
Abstract
Nearly twenty years ago a mutation in the VAPB gene, resulting in a proline to serine substitution (p.P56S), was identified as the cause of a rare, slowly progressing, familial form of the motor neuron degenerative disease Amyotrophic Lateral Sclerosis (ALS). Since then, progress in unravelling the mechanistic basis of this mutation has proceeded in parallel with research on the VAP proteins and on their role in establishing membrane contact sites between the ER and other organelles. Analysis of the literature on cellular and animal models reviewed here supports the conclusion that P56S-VAPB, which is aggregation-prone, non-functional and unstable, is expressed at levels that are insufficient to support toxic gain-of-function or dominant negative effects within motor neurons. Instead, insufficient levels of the product of the single wild-type allele appear to be required for pathological effects, and may be the main driver of the disease. In light of the multiple interactions of the VAP proteins, we address the consequences of specific VAPB depletion and highlight various affected processes that could contribute to motor neuron degeneration. In the future, distinction of specific roles of each of the two VAP paralogues should help to further elucidate the basis of p.P56S familial ALS, as well as of other more common forms of the disease.
Keywords
Introduction: Amyotrophic Lateral Sclerosis and VAPB
Amyotrophic lateral sclerosis (ALS) is a fatal neurodegenerative disease of the human motor system, characterized by the degeneration of motor neurons (MNs) in the cerebral cortex, brain stem and spinal cord. Since these neurons regulate the function of skeletal muscles to control essential voluntary movements, their progressive loss leads to peripheral and/or central paralysis, loss of muscle mass (amyotrophy) and death within a few years of onset. Although motor manifestations are the main clinical manifestations of ALS, up to half of patients have non-motor symptoms, such as cognitive impairment (Brown and Al-Chalabi, 2017; Hardiman et al., 2017). An adulthood disease with middle to late age onset, ALS appears in both inherited and sporadic forms, often clinically indistinguishable. Only about 10% of ALS cases have a familial background (fALS), while the vast majority of them shows no apparent history of the disorder in families and, thus, are classified as sporadic (sALS).
The familial forms of ALS have been linked to a myriad of distinct genes. Indeed, since the discovery in 1993 of the first causative gene, the one coding for Cu/Zn superoxide dismutase (SOD1) (Rosen et al., 1993), more than 50 others associated with disease pathogenesis and susceptibility have been identified (Al-Chalabi et al., 2017; Brenner and Weishaupt, 2019), and research continues to uncover at an incredibly rapid pace novel monogenic disease genes, genetic risk factors, as well as disease-modifying genes. Due to the extremely heterogeneous and complex genetic architecture of ALS, the distinction between fALS and sALS is often blurred, the etiology of the disease not yet clearly defined for most patients and, as a consequence, effective treatments remain elusive (Morello et al., 2020). Nevertheless, the discovery and investigation of genetic forms of ALS has provided and continuously provides paramount information about the molecular mechanisms that cause MN death in ALS. More specifically, studies of ALS genes have given insight into the pathogenic roles of perturbations in protein homeostasis, altered RNA metabolism and DNA-repair, impaired membrane trafficking, axonal transport and cytoskeleton function (Mejzini et al., 2019).
One example of how the characterization of specific pathways and biological processes affected by ALS-linked genes in fALS has improved understanding of ALS pathogenesis, has been that of the VAPB (
VAPB Mutations linked to ALS and Clinical Significance.

aFurther references given in Table 2 and in the text.
bIdentified in a fALS patient carrying a C9orf72 repeat expansion.
A first missense mutation in the VAPB gene, consisting in a transition from C to T in exon 2 and resulting in substitution of proline 56 (within the Major Sperm Protein (MSP) domain - see “The VAPs: Structure and Functions” section) with serine (
In addition to the substitutions at position P56, three further missense mutations have been described at other positions of the VAPB sequence in single ALS patients and not in controls (
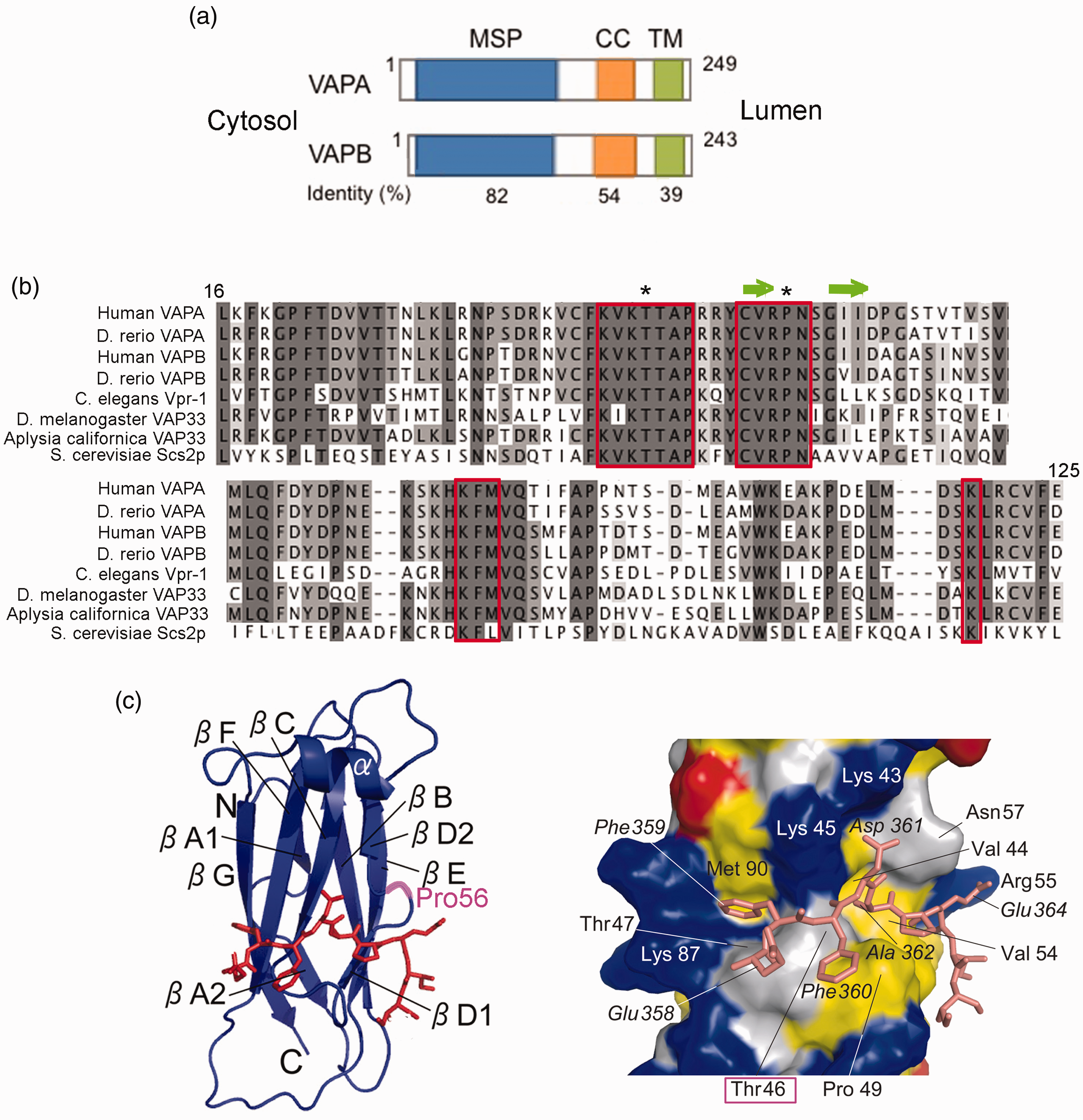
The VAP proteins. (a): Domain organization of VAPA and B. The orientation of the two proteins with respect to the ER membrane and the percent of shared sequence between the two human isoforms are indicated. (b) Multiple sequence alignment (https://www.ebi.ac.uk/Tools/msa/clustalo/) of MSP of mammalian, invertebrate and fungal VAP homologues. Note the remarkable conservation of residues (enclosed in red boxes) at the binding interface with an FFAT- containing peptide (Shi et al., 2010). The asterisks indicate the Thr and Pro residues substituted in the p.T46I and p.P56S mutants. The green arrows indicate the D1 and D2 β strands (nomenclature from (Kaiser et al., 2005)) bridged by an S-shaped loop centred on P56. (c) Images are slightly modified from (Furuita et al., 2010). Left: ribbon representation of the VAPA MSP (nomenclature of the β strands according to (Kaiser et al., 2005)) in complex with an FFAT-containing peptide derived from OSBP, shown in orange as a stick model. The position of Pro56 is highlighted in cyan. Right: Details of the interaction between VAPA-MSP and OSBP FFAT peptide. Residue numbers of OSBP peptide, shown as stick model, are indicated in italics. The surface of VAPA MSP is represented with acidic (red), basic (blue), and hydrophobic (yellow) residues. Thr46 is boxed.
In this review we will discuss possible pathogenic mechanisms of the best characterized, p.P56S, VAPB mutation. We will limit our discussion to the cell-autonomous roles of the VAPs, and will not address possible extracellular effects of cleaved VAPB fragments, a phenomenon studied mainly in invertebrates (Tsuda et al., 2008). After an overview of the manifold functions of the versatile VAP proteins, we will review the results of cellular, animal, and patient studies on mutant VAPB, focussing on possible mechanisms of its dominant inheritance, as well as on how its loss of function could affect MN survival.
The VAPs: Structure and Functions
VAPs are ubiquitously expressed ER resident tail-anchored transmembrane (TM) proteins composed of an N-terminal MSP domain that faces the cytosol, followed by a predicted coiled coil (CC) region, and C-terminal TM helix (Figure 1(a)). The MSP domain is so-named because of its homology to the most abundant nematode sperm proteins, which function both in cell locomotion and as extracellular signalling molecules (Miller et al., 2001; Bottino et al., 2002). The VAPs are evolutionarily conserved from yeast to mammals, and remarkable conservation of the MSP domain is observed among different species (Figure 1(b)), highlighting the significance of this domain for VAP functions (reviewed in Lev et al., 2008; Murphy and Levine, 2016; Dudas et al., 2021).
The first member of the family, VAP33, was identified as an interacting partner of the SNARE protein VAMP in nerve terminals of Aplysia californica (Skehel et al., 1995); hence it was named
Mammals express two VAPs, VAPA and VAPB, which share 82% identity in the MSP domain (Figure 1(a) and (b)). Splicing variants of VAPB have been identified at the mRNA level, however, the translation products of these transcripts have not been detected in tissues (Nishimura et al., 1999; Nachreiner et al., 2010). Recently three additional mammalian ER proteins that harbour an MSP domain have been identified and named
VAPA and B are both able to form homodimers and also to heterodimerize with each other, a process driven by interactions between the TM helices (Nishimura et al., 1999; Kanekura et al., 2006), which both contain a dimerization motif (GXXXG; Russ and Engelman, 2000). The CC regions are also involved in dimerization (Kim et al., 2010), whereas the MSP domains, unlike the nematode homologs, are monomeric in solution (Kaiser et al., 2005; Shi et al., 2010). Dimerization may allow high avidity bivalent interactions of VAP with some of its ligands and/or bring different VAP ligands in close proximity to each other.
The VAPs function as adaptors, recruiting a myriad of binding partners to the cytosolic surface of the ER. This recruitment is often mediated by the binding of a so-called FFAT (two phenylalanines in an acidic tract) motif (consensus sequence: EFFDAXE) in the ligand to VAP's MSP domain (Loewen et al., 2003). Extensive research aimed at identifying and characterizing VAP binding partners (the VAPome) has revealed that deviations from this motif are tolerated, leading to a bewildering expansion of potential VAP interactors (Murphy and Levine, 2016; Slee and Levine, 2019), and to an increased need to experimentally verify predicted interactions as well as to define additional features of VAP ligands that strengthen their binding and confer selectivity for different MSPs (Furuita et al., 2010; Cabukusta et al., 2020). To be noted, FFAT motifs are not present in all cytosolic VAP binding partners, which may interact with regions of the MSP domain distinct from the FFAT binding site. In addition, the VAPs engage in functionally relevant interactions with ER membrane proteins via their TM helices (see “VAP Interactions in the ER” section below).
Structural studies have revealed the basis for the interaction of FFAT motifs with the VAP MSP domain, which is characterized by an immunoglobulin-like fold based on a seven-stranded β sandwich and one α helix. The FFAT binding site within the MSP consists of a conserved electropositive surface running transversally across four of the seven β strands, and two hydrophobic pockets that accommodate the second and fifth residue of the consensus (Figure 1(c); Kaiser et al., 2005; Furuita et al., 2010; Shi et al., 2010).
Many FFAT-bearing VAP ligands engage in additional interactions with partners, which are often specific lipids, exposed on the cytosolic face of membrane-bounded organelles other than the ER, and can thus serve as bridges between the ER and these other compartments. Indeed, the VAPs are involved in the generation of the majority of ER membrane contact sites (MCSs), key structures for interorganellar communication and integration (reviewed in Phillips and Voeltz, 2016; Prinz et al., 2020).
In the following paragraphs, and in Figure 2, we summarize the main VAP functions, identified up till now. The necessarily incomplete account is intended to provide a framework for the subsequent discussion of the cellular basis of the pathology caused by the p.P56S VAPB mutation.

Summary of the functions of the VAP proteins. See text for further explanation.
Lipid Transfer
Numerous FFAT-containing VAP-interacting partners are Lipid Transfer Proteins (LTPs), which exchange single, or a small number of, lipid molecules between the ER, where most are synthesized, and other organelles, thereby regulating the intracellular distribution of diverse lipid species and playing key roles in generating and maintaining the molecular identity of organelles (reviewed in Wong et al., 2018).
Among the mammalian LTPs, oxysterol-binding protein (OSBP) was first identified as a binding partner of VAPA (Wyles et al., 2002), a finding subsequently extended to a number of OSBP-related proteins (ORPs), and leading to the identification of the FFAT binding motif (Loewen et al., 2003). Although OSBP was first identified as an oxysterol-binding protein, it was later demonstrated to couple cholesterol export from the ER to the Golgi with the transport of phosphatidylinositol-4-phosphate (PI4P) in the opposite direction (Mesmin et al., 2013). At contact sites between the ER and the trans-most Golgi cisternae (Ladinsky et al., 1999), OSBP binds VAP on ER membranes, and phosphatidylinositol-4-phosphate (PI4P) in combination with the small GTPase Arf1 at the Golgi (Levine and Munro, 2002). OSBP regulates PI4P levels also in endosomes and lysosomes, acting at contact sites between the ER and these organelles (Dong et al., 2016; Lim et al., 2019).
Other FFAT-containing, VAP-interacting, LTPs, listed in Figure 2, effect non-vesicular transfer from the ER of sphingolipids (Kawano et al., 2006; D'Angelo et al., 2007; Mikitova and Levine, 2012) and glycerophospholipids (Kumar et al., 2018; Guillen-Samander et al., 2021), among which phosphatidylinositol (PI) (Kim et al., 2015; Cockcroft and Lev, 2021), and regulate the back-transport of cholesterol from the lysosomal/late endosome (LE) compartment to the ER (Alpy et al., 2013).
Calcium Regulation
Because of the VAP's pivotal role in phosphoinositide metabolism and because of the role of this lipid class in Ca2+ signalling, the VAPs are important players in many Ca2+ dependent pathways (Balla et al., 2020). In addition, direct protein-protein interactions may mediate VAP's role in regulating cytosolic [Ca2+] and Ca2+ stores. For example, the interaction between VAP and the outer mitochondrial membrane protein, protein tyrosine phosphatase-interacting protein 51 (PTPIP51) is critical for establishment of ER-mitochondria contacts and for the uptake of Ca2+ by mitochondria following its release from ER stores (De Vos et al., 2012; Stoica et al., 2014). The molecular basis of the interaction of PTPIP51 with VAP has not been fully elucidated so far: a functional FFAT-like motif is present in the central part of the sequence (Mikitova and Levine, 2012; Cabukusta et al., 2020; Yamanaka et al., 2020), however VAP regions outside of the MSP also contribute to the binding (Stoica et al., 2014). Notably, however, the VAP- PTPIP51 interaction is affected by TDP-43 and α-Synuclein intracellular aggregates, which are associated with ALS and Parkinson's disease, respectively (Stoica et al., 2014; Paillusson et al., 2017). These pathological aggregates perturb the ER-mitochondria association, in parallel with the disruption of Ca2+ exchange between the two organelles.
An additional role of the VAPs in Ca2+ signalling has been recently demonstrated in neurons, at presynaptic sites, where VAP interacts with the cytosolic protein Secernin-1 (SCRN1) via a FFAT-like motif. This interaction modulates ER dynamics and is required for the integrity of the organelle at nerve terminals; its loss results in alterations in presynaptic Ca2+ regulation and synaptic vesicle cycling (Lindhout et al., 2019).
Organelle Integrity and Dynamics
Through their multiple interactions, the VAPs affect the morphology and dynamics of the organelles that they contact. Such effects may be mediated by the LTPs, with which VAPs interact, including OSBP at endosomes (Dong et al., 2016), and the glycerophospholipid LTPs VPS13A and D at ER-mitochondria and ER-lipid droplet contact sites (Kumar et al., 2018; Yeshaw et al., 2019; Guillen-Samander et al., 2021). In other cases, direct interactions between the VAPs and integral membrane proteins exposed on the surface of interacting organelles are involved. For instance, the VAPs bind the peroxisome membrane protein, acyl-coenzyme A-binding domain protein 5 (ACBD5), and the resulting tethering is critical for restriction of peroxisome motility, while it enhances membrane expansion and peroxisome growth (Costello et al., 2017; Hua et al., 2017). Regulation of organelle motility by VAP is observed also for the lysosome/LE compartment: the direct, cholesterol-regulated interaction of VAP with ORP1L, by preventing the interaction of lysosomes/LEs with the dynein-dynactin complex, blunts their microtubule-dependent, centripetal transport (Rocha et al., 2009). At the same time, VAP facilitates kinesin-dependent transport of endosomal vesicles towards the periphery (see “VAP Interactions in the ER” section, below). Finally, at the neuronal plasma membrane, the voltage sensitive K+ channel Kv2.1 interacts with VAP via a phosphorylated C-terminal FFAT-like motif; the interaction determines clustering of the channels at ER-plasma membrane contacts and also facilitates recruitment to the plasma membrane of the VAP-interacting Nir2/3 PI transfer proteins; these help to return the plasma membrane phospholipid composition to basal values after activation of signalling cascades (Johnson et al., 2018; Kirmiz et al., 2018).
Autophagy
VAP regulates several steps of autophagic processes. VAP (A+B) directly interacts with multiple autophagy proteins, thereby contributing to formation of ER-isolation membrane contacts and autophagosome biogenesis (Zhao et al., 2018). At a later step, the cholesterol-regulated interaction of VAP with the aforementioned ORP1L (“Organelle Integrity and Dynamics” section) on the lysosome/LE membrane, regulates their fusion with autophagosomes (Wijdeven et al., 2016). In addition, ER-mitochondria tethering by the VAPB-PTPIP51 interaction reduces autophagosome formation; delivery of Ca2+ to mitochondria from ER stores, as well as transcriptional responses, appear to be involved in this phenomenon (Gomez-Suaga et al., 2017; Wu et al., 2018).
The size of the ER compartment itself is known to be regulated by a selective form of autophagy called ER-phagy. Several ER membrane proteins have been identified as receptors that mediate interaction of the ER with autophagy-related proteins of the ATG8/LC3 family (reviewed in Wilkinson, 2020). Recently, a cytosolic protein CALCOCO1 was also identified as an additional ER-phagy receptor, and in this case, its binding to VAP via the FFAT motif is critical for its role in promoting ER degradation (Nthiga et al., 2020).
VAP Interactions in the ER
In addition to their connections with membrane proteins of other organelles and with cytosolic proteins that serve as interorganellar bridges, the VAPs also engage in functionally important interactions with ER-resident proteins. These interactions may be mediated by FFAT motifs in the ligand, or may occur within the bilayer via interaction between the TM helices of the VAPs and the interacting protein. As illustrated below, some of these interactions appear to be particularly important in neurons.
Neurite extension involving the VAP-protrudin interaction. Protrudin is an ER membrane protein that bears a FFAT-like motif and binds the microtubule motor kinesin. It interacts with LEs by coincidence detection of the small GTPase Rab7 and Phosphatidylinositol-3-phosphate (Raiborg et al., 2015). This allows the handover of kinesin to the LEs and drives their transport towards the periphery, a process that provides the membrane required for neurite elongation. Since protrudin is an integral membrane protein, VAP is not required for its localization to the ER. Nonetheless, VAPA binds protrudin via the FFAT motif and is required for protrudin's function in promoting neurite elongation (Saita et al., 2009). Notably, VAPA binds protrudin much more strongly than does VAPB, providing one of the few examples of different roles of the two paralogues.
ER-to-Golgi transport and neurite extension involving YIF1A. Yip1-interacting factor homologue A (YIF1A) is a transmembrane protein that recycles between the ER and Golgi, and that facilitates secretory transport from the ER to the Golgi in neurons, enhancing the generation of COPII-coated transport vesicles by an incompletely understood mechanism. The VAP-YIF1A interaction, mediated by the TM domains of the two proteins, appears to be functionally relevant: in cultured hippocampal neurons VAP depletion results in displacement of YIF1A from the ER to the ER-Golgi intermediate and cis Golgi compartments, implicating that its recycling at the ER-Golgi interface is disturbed; the altered recycling correlates with a delayed transport of membrane vesicles into neurites, an effect caused also by YIF1A depletion, and which results in reduced dendrite extension and axon growth (Kuijpers et al., 2013b).
Phosphoinositide phosphatase Sac1 and phosphoinositide regulation. The ER-localized transmembrane protein Sac1 converts PI4P to PI, thereby regulating the levels of this phosphoinositide, a prominent player in lipid exchange at MCSs (“Lipid Transfer” section), in membrane trafficking and in cytoskeletal dynamics (Hammond and Burke, 2020). In Drosophila, the single VAP ortholog, Vps33, binds Sac1 via interaction between the TM domains and appears to be required for Sac1's hydrolytic activity (Forrest et al., 2013). The interaction of the VAPs with Sac1 is also observed in yeast (Stefan et al., 2011) and in mammals (Wakana et al., 2015), where, as in the case of protrudin, VAPA is the preferred partner.
Hyperpolarization-activated cyclic nucleotide
ATF6, FAF1 and the Unfolded Protein Response (UPR). The VAPs have been implicated in the regulation of proteostasis and of the UPR, although different conclusions have been reached in different experimental models. Two described protein-protein interactions could contribute to VAP's role in these processes: one that involves the UPR sensor/transcriptional regulator ATF6 (Gkogkas et al., 2008) and the other the p97 adaptor FAF1/UBXN3A/UBXD12 (Baron et al., 2014). ATF6 interacts with VAP's MSP via an unidentified portion of its N-terminal, cytosolic domain; the interaction, by blunting ATF6's transcriptional activity, is thought to negatively regulate this arm of the UPR. The FAF1 adaptor belongs to a family of ubiquitin-binding adaptors for p97/Valosin Containing Protein (VCP)1, an AAA ATPase that, by extracting misfolded proteins from the ER for proteasomal degradation, is a central player in the ER Associated Degradation pathway (ERAD) (reviewed in Qi et al., 2017). Through the interaction with FAF1, mediated by a non-canonical FFAT motif, VAP is suggested to be recruited to a complex of proteins that mediate the ERAD pathway and to negatively regulate their activity (Ernst et al., 2016).
Possible Mechanisms for the Dominance of the p.P56S-VAPB Allele: Gain Versus Loss of Function
Whereas haplosufficiency, i.e., sufficiency of the product of one wild-type allele for normal function, is the single cause of the recessive inheritance of many diseases, different, often not mutually exclusive, mechanisms underlie the phenotypic effects of dominant mutations (Wilkie, 1994). These can be roughly classified as gain-of-function mutations, meaning that the mutant protein acquires a new, generally harmful, function not possessed by its wild-type counterpart, or loss-of-function, meaning that the functionality of the product of the single wild-type allele is reduced or insufficient in the heterozygote. As mentioned in “Introduction: Amyotrophic Lateral Sclerosis and VAPB” section, the P56S-VAPB phenotype is dominantly inherited, and a number of possible mechanisms for this mode of transmission are conceivable, as schematized in Figure 3.

Possible mechanisms of dominance of the p.P56S mutation. Top: heterozygous individuals produce P56S-VAPB (red) and the wt protein (green). Left: toxic gain of function of the mutant: P56S-VAPB aggregates, or smaller oligomeric assemblies, could be harmful to the cell by sequestering proteins involved in proteostasis or other basic cellular processes, or by unphysiologically trapping normal binding partners (sequestered proteins are represented by blue circles). Right: Loss of function of the wt allele: this could occur by negative dominance, i.e., inactivation of the wt protein by sequestration into mutant aggregates; alternatively, P56S-VAPB inclusions could be without effect and easily degraded (red dots), but the amount of protein generated from the single wt allele would be insufficient to sustain normal function (haploinsufficiency).
P56S-VAPB, when transfected into cultured cells or expressed in some model organisms, forms intracellular aggregates (Nishimura et al., 2004b; Tsuda et al., 2008; Fasana et al., 2010 - discussed in more detail in “The Intracellular Inclusions Formed by P56S-VAPB” section]. Because intracellular inclusions are hallmarks of neurodegenerative diseases, including ALS (Blokhuis et al., 2013), and because large aggregates or smaller oligomers of mutant proteins are the suspected culprits of many of these pathologies (Haass and Selkoe, 2007), much work has focussed on the possible role of the P56S-VAPB inclusions in ALS8 pathogenesis. A priori, it could be hypothesized that the mutant protein is endowed with a gain of toxic function, because the intracellular inclusions that it generates could unphysiologically trap key proteins involved in pathways essential for cell survival (Powers et al., 2009; Olzscha et al., 2011), and/or lead to the accumulation of reactive oxygen species, thereby inflicting oxidative damage to cellular macromolecules (Barber et al., 2006). In addition, the binding by the inclusion-localized mutant VAPB to any of its numerous physiological interaction partners in a way that would not occur under normal conditions, should also be classified as gain-of-function. Instead, sequestration by the inclusions of the product of the wild-type VAPB allele would lead to loss-of-function by a classical mechanism of negative dominance. Additionally, loss-of function caused by haploinsufficiency could underlie the dominance of the p.P56S mutation. Haploinsufficiency, i.e., the intolerance of a diploid organism to the loss of one functional allele, occurs in the case of dosage-dependent genes, whose products, under physiological conditions, operate at levels close to the threshold for normal function (Wilkie, 1994; Johnson et al., 2019). It is the causal factor of a number of dominantly inherited diseases (Johnson et al., 2019), and contributes, for instance, to the pathogenic mechanism of the ALS-linked genes, C9ORF72 (Shi et al., 2018) and TBK1 (Freischmidt et al., 2015).
We have presented the above summary as a premise to the following sections of this review, in which we will discuss the evidence in support of, or against, each of the possible pathogenic mechanisms of the p.P56S mutation.
The Intracellular Inclusions Formed by P56S-VAPB
Concurrently with the pedigree analysis of the p.P56S mutation in Brazilian families, in vitro studies revealed that, differently from the wild-type protein, P56S-VAPB expressed in mammalian cultured cells clusters into large aggregates that can be detected by immunofluorescence (Nishimura et al., 2004b). The presence of such intracellular inclusions was subsequently confirmed both in transfected cells (Figure 4(a); Kanekura et al., 2006; Teuling et al., 2007; Prosser et al., 2008; Suzuki et al., 2009; Fasana et al., 2010; Papiani et al., 2012) and in transgenic animals (see Table 2 for references) and correlated well with the biochemical experiments showing the insolubility of mutant VAPB in neutral detergents. Further analysis revealed that, while the P56S substitution in the MSP is required for aggregation, the CC domain as well as the TM bearing the GXXXG dimerization motif are essential contributing factors (Kim et al., 2010).

Characterisation of inclusions formed by P56S-VAPB. (A) Immunofluorescence of HeLa cell expressing wt or P56S-VAPB. (B and C) Transmission electron micrographs of HeLa cells transiently transfected with wt (B) or P56S-VAPB (C). N, nucleus, M, mitochondrion; the arrows in (B) indicate normal ER cisternae. (D) high magnification view of altered ER induced by P56S-VAPB. The asterisks mark the ER lumen and the arrows indicate the continuity of the electron dense layer between the cisternae with the surrounding cytosol. The five small arrowheads indicate membrane-attached ribosomes. (E) Tomographic 3D reconstruction of P56S-VAPB inclusion in transfected HeLa cell, confirming the presence of ER cisternae in the structures. Panels (A) to (D): modified from (Fasana et al., 2010), panel (E) is from (Papiani et al., 2012). Scale bars: (A), 5 μm; (B) and (), 1 μm; (D) and (E), 200 nm.
Animal Models and Patient Studies on the Consequences of VAPB Depletiona or of P56S-VAPB Expression.

aThe knock-out animals included in this table are relevant to effects specific for VAPB deletion alone. Knock-out fly and worm lines are not listed, because these organisms harbour a single VAP homolog; hence its deletion is equivalent to the double knockout of the two VAP isoforms in mammals. To be noted, VAPA knockout in mice is embryonic lethal (McCune et al., 2017).
bInclusions are positive for autophagy, and not ER, markers.
cThe table reports only the results obtained with transgenic flies, in which mutant VAP was expressed in the presence of the wt protein; results were different when the mutant was expressed in a null background (see text).
dIn this study, mutant and wt VAP were co-expressed at similar, physiological, levels.
Although it was initially anticipated that the P56S-VAPB inclusions would share features with other mutant protein aggregates involved in neurodegenerative diseases (reviewed in Chung et al., 2018), biochemical and high resolution morphological analyses revealed that, differently from other investigated pathological aggregates, P56S-VAPB inclusions in transfected cells correspond to dramatically restructured ER domains. These form after normal integration of the newly synthesized mutant protein into the ER membrane (Fasana et al., 2010). Unlike the multilamellar structures of classical Organized Smooth ER (OSER; Snapp et al., 2003), the ER subdomains in which these aggregates reside consist in a limited number (generally two or three) of undulating cisternae separated by a ∼30 nm-wide electron-dense layer of cytosol (Figure 4(c) to (e); Fasana et al., 2010; Papiani et al., 2012). In agreement with these EM studies, proteomic analysis of immuno-isolated cellular subfractions, as well as in situ proximity ligation assays recently demonstrated the delocalization of mutant VAPB from regions of tubular smooth ER (Yamanaka et al., 2020).
Confocal analysis showed the presence of some ER resident proteins within the P56S-VAPB inclusions. Furthermore, GFP, expressed in the cytosol as a soluble protein or anchored to the ER membrane, can move in and out of them (Teuling et al., 2007; Fasana et al., 2010), indicating that the inclusions do not have the compact structure characteristic of some other mutant protein aggregates (Matsumoto et al., 2006), and demonstrating their continuity with the rest of the ER. Like other mutant proteins involved in neurodegeneration, however, aggregated P56S-VAPB is polyubiquitinated, both in transfected mammalian cells (Kanekura et al., 2006; Papiani et al., 2012) and in transgenic animals (Ratnaparkhi et al., 2008; Tsuda et al., 2008; Tudor et al., 2010; Kuijpers et al., 2013a; Larroquette et al., 2015).
An unexpected discovery for us was the ease with which the cell can eliminate the mutant VAPB inclusions (Papiani et al., 2012). In an inducible HeLa cell model, we found that aggregated P56S-VAPB was eliminated with a half-time of ∼ 7 h versus 17 h for the wt protein. Clearance of the inclusions was operated by the proteasome, with no involvement of macroautophagy, and was inhibited by a dominant negative form of p97/VCP, suggesting that this AAA ATPase extracts single P56S-VAPB molecules from the aggregates for delivery to the proteasome (Papiani et al., 2012; Genevini et al., 2014). In agreement with these results, p97/VCP was detected by immunofluorescence associated with mutant VAPB inclusions (Kuijpers et al., 2013a). The observed instability of P56S-VAPB most likely underlies its generally lower steady state levels compared to the wild-type protein both in transfected cultured cells (e.g., (Kanekura et al., 2006; Suzuki et al., 2009; Langou et al., 2010) and in transgenic animals (Aliaga et al., 2013; Kuijpers et al., 2013a; Qiu et al., 2013).
The precise structural modifications that underlie the aggregation propensity of the P56S-VAPB MSP have not been entirely clarified. As shown in Figure 1(b) and (c), Pro56 lies in a highly conserved region of the MSP domain, next to the FFAT peptide binding domain; within this region it adopts the cis-peptide bond conformation, which appears to be required for stabilization of the S-shaped loop that connects the D1 and D2 β strands (Kaiser et al., 2005). Arg55 within the loop and Val54 of the D1 strand directly contact FFAT residues (Furuita et al., 2010; Shi et al., 2010). It might be predicted that substitution of Pro with Ser, which is expected to adopt the energetically more favourable trans-peptide bond conformation, would destabilize the functional conformation, however, there are only few studies to directly assess the structural changes caused by the P56S substitution. NMR and far UV-CD analysis of bacterially expressed P56S-VAPB MSP extracted from inclusion bodies revealed a predominant random-coil conformation of mutant MSP in aqueous solution (Shi et al., 2010), and adoption of an alpha helical conformation in the presence of lipids (Qin et al., 2013). Results of another study, instead, indicated that the mutation has no severe impact on the fold of recombinant MSP, but that solvent-exposed hydrophobic patches on the surface of the mutant drive its temperature-dependent aggregation (Kim et al., 2010). The reported temperature-dependence is consistent with the observation that the mutant is largely non-aggregated when expressed in mammalian cells at 20°C (Teuling et al., 2007). The conclusion that the mutation does not cause gross structural changes was reached also for the P56S-VAPA MSP (Furuita et al., 2010).
Relatively mild conformational alterations induced by the P56S substitution would also be consistent with the capability of the mutant MSP, under conditions in which it is not aggregated, to bind FFAT-bearing proteins (Furuita et al., 2010; Kim et al., 2010; Baron et al., 2014). This may explain why the co-expression of mutant VAPB with an FFAT-bearing construct rescues the inclusion phenotype in mammalian cells (Prosser et al., 2008); it may also underlie the surprising observation that mutant VAP (either human VAPB or the Drosophila orthologue VAP33A) rescues lethality and other milder phenotypic effects caused by VAP deletion in flies (Chai et al., 2008; Moustaqim-Barrette et al., 2014; Kamemura et al., 2021). Nevertheless, mutant VAPB within aggregates is unable to interact, or interacts poorly, with the FFAT motif (Teuling et al., 2007; Suzuki et al., 2009; Kim et al., 2010; Kuijpers et al., 2013a); in agreement, the recent study of two of us showed that the P56S substitution caused VAP to lose interactions with multiple partners, including ones that mediate contacts with other organelles in mammalian cells (Yamanaka et al., 2020). Furthermore, in yeast and in zebrafish the mutant protein, unlike in flies, is unable to compensate for VAP loss (Suzuki et al., 2009; Kabashi et al., 2013). These results support the conclusion that the mutation, by driving the aggregation of P56S-VAPB, de facto generates a non-functional protein.
Are P56S-VAPB Inclusions Pathogenic?
Evidence for Gain-of-Function
The discovery and characterization of P56S-VAPB-containing aggregates led, not surprisingly, to the hypothesis that the aggregates could harm cells by sequestering, through aberrant interactions, wt VAPB expressed from the normal allele (negative dominance - Figure 3) and/or other proteins (gain-of-function). Indeed, despite loss of binding to many of its partners (Yamanaka et al., 2020), proteomic analysis has shown that P56S-VAPB retains or shows even increased binding to some of its normal interactors (Huttlin et al., 2015). Accordingly, a number of proteins described in “The VAPs: Structure and Functions” section of this review have been observed to accumulate in P56S-VAPB inclusions: (i) recruitment of Yif1A to VAPB inclusions in transfected mammalian cells results in inhibition of dendritic and axonal growth and maintenance (Kuijpers et al., 2013b); (ii) sequestration by the aggregates of the PI4P phosphatase Sac1 is paralleled by increased PI4P levels in transgenic fly tissues and by consequent neurodegeneration (Forrest et al., 2013)). Recruitment of transfected GFP-Sac1 to P56S-VAPB aggregates has also been observed in mammalian cells (Darbyson and Ngsee, 2016); (iii) the outer mitochondrial membrane protein PTPIP51 drives clustering of mitochondria near the aggregates in cultured cells that strongly overexpress the mutant, in parallel with perturbed Ca2+ handling (De Vos et al., 2012); (iv) similarly, the peroxisomal membrane protein ACBD5 mediates recruitment of peroxisomes to aggregated P56S-VAPB (Hua et al., 2017); (v) the HCN2 channel is trapped in P56S-VAPB aggregates in the cell body of transfected primary cultured cortical neurons and consequently hindered from reaching its normal localization on the cell surface (Silbernagel et al., 2018).
Other proteins reported to accumulate in the inclusions are autophagy and ER stress markers (Tsuda et al., 2008; Aliaga et al., 2013; Larroquette et al., 2015), as well as proteins of the ERAD pathway (Kuijpers et al., 2013a), indicative of disturbed proteostasis and pathological chronic ER stress. In two transgenic mouse lines, the neuronal levels of the ubiquitin-binding autophagy receptor p62/sequestosome are increased (Aliaga et al., 2013; Larroquette et al., 2015), suggesting that alterations in autophagic flux, induced by mutant VAPB, might contribute to neurodegeneration.
Evidence for Negative Dominance
There is ample evidence that P56S-VAPB aggregates are able to sequester the wt protein. Indeed, both in transfected cultured mammalian cells (Kanekura et al., 2006; Teuling et al., 2007; Suzuki et al., 2009; Kim et al., 2010), and in tissues of transgenic flies (Chai et al., 2008; Ratnaparkhi et al., 2008; Tsuda et al., 2008), wt VAPB is recovered within Triton-insoluble P56S-VAPB aggregates and/or is enriched in the inclusions observed by immunofluorescence. The TM domain of wt VAPB is necessary and sufficient for its sequestration (Teuling et al., 2007; Suzuki et al., 2009), but the affinity of the binding is increased by interactions between the wt and the mutant MSPs (Suzuki et al., 2009). In contrast to VAPB, and perhaps surprisingly, given the high degree of sequence conservation between the two VAP paralogues and their capacity to engage in heterodimerization, VAPA is only weakly recruited into the inclusions, if at all (Kanekura et al., 2006; Teuling et al., 2007; Suzuki et al., 2009; Qiu et al., 2013; Yamanaka et al., 2020).
The negative dominance hypothesis has been strengthened by results with animal models, showing that the phenotypic consequences of VAPB (or Drosophila VAP33) deletion are the same as those of mutant VAPB overexpression in a wt background. This is illustrated by the studies on the organization of the neuromuscular junction (NMJ) in the fly model: VAP33 null or hypomorphic mutants and transgenic animals with overexpressed P58S-VAP33 (equivalent to the P56S substitution in humans) in a wt background have a similar phenotype, consisting in a decreased number of synaptic boutons and an increase in their size (Pennetta et al., 2002; Chai et al., 2008; Ratnaparkhi et al., 2008; Forrest et al., 2013) (see Table 2). In addition, P58S-VAP33 overexpression or VAP33 silencing both result in decreased Sac1 activity and increased PI4P levels in MNs (see “The VAPs: Structure and Functions” section; Forrest et al., 2013).
Evidence in Favour of Haploinsufficiency
Despite the above summarized results in support of a pathogenic role of P56S-VAPB, we argue that the p.P56S mutation is unlikely to play a major direct role in ALS pathogenesis either by gain-of-function or by dominant negative loss-of-function mechanisms.
The first, obvious, consideration is that the majority of the studies on the effects of P56S-VAPB inclusions, summarized in Table 2, were carried out in models with expression levels of the mutant well over those supported by one mutant allele as in ALS8 patients. In the one study in Drosophila, in which mutant and wt VAP33 were co-expressed at levels close to the physiological one, no motor deficit, nor neurodegeneration, nor changes in longevity of adult flies were observed (Moustaqim-Barrette et al., 2014). Furthermore, how the effects of mutant VAP on NMJ structure of flies relate to MND is not clear, as overexpression of wt VAP also causes motor dysfunction in adult flies (Tsuda et al., 2008; Sanhueza et al., 2014).
Considering the murine models, of the four reported p.P56S-VAPB transgenic mouse lines, only one, in which the mutant protein was highly overexpressed, developed mild motor abnormalities and loss of cortical, but not spinal, MNs (Aliaga et al., 2013). The other three strains, although presenting P56S-VAPB-containing inclusions in MNs, showed no motor abnormalities (see Table 2).
ALS8 patients' cells have also been analysed for the presence of inclusions, which were, however, not detected either in MNs generated from induced pluripotent stem cells (IPSCs) from Brazilian subjects (Mitne-Neto et al., 2011) or in skin fibroblasts from a North American patient (Guber et al., 2018). In the Brazilian study, immunoblotting with an antibody that recognizes both the wt and the mutant protein revealed reduced levels (∼50% of controls) of VAPB, suggesting that the product of the mutant allele is unstable and eliminated, in agreement with our studies in cultured cells (Papiani et al., 2012; Genevini et al., 2014).
It might be hypothesized that the lack of pathogenicity of the inclusions in transgenic mice is due to differences in metabolism and lifespan between mice and men; however, the results obtained with knock-in mice argue against this idea: the heterozygote knock-in mice did develop mild, late onset motor deficits and partial denervation of lower MNs, consistent with the slow progression of ALS8 (Larroquette et al., 2015). Comparison of these results with those of the transgenic animals, in which the full dose of wt VAPB is present, argues strongly in favour of the concept that halving the gene dosage of wt VAPB is necessary to drive ALS8 pathology.
If haploinsufficiency, rather than P56S-VAPB expression, is the main disease-causing mechanism, one should expect that a VAPB knockout heterozygote is equivalent to the p.P56S-VAPB heterozygote knock-in animal. Although VAPB knockout homozygote mice did develop a very mild, but significant, deficit in the rotarod test (Kabashi et al., 2013), the performance of the heterozygotes in this test, although slightly impaired, was not reduced in a statistically significant manner. The better motor performance of the heterozygote knock-out than the heterozygote knock-in mice suggests that the expression of P56S-VAPB does contribute to the disease. However, a study aimed at directly comparing the knockout and knock-in animals has not, to our knowledge, been performed.
In summary, the poor correlation between the presence of P56S-VAPB aggregates and motor dysfunction in transgenic animals and in patients, and the development, instead, of pathology in heterozygote knock-in animals support the conclusion that the reduced expression of the functional protein driven by a single allele is required to cause MN disease, as previously suggested by us (Papiani et al., 2012; Genevini et al., 2014) and others (Mitne-Neto et al., 2011; Qiu et al., 2013). It cannot, however, be excluded that P56S-VAPB aggregates, despite not being detected in patients' fibroblasts or iPSC-derived MNs, do accumulate to some extent in aged ALS8 MNs, thereby aggravating VAPB haploinsufficiency. A harmful effect of the p.P56S mutation is further suggested by the lack of detection, so far, of loss-of-function mutations of VAPB, not involving the generation of an aggregate-prone protein, in ALS8 patients. Such mutations (e.g., early truncations or frameshift mutations) would strongly support the “pure” haploinsufficiency hypothesis. Future work on animals, cells, and patients will likely resolve the issue of whether haploinsufficiency alone or haploinsufficiency aggravated by the p.P56S mutation drives fALS8 pathology.
Loss of VAPB Function: What Function?
Given the extraordinarily large number of vital processes in which the VAPs are involved (“The VAPs: Structure and Functions” section) it is impossible to predict which of these could be impaired in MNs by VAPB deficit. Most likely, more than one process is affected, and the summation of the resulting deficits drives MNs to their demise. In agreement with a pleiotropic effect of VAPB loss, a number of different processes have been reported to be affected in cellular or animal models of VAPB deficiency.
In this section, we will summarize the results of exclusive VAPB deletion/reduction in cellular or animal models; we will not discuss the effects of overexpressed mutant VAPB, nor those of combined VAPA/B knockdown or of VAP deletion/reduction in animals that harbour only one VAP isoform. Whist the dramatic phenotypes observed as a result of total VAP depletion have yielded important mechanistic information on VAP function, we focus here on specific effects of VAPB downregulation as more directly relevant to ALS8 pathogenesis.
ER-Mitochondria Contact Sites, Ca2+ Regulation, Autophagy
Silencing of VAPB in the MN-like NSC34 cell line reduces the extension of mitochondria-ER close contacts by 30-40%, and results in a delay in the uptake of cytosolic Ca2+ by mitochondria upon the stimulated release of the cation from ER stores (De Vos et al., 2012; Stoica et al., 2014). The loss of contacts is attributed to the deletion of the VAPB-PTPIP5 tether (“The VAPs: Structure and Functions” section), as VAPB or PTPIP51 silencing produce superimposable effects. Considering that multiple protein complexes are responsible for tethering mitochondria to the ER (Zung and Schuldiner, 2020), the 40% decrease in contact extension obtained by VAPB knockdown is a dramatic effect, implicating the VAPB-PTPIP51 interaction as a prominent contributor to ER-mitochondria contacts. Importantly, such VAPB-PTPIP5-mediated contacts are present also in nerve terminals, and VAPB or PTPIP51 silencing in primary hippocampal neuron cultures reduces ER-mitochondria contacts concomitantly with synaptic activity (Gomez-Suaga et al., 2019).
Another consequence of VAPB silencing, directly linked to the reduction of ER-mitochondria contacts and to impaired Ca2+ regulation, is an increase of basal autophagy and autophagic flux, an effect that can be reversed by expression of an artificial ER-mitochondrion tether in VAPB-silenced cells (Gomez-Suaga et al., 2017). Since autophagy deregulation is implicated in ALS pathogenesis (Navone et al., 2015; Deng et al., 2017), this observation provides a further possible connection between VAPB deficit and MN degeneration.
HCN Channels
The discovery of the interaction of the VAPs with HCN1 and 2 channels (“The VAPs: Structure and Functions” section), prompted investigation of the electrophysiological modifications in the heart and in the ventrobasal thalamus of VAPB knockout mice (Silbernagel et al., 2018). In slice patch-clamp experiments, a reduction in the amplitude of the hyperpolarization-induced cation-mediated current characteristic of HCN channels was observed in the neurons of the ventrobasal thalamus of VAPB knock-out mice in comparison to controls. Severe bradycardia, more difficult to relate to ALS, were also observed in the knockout mice. The decreased current amplitude observed in neurons was consistent with decreased surface expression of the HCN1 and 2 channels, a phenomenon attributed to the facilitatory effect of VAPB on transport of the channel through the secretory pathway. As noted in “The VAPs: Structure and Functions” section, the interaction with HCN1 and 2 is mediated by VAPB's TM helix, and therefore occurs within the plane of the bilayer, a finding difficult to reconcile with the different localisations (ER and plasma membrane) of the interacting proteins. The authors suggest that VAPB is a subunit of HCN channel complexes, however, this hypothesis awaits rigorous proof. The finding that VAPB deletion induces changes in neuronal excitability provides a potential link to ALS8 pathogenesis, of particular interest in light of the implication of ion channel dysfunction in other neurodegenerative diseases (Chang et al., 2019).
Dysregulation of Phosphoinositide Metabolism
A general finding of the past decade has been that VAP depletion causes elevation of intracellular PI4P levels; this phenomenon is observed also in cells with exclusive VAPB knockdown and intact VAPA levels (Darbyson and Ngsee, 2016; Genevini et al., 2019), demonstrating a critical role of VAPB in preserving phosphoinositide homeostasis. Interestingly, in the MN-like NSC34 cell line, even a partial reduction of VAPB, to levels mimicking those of a p.P56S-VAPB heterozygote, is sufficient to affect PI4P levels in the Golgi complex and to cause the expansion of a PI4P-positive population of peripheral vesicles (Genevini et al., 2019). Increased levels of PI4P have been directly linked to neurodegeneration in Drosophila and C. elegans ALS8 models, in which downregulation or pharmacological inhibition of PI4 kinases, the enzymes that generate PI4P from PI, improves neuronal survival (Forrest et al., 2013; Zhang et al., 2017).
There are several plausible mechanisms that could underlie the neurotoxic effect of elevated PI4P. High levels of the phopshoinositide could excessively or aberrantly recruit PI4P-binding proteins to compartments of the exo-endocytic pathway (De Matteis et al., 2013; Dong et al., 2016; Tan and Gleeson, 2019) or might be linked to disturbances in the distribution of other lipids, whose transport is coupled to that of PI4P. Such alterations could result in impairment of lysosomal degradation and autophagy, as reported by two recent studies (Mao et al., 2019; Zhang et al., 2020) and/or in a deficit in exocytic transport in neurons. Indeed, in the case of the NSC34 cell model, a significantly reduced rate of neurite elongation was observed in VAPB-downregulated cells induced to differentiate (Genevini et al., 2019), suggesting a defect in the trafficking of transport vesicles to the growing neurite; this deficit could as well be relevant to the ability of mature MNs to replenish their axons and dendrites with membrane components. A causal relationship between delayed neuritogenesis and elevated PI4P in the NSC34 model was indicated by the rescue of the phenotype when PI4P levels were reduced by pharmacological inhibition of PI4 kinase III
Another contributor to PI4P-induced neurodegeneration could be the disruption of the transport of nuclear envelope and pore proteins to their correct destination, in parallel with nuclear envelope abnormalities, observed as a consequence of VAPB depletion and PI4P elevation (Tran et al., 2012; Darbyson and Ngsee, 2016). These abnormalities could negatively affect nucleo-cytoplasmic transport, a phenomenon implicated in ALS pathogenesis (Kim and Taylor, 2017), and observed in cultured fibroblasts of an ALS8 patient (Guber et al., 2018).
An open question is the identity of the VAPB-sensitive step(s) within the complex cycle of events that regulate PI4P intracellular levels. Two key players of the cycle are OSBP, which transports PI4P, generated in the Golgi by PI4 kinases, to the ER in exchange for cholesterol, and the phosphatase Sac1, which hydrolyses PI4P in the ER, thus maintaining a Golgi-ER concentration gradient of the phosphoinositide that fuels the OSBP-mediated cholesterol transport out of the ER against its concentration gradient (reviewed in (Antonny et al., 2018)). Sac1 can also act in trans to locally hydrolyse PI4P in the Trans Golgi Network (Venditti et al., 2019). Both OSBP and Sac1 are VAP-dependent (see “The VAPs: Structure and Functions” section), however, OSBP associates with both VAP paralogues (Huttlin et al., 2015; Murphy and Levine, 2016; Cabukusta et al., 2020), and Sac1 has a strong preference for VAPA over VAPB (Wakana et al., 2015). Thus, it is unclear whether either OSBP or Sac1 are affected by the selective downregulation of VAPB, or whether other VAPB-dependent proteins are involved in generating the phosphoinositide imbalance (Darbyson and Ngsee, 2016). The problem of the contribution of VAPB to global VAP (A+B) function will be further discussed in “Why Motor Neurons and Why VAPB?” section.
ER Stress
Although indicators of the Unfolded Protein Response (UPR) have been reported to be upregulated in some p.P56S-VAPB transgenic animals (Tsuda et al., 2008; Aliaga et al., 2013), there is relatively little information on the effects of the reduction of endogenous VAPB on ER stress. ER stress takes stage when the protective (adaptive) UPR fails to resolve problems due to excessive load on the ER; chronic ER stress (or maladaptive UPR) can drive cells into apoptosis, hence its involvement in degenerative diseases (Lindholm et al., 2017), including ALS (Kikuchi et al., 2006; Nagata et al., 2007; Saxena et al., 2009).
Kanekura et al. (Kanekura et al., 2006) investigated the role of VAPB in one of the three branches of the UPR, the Ire1-XBP1 axis, which mediates the upregulation of ER chaperones and ERAD (Ron and Walter, 2007). This pathway was blunted in VAPB-silenced NSC34 cells treated with ER stressors, suggesting a role of VAPB in its activation. In another study (Gkogkas et al., 2008), instead, VAPB depletion resulted in increased expression of a UPR reporter under the control of two of the three UPR branches (the transcription factors XBP1 and ATF6), suggesting a role for VAPB in limiting the UPR, a role that was attributed to the direct interaction between VAPB and ATF6. It is noteworthy, however, that in the spinal cord and tibialis anterior muscle of a knockout mouse model, no alterations of any of the transcriptional readouts for the UPR (the master regulator BiP, and the transcription factors ATF4, ATF6, and CHOP) were detected (Kabashi et al., 2013).
A stronger argument for the involvement of ER stress in the development of ALS8 derives from the work on the knock-in mouse model (Larroquette et al., 2015). Before onset of motor symptoms, the homozygous knock-in mouse showed increased levels of BiP and of protein disulfide isomerase, which both report on an activated UPR. The authors hypothesized that the observed ER stress could be a consequence of deregulated autophagy, as the autophagy receptor p62/SQSTM was upregulated and accumulated in VAPB-containing foci in the MNs of these animals. The two UPR reporters were not, however, increased in heterozygous, age-matched, knock-in mice, which are of greater direct relevance to ALS8. Instead, and interestingly, a significant increase in the phosphorylation of the translation initiation factor eIF2 was observed in the spinal cord of these heterozygotes. Phosphorylation of eIF2α is the direct consequence of activation of the third UPR branch, mediated by the PKR-like ER kinase (PERK), which plays a key role in the maladaptive UPR. Although there was no increase in the expression of the transcription factors (CHOP and ATF4) involved in the PERK-triggered apoptotic pathway, the possibility that this pathway is upregulated in ALS8 is supported also by studies on patient-derived cultured fibroblasts (Guber et al., 2018).
In summary, UPR induction as a direct consequence of VAPB loss is, at present, poorly supported. However, it is feasible that ER stress occurs as an indirect consequence of the VAPB deficit-driven impairment of other processes (“ER-Mitochondria Contact Sites, Ca2+ Regulation, Autophagy,” “HCN Channels,” and “Dysregulation of Phosphoinositide Metabolism” sections) combined with the expression of the P56S mutant from a single allele. Such chronic ER stress could, in the long run, contribute to the deterioration of MN health.
Why Motor Neurons and Why VAPB?
A poignant question that concerns many genetic diseases is why only one or a few cell populations are harmed by the mutation of a ubiquitously expressed gene. In the case of VAPB, the question is particularly relevant, because, based on the proteomic analysis of VAP interactors, one might anticipate that the functions of VAPB are largely redundant with those of VAPA, so that loss of function of VAPB might be compensated by the A paralogue. The more so, because in all analysed tissues, including spinal cord, VAPA is expressed at higher levels than VAPB, both at the mRNA and at the protein level (www.genecards.org; www.ebi.ac.uk/gxa; www.gtexportal.org).
The vulnerability of MNs to VAPB loss of function has been attributed to the relatively elevated concentration of VAPB in spinal cord homogenates (Teuling et al., 2007); among spinal cord cells, particularly high levels are observed in MNs (Teuling et al., 2007; Larroquette et al., 2015). However, this reasoning does not take into account that also VAPA levels are high in spinal cord, where, like elsewhere, there is more VAPA than VAPB. To be noted, unlike the case of VAPB, VAPA knockout is embryonic lethal in mice (McCune et al., 2017).
In MN-like NSC34 cells, direct comparison of VAPA and VAPB protein levels by quantitative immunoblotting of cell lysates in parallel with standard recombinant proteins revealed an over 4-fold excess of VAPA over B (Genevini et al., 2019). On this basis, complete depletion of VAPB is expected to produce a mere 20% reduction of the global VAP pool. Nonetheless, even partial (50-60%) silencing of VAPB in these cells produced significant phenotypic effects (described in “Loss of VAPB Function: What Function?” section). It seems unlikely that these effects were due to a generalized diminution of the global VAP (A+B) pool: this would imply that the pool operates at levels so close to threshold that even a minor reduction in its levels is poorly tolerated. Instead, it seems more plausible that some specific function(s) of VAPB, which cannot be carried out by VAPA and which are particularly important for MN health and survival, underlie the effects of its depletion.
As noted above, because of the high conservation of the sequences of the two VAP isoforms, particularly of the structurally similar MSP domains, and in view of the results of proteomic analyses showing widely overlapping interaction profiles (e.g., Cabukusta et al., 2020), the two isoforms are considered to share most of their binding partners and to regulate common biological pathways. Nevertheless, as mentioned in previous sections of this review, two binding partners are described to interact more strongly with VAPA than with VAPB; one of these, protrudin (Saita et al., 2009), is a FFAT-bearing protein, while the other one, the Sac1 phosphatase, interacts with VAP's TM helix (Forrest et al., 2013). The converse situation has not been demonstrated so far: while 2-Hybrid screening has identified VAPB as binding partner of PTPIP51 (De Vos et al., 2012) and of HCN1 and 2 (Silbernagel et al., 2018), interactome studies identify also VAPA and MOSPD1-3 as PTPIP51 interactors (Huttlin et al., 2015; Cabukusta et al., 2020); furthermore, PTPIP51 as well as HCN2 are pulled down by both VAP paralogues.
Further differences between the two isoforms are suggested by different effects of overexpression of the wt VAPA or B on secretory pathway function (Prosser et al., 2008) and by the different effect that the P56S substitution has on their structure: introduction of the p.P56S mutation in the VAPA gene does not drive the dramatic aggregation of the overexpressed protein product, as is the case for VAPB (Teuling et al., 2007; Prosser et al., 2008). The future elucidation of additional features that distinguish the two paralogues will be key to unravelling the molecular basis of VAPB's role in ALS.
Conclusions and Outlook
The discovery of VAPB as a gene linked to MND occurred in the same time period in which the VAP proteins and the repertoire of their binding partners were beginning to be characterized. During the following years, research on the pathogenic mechanism of the VAPB mutation(s) proceeded in parallel with advances in the understanding of the functions of the VAP proteins and of their prominent role in the generation of MCSs.
Analysis of the sum of the studies on p.P56S-driven pathology leads to the conclusion that expression of P56S-VAPB is insufficient to cause disease, and that loss of one VAPB wt allele is necessary to trigger pathological changes in MNs. However, it still remains to be elucidated which of the many VAPB functions/binding partners are critical for MN health and survival; a number of very interesting leads will hopefully be further investigated in the coming years.
Because of the involvement of VAPB in MND, many interaction studies have focussed on the B isoform, disregarding a comparison with VAPA, perhaps because there are no known VAPA-linked diseases. Nevertheless, since there is more VAPA than B in MNs, and since the repertoire of binding partners between the two paralogues is largely overlapping, we feel that an important step in the direction of fully elucidating the pathogenic mechanism of VAPB loss will be to identify those interactors that crucially depend on the entire VAP pool or, alternatively, prefer VAPB over the A paralogue. The discovery of functions carried out preferentially by VAPB could lead to the identification of the principal culprits of the MN degeneration caused by VAPB loss, and to the development of novel therapeutic approaches.
Understanding the role of VAPB deficit in ALS8 is relevant not only for this rare fALS, but also for the more common sALS forms. Reduced VAP levels, or reduced accessibility to anti-VAPB antibodies, have been observed in the spinal cords of human sALS patients and of a transgenic fALS mouse model expressing a mutant protein not related to VAPB (G93A-SOD1) (Teuling et al., 2007; Anagnostou et al., 2010; Kim et al., 2016; Cadoni et al., 2020). Conversely, highly overexpressed VAPB was found to be protective for MNs in the same fALS mouse model (Kim et al., 2016).
Finally, beyond MNs, the VAPs are relevant to other pathologies: they play a role in infectious diseases, by facilitating the replication of some viruses and by driving formation of MCS between the ER and the inclusion membrane that encloses intracellular bacteria (reviews:(Derré, 2017; Dudas et al., 2021); VAPB (and maybe VAPA - not tested) promotes breast tumour growth (Rao et al., 2012; Jia et al., 2020), stabilizes mutant Cystic Fibrosis Transmembrane Conductance Regulator (Ernst et al., 2016), affects the inflammatory response of cystic fibrosis bronchial cells to pathogens (Rimessi et al., 2020), and regulates cardiac rhythm (Silbernagel et al., 2018). These phenomena further broaden the scope of future research aimed at unravelling the role of VAPB in MN and other disorders.
Footnotes
Acknowledgements
We are grateful to Marianna Leonzino (CNR Neuroscience Institute) for critically reading the manuscript.
Declaration of Conflicting Interests
The author(s) declared no potential conflicts of interest with respect to the research, authorship, and/or publication of this article.
Funding
The author(s) disclosed receipt of the following financial support for the research, authorship, and/or publication of this article: Work in the laboratory of NB and FN was supported by Fondazione Regionale per la Ricerca Biomedica (TRANS-ALS grant no. 2015-0023). Work in the laboratory of TY and NN was supported by a Grant-in-Aid from Ministry of Education, Culture, Sports, Science and Technology (MEXT) of Japan for T.Y. (18H02723, 17KT0131), Takeda Science Foundation, and The Sumitomo Foundation.