3, 2.1), RyRs, and Ca2+-activated potassium channels (both IKCa and BKCa) forms a critical signaling axis in neurons, essential for various neurophysiological processes including learning and memory, regulation of excitability, integration of sensory inputs, and fine-tuning of motor signals (Berkefeld et al., 2006; Fakler and Adelman, 2008; Muller et al., 2010; Irie and Trussell, 2017). This CaV-RyR-KCa complex orchestrates Ca2+-dependent bi-directional control of membrane potential. The signaling cascade begins with CaV channels on the plasma membrane, which are activated by membrane depolarization, allowing Ca2+ influx into the cell. This initial Ca2+ elevation then activates RyRs positioned on the ER membrane, triggering CICR. This amplification of intracellular Ca2+ subsequently activates intermediate/small conductance Ca2+-activated K+ channels (Sahu et al., 2019) (IKCa3.1; Figure 3B) and large conductance Ca2+-activated K+ channels (BKCa, Figure 3C). Both channel types, located on the plasma membrane, mediate K+ efflux upon activation, resulting in membrane hyperpolarization. This process serves as a crucial feedback mechanism to regulate neuronal excitability.
Recent studies have shed light on the structural components that enable the functional coupling within CaV–RyR–KCa complexes (Figure 3B). Junctophilins, particularly JPH3 and JPH4, have emerged as critical tethering components. These proteins are instrumental in generating and maintaining the functional coupling between ion channels at this membrane contact site (MCS). Evidence for this comes from studies on hippocampal neurons from JPH3/4 double knockout animals, which exhibit a lack of apamin-sensitive currents, completely abolished after-hyperpolarizations (AHP), impaired hippocampal plasticity, and deficits in memory formation (Moriguchi et al., 2006). Further structural and functional evidence defining the key role of CaV–RyR–KCa complexes in regulating neuronal excitability has been obtained through super-resolution imaging, Förster Resonance Energy Transfer (FRET), and electrophysiology experiments in CA1 pyramidal cells. These studies have revealed that CaV1.3, RyR2, KCa3.1, and JPH3 are all located within 10–20 nm of one another, highlighting the nanodomain organization of this signaling complex (Sahu et al., 2019). Moreover, knockdown experiments targeting JPH3 and JPH4 have demonstrated alterations in the AHP and spike frequency accommodation, further emphasizing the functional importance of these structural components.
In addition to IKCa channels, BKCa channels have been extensively studied for their functional crosstalk with CaV channels (Berkefeld et al., 2006; Fakler and Adelman, 2008; Muller et al., 2010; Irie and Trussell, 2017). Significant evidence supports the localization of this channel complex with RyR on the ER (Figure 3C), particularly in specialized neuronal subtypes. In cartwheel interneurons of the dorsal cochlear nucleus, triggered opening of plasma membrane CaV2.1 channels facilitates CICR from RyR on ER membranes to activate somatic BKCa channels (Irie and Trussell, 2017). This functional unit controls burst firing in the neuron population and demonstrates tight spatial and functional coupling between these channels. Further evidence for a tripartite arrangement comes from cerebellar Purkinje cells, where CaV2.1 Ca2+ channels and BKCa channels colocalize over somatic subsurface cisternae (Indriati et al., 2013). This colocalization indicates that CaV2.1-RyR-BKCa triads are strategically positioned at ER-PM junctions to ensure rapid and efficient Ca2+ signaling to low affinity BKCa channels. While the molecular tether that anchors this complex is unknow, spatial proteomic analysis of CaV2.1 interactors revealed the presence of BKCa alongside JPH3 suggesting it may help facilitate the organization of this channel complex at ER-PM MCS.
Collectively, these findings underscore the importance of spatial organization in ion channel function and highlight the dynamic nature of ER-PM junctions in neuronal signaling. The precise arrangement of CaV, RyR, and BK channels within these nanodomains allows for highly localized and efficient Ca2+ signaling, enabling fine-tuned control of neuronal excitability. Despite the progress made in identifying some of the key ion channel and tethering proteins at MCS in neurons, there are likely many more involved in neuronal MCS formation and regulation. Advanced proteomic approaches, such as mass spectrometry and proximity labeling, could help identify novel neuronal MCS proteins. Once identified, their specific roles and interactions within MCSs need to be characterized for neurons across multiple brain regions.
Store-Operated Ca2+ Entry (SOCE)
The STIM1–ORAI1 complex at ER–PM MCS is the most well-characterized ion channel complex mediating store-operated calcium entry (SOCE) (for comprehensive reviews, see Majewski and Kuznicki, 2015; Wegierski and Kuznicki, 2018; Tiscione et al., 2019). This complex plays a critical role in immune cell function, particularly in T-cell activation, proliferation, and cytokine production, as well as mast cell degranulation and B cell function. While SOCE has been described in various cell types, including cardiac, skeletal, and smooth muscles, as well as neurons, we briefly focus on its role in immune cells. James Putney first proposed SOCE in 1986 as a crucial mechanism for ER store refilling following receptor activation (Putney, 1986). In this model, depletion of intracellular Ca2+ stores triggers Ca2+ influx across the plasma membrane. The field advanced significantly with the identification of STIM1 as the ER Ca2+ sensor (Liou et al., 2005; Roos et al., 2005) and Orai1 as the pore-forming subunit of the Ca2+ release-activated Ca2+ (CRAC) channel in the plasma membrane (Feske et al., 2006; Vig et al., 2006). These discoveries enabled precise characterization of the localization, kinetics, regulatory mechanisms, and functional roles of this ER–PM MCS complex. SOCE activation is initiated when ER Ca2+ stores are depleted, typically through Gq-receptor activation of phospholipase C, leading to IP3-mediated release. The resulting decrease in ER Ca2+ reduces Ca2+ binding to STIM1's EF-hand domain, causing STIM1 to oligomerize and translocate to ER-plasma membrane junctions. There, it interacts with and activates Orai1 channels, allowing Ca2+ influx (Hogan et al., 2010). Multiple experimental approaches have demonstrated SOCE's importance in immune cell function. Patch-clamp electrophysiology studies revealed the biophysical properties of CRAC channels (Zweifach and Lewis, 1993), while Ca2+ imaging techniques showed sustained Ca2+ elevations in T cells upon antigen stimulation, later linked to SOCE (Lewis and Cahalan, 1995). Genetic approaches, including knockouts and mutations of STIM1 and Orai1, revealed their critical roles in T cell activation, proliferation, and cytokine production (Oh-hora et al., 2008). Clinical evidence further underscores SOCE's significance as patients with mutations in STIM1 or Orai1 genes exhibit severe combined immunodeficiency, demonstrating SOCE's essential role in immune function (Feske et al., 2006; McCarl et al., 2009; Feske et al., 2010). Functionally, SOCE plays a crucial role in lymphocyte activation following antigen recognition. Sustained SOCE activates the phosphatase calcineurin, which dephosphorylates NFAT (Nuclear Factor of Activated T cells). This allows NFAT to translocate to the nucleus and initiate transcription of genes crucial for T cell activation (Yazbeck et al., 2017). By mediating these diverse functions, SOCE helps coordinate and fine-tune the immune response, enabling immune cells to respond effectively to pathogens and other threats.
Endoplasmic Reticulum—Mitochondria MCS
ER—mitochondria contact sites, often referred to as mitochondria-associated membranes (MAMs), have been extensively characterized given their ubiquity and importance for cellular signaling and homeostasis. These specialized nanodomains facilitate the transfer of ions and lipids between the ER and mitochondria, impacting a range of cellular functions.
Membrane Tethers
Several membrane tethers have been implicated in forming ER–Mito MCS (Figure 4A) including: (i) Mitofusins 1/2 (Mfn): ER and mitochondrial residing low-affinity GTPase's that form homo- and hetero- complexes with Mfn1 or Mfn2, respectively, localized on the mitochondrial membrane, likely through dimerization in trans of the Heptated Repeated 2 (HR2) domains (de Brito and Scorrano, 2008; Filadi et al., 2018), (ii) Glucose-regulated Protein 75 (GRP75): a chaperone protein that links the inositol 1,4,5-trisphosphate receptor (IP3R) on the ER to the voltage-dependent anion channel 1 (VDAC1) on the mitochondria (Szabadkai et al., 2006), and (iii) VAPB-PTPIP51: interactions between VAPB on the ER and protein tyrosine phosphatase-interacting protein 51 (PTPIP51) on mitochondria (Gomez-Suaga et al., 2017). These tethering complexes ensure efficient transfer of Ca2+ and lipids between ER and mitochondria which is crucial for energy metabolism, calcium buffering (both in cytoplasm and ensuring sufficient Ca2+ in the mitochondrial matrix), and regulation of apoptosis.

Ion channels in ER–mitochondrial MCS. Tethers and ion channels present at ER–mitochondrial MCS influence mitochondrial Ca2+ levels to regulate bioenergetics and apoptosis.
Molecular Organization of ion Channels, Receptors, and Transporters
At ER–Mito MCS multiple ion channels are located including the IP3R and RyR on ER membranes and the voltage-dependent anion channel (VDAC) on mitochondrial membranes. IP3R–VDAC (Rizzuto et al., 1998; Szabadkai et al., 2006; Bartok et al., 2019) and RyR–VDAC (Hajnóczky et al., 2002; Min et al., 2012) complexes are anchored through the actions of GRP75. When Ca2+ is released from the ER (Giacomello et al., 2010) following cytoplasmic IP3 binding to IP3R or when elevations in Ca2+ facilitate CICR from RyR, causing a Ca2+ increase into the micromolar range, VDAC enables Ca2+ entry across the outer mitochondrial membrane (Colombini, 1979; De Pinto et al., 1987; Blachly-Dyson et al., 1990). This Ca2+ is subsequently taken up into the mitochondrial matrix by the mitochondrial Ca2+ uniporter (MCU; Figure 4A). The constitutive release of IP3R Ca2+ at ER–Mito MCS is essential for ATP production and suppression of autophagy, while the loss of this Ca2+ transfer results in decreased ATP production and activation of AMP kinase to promote autophagy (Cárdenas et al., 2010). In neurons (Wu et al., 2017), these contacts are especially critical due to the high energy demands and complex Ca2+ signaling required for neuronal function. For example, ER–mitochondria MCS help maintain mitochondrial health, ensuring adequate ATP supply necessary for sustained neuronal activity. Further, efficient Ca2+ signaling at ER–mitochondria contacts support synaptic activity and plasticity (Hirabayashi et al., 2017; Lee et al., 2018; Fowler et al., 2019; Gao et al., 2022), essential for learning and memory. Finally, proper functioning of MAMs protects neurons from Ca2+-induced excitotoxicity, which is implicated in neurodegenerative diseases (Toglia et al., 2016; Ludtmann and Abramov, 2018; Verma et al., 2018; Lin et al., 2019; Calvo-Rodriguez et al., 2020; Apicco et al., 2021; Casas et al., 2023). Thus, ER–mitochondria contact sites are integral to cellular and neuronal function. Understanding the molecular intricacies of these contacts, including the roles of ion channels and tethers, is vital for unraveling their contributions to both normal physiology and disease states.
Endoplasmic Reticulum –Endo/Lysosome MCS
ER—lysosome contact sites are critical for maintaining cellular homeostasis and facilitating various signaling pathways. These contacts, enable direct communication between the ER and lysosomes, impacting ion homeostasis, lipid metabolism, and autophagy.
Member Tethers
At ER—lysosome MCS, several tethers have been identified in bringing each organelle membrane into close apposition, including: (i) VAP and ORPs (Du et al., 2011; Zhao and Ridgway ; Lim et al., 2019): here, the ER-resident VAP proteins recruit through FFAT interactions different ORP proteins (i.e., OSBP, ORP1L, ORP5, ORP9, ORP10) which in turn bind to negatively charged phosphoinositide's on the lysosomal membrane to generate ER–lysosome MCS (Kutchukian et al., 2021), and (ii) Protrudin-Rab7-GTPase–PDZD8 (Raiborg et al., 2015): association of the integral ER protein protrudin and PDZD8 contact the endo/lysosomal membrane through binding to Rab7-GTP together with PI(3)P, or to only the Rab7-GTPase through the Rab-Binding Domain (RBD) (Guillén-Samander et al., 2019; Elbaz-Alon et al., 2020; Khan et al., 2021; Gao et al., 2022). Other ER-localized proteins that have been reported to also interact with endo/lysosome membranes include STARD3 (Wilhelm et al., 2017) and VPS13C (Kumar et al., 2018). The majority of current information on ER–lysosome tethers come from experiments in expression system cells, although extremely informative, there is a clear need to extend these datasets into primary cells like neurons or myocytes.
Molecular Organization of ion Channels, Receptors, and Transporters
At ER-lysosomal MCS multiple ion channels and receptors have been identified including the transient receptor potential mucolipin 1 (Dong et al., 2008; Dong et al., 2010) (TRPML1) and two-pore channels (TPC1 and TPC2 Cang et al., 2014, Wang et al., 2012) on the lysosomal membrane and IP3R and RyR on ER membranes (Atakpa et al., 2018). On the ER side of the MCS, it has been reported that endogenous IP3R form small clusters within ER membranes with the majority of IP3R clusters within the ER being mobile (∼75%) however it is a small fraction of immobile IP3R clusters that are responsible for Ca2+ signals (called Ca2+ “puffs”) (Thillaiappan et al., 2017). At ER–lysosome MCS IP3Rs are not required for MCS formation but rather selectively deliver large Ca2+ signals to lysosomes (Atakpa et al., 2018). On the Endosome site of the MCS, TPC1 localizes to ER-endosome contact sites and appear essential for MCS formation as knocking down its expression, or reducing is ligand NAADP reduces the formation of these contact sites (Kilpatrick et al., 2017). At ER–endosome contacts release of Ca2+ from IP3R on ER membranes has been reported to amplify TPC Ca2+ release (Ruas et al., 2010). While there is evidence that stimulating Ca2+ release from acidic endocytic vesicles can stimulate ER Ca2+ mobilization and vice versa (Lopez-Sanjurjo et al., 2013; Morgan et al., 2013), more investigations are required to disentangle the substantial mechanisms of crosstalk at ER-endosome MCS and how they influence organelle function and position within cells. In addition to TPC1, endo/lysosome membranes also have TRPML1 Ca2+ release channels. TRPML1 Ca2+ release is important for regulating a host of different pathways including: (i) Ca2+ dependent activation of the protein phosphatase, calcineurin, which dephosphorylates the transcription factor TFEB, allowing it to enter the nucleus and switch on the expression of autophagy regulating proteins (Medina et al., 2015). An additional layer of regulation of TRPML1 complex comes in the form of the endo/lysosomal phosphoinositide, PI(3,5)P2, which acts as a positive cofactor for channel function (Dong et al., 2010; Zhang et al., 2012). Loss of function of TRPML1 function is associated with mucolipidosis type IV (Dong et al., 2008). While the specific localization of TRPML1 at ER–endosome contacts is open for debate, it is worth noting that activation of TRPML1 channels reduces cholesterol accumulation in NPC1 disease (Shen et al., 2012). Given that cholesterol egress at lysosomes occurs at MCS presents good evidence that TRPML1 may also be present at these contacts. While many ion channels have been detailed to be important and regulated at ER–Lysosome MCSs, further research is needed to better understand how these complexes collaborate to shape organelle function and how lipid transport at ER-lysosomal contacts interfaces with these Ca2+ signaling nanodomains. Understanding these interactions is crucial for elucidating the roles of ER-lysosome contact sites in both normal physiology and disease states.
Non-ER MCS
The majority of MCS have been identified as being between the ER and another closely apposed organelle. That said, other non-ER MCS have been identified. It should be noted that given the importance of Ca2+ signaling for MCS, the extensive and dynamic nature of the ER, and reports of 3-way MCSs (Wang et al., 2016), it seems that the ER could very well also be closely apposed and add a further layer of regulation to these MCS.
Mitochondria-Lysosome
Dynamic contacts between mitochondria and lysosomes have been reported as key regulators of mitochondrial positioning, dynamics and fission (Aston et al., 2017; Valm et al., 2017; Wong et al., 2018). The molecular tether facilitating contact between these two organelles appears to at least involve the small guanosine triphosphatase (GTPase), RAB7 (Wong et al., 2018). In the GTP-bound form, lysosomal RAB7-GTP promotes MCS formation with mitochondria which positions lysosomal TRPML1 and mitochondrial VDAC1 to coordinate to transfer Ca2+ into mitochondria (Peng et al., 2020). Loss of function or disease mutations in TRPML1 decrease lysosome–mitochondrial Ca2+ transfer (Peng et al., 2020). The mechanism that facilitates interaction between RAB7-GTP and mitochondria to enable Ca2+ transfer has yet to be fully elucidated. The uncoupling of lysosomes and mitochondria MCS is regulated by the mitochondrial protein FIS1 which recruits the GTPase-activating protein (GAP) TBC1D15 to hydrolyze RAB7-GTP and release the contacts. Formation of mitochondrial-lysosome MCS spatially organizes lysosomal TRPML1 and mitochondrial VDAC1 to coordinate to transfer Ca2+ into mitochondria (Peng et al., 2020). Functionally, these contacts appear to mark sites of mitochondrial fission, allowing regulation of mitochondrial networks by lysosomes. The role of TRPML and VDAC in mitochondrial division, or bioenergetics requires further investigation.
Plasma Membrane—Mitochondrial Contacts
Mitochondria can also form associations with the plasma membrane, called plasma membrane–associated mitochondria (PAM) (Kakizawa et al., 2007; Giacomello et al., 2010; Suski et al., 2014; Montes de Oca Balderas, 2021). In mammalian cells, the complete molecular identity of the tethering complex responsible for ER-mitochondria association remains elusive. In contrast, work in yeast has identified the Num1/Mdm36 complex as a key player in this process (Lackner et al., 2013). While the exact spatial arrangement of ion channels and transporters at these contact sites in mammalian cells is not fully characterized, the strategic positioning of mitochondrial Ca2+ channels near other Ca2+ handling proteins would facilitate localized Ca2+ signaling and regulation of cellular Ca2+ dynamics. Further research is needed to elucidate the precise molecular architecture and functional implications of these putative arrangements.
Discussion and Future Directions
Over the past 70 years, our understanding of ion channel and transporter activity at MCSs has advanced significantly. We now possess considerable knowledge about the tethering proteins that establish these contacts, the lipid and Ca2+ handling proteins within them, and their critical importance for cellular homeostasis. However, numerous questions remain in this rapidly evolving field of cell biology. Here, we outline key areas for future research:
Identification and Characterization of Molecular Tethers
While progress has been made in identifying key MCS-forming proteins, many more likely remain undiscovered. Advanced proteomic approaches, such as quantitative mass spectrometry and proximity labeling techniques (e.g., BioID, APEX), will be crucial in identifying novel MCS proteins. Once identified, their specific roles, interactions, and regulatory mechanisms within MCSs across different cell types need to be elucidated. This includes investigating their binding partners, post-translational modifications, and how they respond to various cellular stimuli. Uncovering and characterizing novel member tethering proteins will undoubtedly expand our understanding of ion channels within MCSs.
Mechanisms of Protein Recruitment and Localization
A critical question is how proteins, particularly ion channels, are recruited to specific MCSs across distinctive cellular subdomains (e.g., soma, dendrites, synaptic regions). What molecular signals govern the recruitment of ion channels to MCSs, and how is this process regulated? Insights from lipid transport at MCSs suggest the need for recruiting signals at each membrane, such as VAPs on ER membranes and complementary lipid or protein effectors on the apposing membrane. This mechanism may be conserved for ion channels, as exemplified by KV2.1–VAP interactions at somatic ER-PM junctions and IP3R–GRP75–VDAC complexes at ER-mitochondria contacts. Future studies should focus on identifying these recruiting signals and elucidating their regulatory pathways.
Detailed Analysis of MCS Proteins Across Cell Types
Most studies identifying MCS proteins have been conducted in immortalized cell lines like HEK293 and HeLa. While this work is invaluable, it is evident that not all MCS proteins are present in primary human cells, especially those with specialized functions such as neurons or myocytes. Detailed characterization of MCS organization and function in primary cells is essential, as differences in cellular architecture and function suggest that the repertoire and roles of MCS might vary significantly across cell types. This research should employ a combination of proteomics, high-resolution imaging, and functional assays to provide a comprehensive understanding of cell type-specific MCS composition and function.
Structural and Functional Plasticity of MCSs
Further high-resolution structural analysis, such as cryo-electron microscopy (cryo-EM), super-resolution imaging techniques (e.g., STORM, PALM), or correlative light and electron microscopy (CLEM) is essential to characterize MCSs at the molecular level. These approaches will help elucidate how dynamic interactions between different proteins change in response to post-translational modifications (e.g., phosphorylation, ubiquitination) and cellular perturbations.
Impact of MCSs on Physiology and Pathophysiology
Exploring how different MCSs integrate their functions to contribute to cellular signaling will enable a more comprehensive understanding of their physiological roles. This includes investigating how MCS coordinate responses to various stimuli, such as changes in metabolic state or cellular stress. Additionally, studying how disruptions in MCS proteins contribute to pathophysiological conditions, such as neurodegenerative disorders or metabolic diseases, could provide insights into disease mechanisms and potential therapeutic targets.
Role of Lipid Composition in MCS Formation and Function
Investigating how the lipid composition of apposing membranes within an MCS influences its formation and function is crucial. Given that common membrane tethers form both lipid and Ca2+-signaling nanodomains at MCSs, and ion channels are regulated by lipids (Hille et al., 2015) means these two instructional signals may be intimately related and shape each other's transfer abilities. Key questions include: could elevations in Ca2+ at MCSs drive or halt lipid signaling? Conversely, could lipid transfer at MCSs enhance or diminish the release of Ca2+ into the MCS space? Understanding the interplay between these two exchange processes is important for comprehending how MCS signaling may be integrated and controlled. Future studies should employ lipidomics approaches, coupled with Ca2+ imaging and functional assays, to dissect the relationship between lipid composition and calcium signaling at MCS.
Longitudinal Mapping of MCS Dynamics
Conducting longitudinal studies to monitor changes in MCS during development, aging, and in response to various physiological and pathological stimuli will provide insights into their roles over time. This could involve the use of long-term live-cell imaging techniques, coupled with genetic manipulations, to track MCS formation, dissolution, and remodeling in various cellular contexts. For example, studying how MCSs between the ER and PM change during neuronal development and synaptic plasticity could reveal new insights into the role of these structures in brain function and cognition.
With the development of new technologies, such as improved super-resolution microscopy techniques, more sensitive mass spectrometry approaches, and advanced genetic tools for manipulating MCS proteins, researchers can gain a more comprehensive understanding of the molecular underpinnings of MCSs in different cell types, their physiological significance, and their potential as targets for therapeutic intervention in disease. By addressing these key areas of research, we can expect significant advances in our understanding of MCS biology and its implications for human health and disease in the coming years
Conclusion
The study of MCS and their associated ion channels, transporters, and receptors has significantly advanced our understanding of cellular organization and function. These specialized regions facilitate critical processes such as Ca2+ signaling, lipid transfer, and organelle communication. The intricate interplay between various ion channels at MCSs, particularly those involving the ER, highlights the complexity and importance of these structures in maintaining cellular homeostasis and responding to diverse stimuli. The field of MCS, particularly concerning tethers and ion channels at these sites, represents a vibrant and rapidly evolving area of cell biology. Continued research in this field promises to yield valuable insights into fundamental cellular processes and opportunities to treat a wide range of diseases.
Footnotes
Acknowledgements
We are very grateful to Dr. Rose Dixon, Dr. Manuel Navedo, Dr. Fernando Santana, and members of the Dickson laboratory for discussions, reading and/or providing comments about this review. We thank the large number of laboratories who made significant contributions to our understanding of ion channels at membrane contact sites and apologize to those groups that were not cited. Figures were made using Biorender. This work was support by NIH grants R35GM149211 and RF1NS131379 (to E.J.D).
Declaration of Conflicting Interests
The authors declared no potential conflicts of interest with respect to the research, authorship, and/or publication of this article.
Funding
The authors disclosed receipt of the following financial support for the research, authorship, and/or publication of this article: This work was supported by the National Institute of Neurological Disorders and Stroke, National Institute of General Medical Sciences, (grant number RF1NS131379, R35GM149211 ).