
Abstract
Mitochondria-endoplasmic reticulum contacts (MERCs), also called endoplasmic reticulum (ER)-mitochondria contact sites (ERMCS), are the membrane domains, where these two organelles exchange lipids, Ca2+ ions, and reactive oxygen species. This crosstalk is a major determinant of cell metabolism, since it allows the ER to control mitochondrial oxidative phosphorylation and the Krebs cycle, while conversely, it allows the mitochondria to provide sufficient ATP to control ER proteostasis. MERC metabolic signaling is under the control of tethers and a multitude of regulatory proteins. Many of these proteins have recently been discovered to give rise to rare diseases if their genes are mutated. Surprisingly, these diseases share important hallmarks and cause neurological defects, sometimes paired with, or replaced by skeletal muscle deficiency. Typical symptoms include developmental delay, intellectual disability, facial dysmorphism and ophthalmologic defects. Seizures, epilepsy, deafness, ataxia, or peripheral neuropathy can also occur upon mutation of a MERC protein. Given that most MERC tethers and regulatory proteins have secondary functions, some MERC protein-based diseases do not fit into this categorization. Typically, however, the proteins affected in those diseases have dominant functions unrelated to their roles in MERCs tethering or their regulation. We are discussing avenues to pharmacologically target genetic diseases leading to MERC defects, based on our novel insight that MERC defects lead to common characteristics in rare diseases. These shared characteristics of MERCs disorders raise the hope that they may allow for similar treatment options.
Introduction
The Discovery of Mitochondria-Endoplasmic Reticulum Contact Sites (MERCs) as Mitochondria-Associated Membranes (MAMs)
The original discovery by the Bernhard lab of mitochondria and endoplasmic reticulum (ER) contact sites (MERCs) showed over 70 years ago that these interorganellar contacts respond acutely to distinct metabolic and feeding states (Bernhard et al., 1952). We have reviewed the history of MERCs research recently (Herrera-Cruz and Simmen, 2017). This and other early findings remained obscure for a long time but suggested that MERCs intricately adapt to a variety of conditions, and that their putative functions could be compromised in a disease setting. Close to 40 years passed until MERC functions found solid experimental evidence with the discovery of MAMs as a lipid transfer platform (Vance, 1990) and a site of Ca2+ flux (Rizzuto et al., 1998). Both findings were key to the acceptance of MERCs as a true functional entity. The discovery of inflammasome formation and reactive oxygen species (ROS) nanodomains at MERCs added their connection to oxidative stress signaling (Zhou et al., 2011; Booth et al., 2016). From these seminal studies, many additional functions have emerged, which we will summarize in the following sections.
MERCs as Synthesis and Transfer Hubs for Lipids
The first function detected on MERCs was lipid metabolism (Figure 1, top panel) through the presence of enzymes synthesizing phosphatidylserine (PS; PS synthases 1 and 2 [PSS1, PSS2]), and phosphatidylcholine (PC; phosphatidylethanolamine methyltransferase [PEMT]) (Vance, 1991). The expression of these enzymes is highest in liver tissue, where they are frequently used as MERCs/MAMs marker proteins (Stone and Vance, 2000). A MAM marker that is more widely expressed is acyl-CoA synthetase long-chain family member 4 (ACSL4), also known as fatty acid-CoA ligase 4 (FACL4), which ligates fatty acids to coenzyme A (CoA) and controls polyunsaturated fatty acid (PUFA) synthesis (Lewin et al., 2001, 2002). These enzymes will be discussed in detail regarding their involvement in MERC rare diseases.

Functioning of MERC tethers and regulatory proteins associated with rare diseases. The schematics of the most important MERCs proteins and their functions are presented. For detail, please refer to the corresponding main text. The genes whose mutations have been known to cause rare diseases are shown in red. The proteins are categorized by their functions. (Top) The proteins involved in phospholipid transfer/metabolism at MERCs. (Second) The proteins involved in Ca2+ signaling and its regulation. (Third) The proteins involved in ROS signaling. (Bottom) The proteins involved in MERC tethering and mitochondrial membrane dynamics.
Following the identification as a lipid synthesis hub, MAMs were identified as the ER membrane, where PS is transferred to mitochondria, followed by its transformation into phosphatidylethanolamine (PE) on the inner mitochondrial membrane (Vance, 2003). PS-PE conversion is catalyzed by mitochondrial phosphatidyl decarboxylase (PISD), in parallel to ER-localized ethanolamine phosphotransferase 1 (EPT1) (Vance, 2015). While vesicular or direct cytosolic transport could not be detected for this lipid flux (Voelker, 1989), it requires ATP in mammalian liver cells, suggesting ongoing mitochondrial oxidative phosphorylation (OXPHOS) is required for this MERC function (Tatsuta et al., 2014). Recent findings indicate that MERC bulk lipid flux in general is mediated by the Vps13D lipid channel (Guillen-Samander et al., 2021). Smaller scale lipid exchange occurs through the pairing of ER oxysterol binding protein-related proteins 5 and 8 (ORP5, ORP8) (Guyard et al., 2022) with the mitochondrial contact sites and cristae junction organizing system (MICOS) (Monteiro-Cardoso et al., 2022). Given the importance of lipid synthesis and metabolism for liver tissue (Nguyen et al., 2008), the knockout of MERC-associated PSS1 and PSS2 was initially found to compromise liver functions (Bergo et al., 2002; Arikketh et al., 2008).
Another lipid transiting from the ER to mitochondria is phosphatidic acid (PA), which subsequently converts into mitochondrial cardiolipin (CL) with the help of a cascade of enzymes that culminates in CL synthase (Yeo et al., 2021). This cone-shaped lipid is critical for the classic curvature of mitochondrial cristae (Beltran-Heredia et al., 2019) and essential for the supercomplex assembly mediating OXPHOS (Zhang et al., 2002; Pfeiffer et al., 2003). The three-dimensional structure of cristae, as dictated by their enrichment of cone-shaped lipids, is thus a key requirement for the efficiency of mitochondrial respiration and cellular bioenergetics overall (Cogliati et al., 2013). This function is particularly evident in tissues with high energy demand (Finsterer, 2019). To a lesser extent, cone-shaped PE also promotes the mitochondrial OXPHOS capacity (Boyd et al., 2017; Funai et al., 2020). Since CL and PE are so important for mitochondrial ATP production, their synthesis is expected to predominantly impact mitochondrial functions, and thus predict weak MERC dependence of these pathologies. Indeed, defective CL synthesis in Barth syndrome (Mendelian Inheritance in Man, MIM#302060) is considered a mitochondrial disease (Finsterer, 2019). The rare X-linked pathology is characterized by cardiac and skeletal myopathy, as well as growth retardation in early life (Clarke et al., 2013). Unlike typical MERC-associated diseases (see later), Barth syndrome only affects the brain to a minor extent, compromising sensory perception, and leading to fatigue and minor cognitive defects (Olivar-Villanueva et al., 2021). In contrast, the importance of PE for the brain is more pronounced, maybe because it is not as essential for mitochondria and also, since PE is particularly abundant in this tissue (Vance and Tasseva, 2013). Within the brain, the significance of EPT1-associated PE synthesis is particularly obvious within motor neurons, where EPT1 mutations lead to hereditary spastic paraplegia (MIM#618768; Ahmed et al., 2017). However, similar to CL, its promotion of mitochondrial OXPHOS is also important for the functioning of skeletal muscle (Heden et al., 2019), and brown adipose tissue (Johnson et al., 2023). Together, MERC lipid synthesis and flux emerge as a key factor for mitochondrial ATP production. This function is tightly associated with the role of lipids like CL and PE for membrane curvature, a key feature needed for mitochondrial OXPHOS on their cristae. Thus, the functional readout of MERC-associated lipid metabolism is predominantly mitochondrial and is critical for liver, brain, and muscle tissue, leading to broad-spectrum disease if the genes encoding these proteins are mutated.
MERCs as a Ca2+ Signaling Center
The metabolic significance of MERCs is perhaps even more obvious from their role as a center of intracellular Ca2+ signaling (Figure 1, second panel). On MERCs, ER inositol 1,4,5-trisphosphate receptors (IP3Rs) interact with mitochondrial voltage-dependent ion channels (VDACs), assisted by the mitochondrial chaperone GRP75 (Szabadkai et al., 2006). IP3Rs can also interact with mitochondrial TOM70 (Filadi et al., 2018a). Therefore, the formation of these multimeric protein complexes is essential not only for the actively triggered Ca2+ transfer from the ER to mitochondria, but also for MERC tethering, suggesting the two functions overlap. Potentially, this is further promoted by mitochondrial arrest in locations with high [Ca2+] (Saotome et al., 2008; Wang and Schwarz, 2009). More importantly for the topic of this review, IP3R-originating Ca2+ flux controls the activity of mitochondrial Krebs cycle enzymes and, thus, mitochondrial respiration and ATP production (Cardenas et al., 2010).
To our knowledge, the relative importance of MERC lipid and Ca2+ flux for mitochondrial metabolism has never been quantified and such an endeavor might prove challenging. As we will discuss further below and in contrast to some of the lipid metabolic enzymes, mutations in proteins mediating or regulating MERC Ca2+ flux do not fully compromise mitochondria, but rather reduce their relative contribution to cellular bioenergetics and increase compensatory glycolysis, thus providing a hallmark change upon MERC interference (Tarasov et al., 2012). Another emerging theme of this review is that MERCs are critical for the central and peripheral nervous systems (CNS/PNS). This is also highlighted by the large number of MERC Ca2+-handling proteins associated with neurodegeneration (Krols et al., 2016). Prominent members of this group are the presenilins, genetic loci for Alzheimer's disease (AD) (Area-Gomez et al., 2009), which control mitochondrial Ca2+ uptake at the synapse, where these organelles provide much of the energy needed for neurotransmitter release (Walters and Usachev, 2023). Accordingly, mutant presenilin-2 decreases the ER Ca2+ content by reducing the activity of sarco-endoplasmic reticulum Ca2+ ATPase (SERCA; Green et al., 2008), which increases Ca2+ flux from the ER to mitochondria (Zampese et al., 2011). Through this Ca2+ imbalance, MERCs increase in AD patient and mouse model tissue (Hedskog et al., 2013). However, presenilin mutant proteins also increase PE synthesis and promote MERC formation, again suggesting a tight interweaving of these two functions (Area-Gomez et al., 2012). Given that mutations within the presenilin genes are not considered rare, we will not further discuss them in this review. Similarly, MERC Ca2+ flux, dependent on IP3Rs, VDACs and the mitochondrial Ca2+ uniporter (MCU) plays an important role in many tissues and diseases, for instance diabetes (Rieusset, 2017). In terms of mutations leading to disease, gain-of-function mutations of IP3Rs that result in spinocerebellar ataxia are the basis of rare diseases, discussed in detail later (Casey et al., 2017). Therefore, MERC Ca2+ flux controls the qualitative contribution of mitochondria to bioenergetics, which leads to a less mitochondria-centric disease array with implications for a wide variety of tissues such as the pancreas, but most critically for the CNS.
MERCs as a Target and Source of Oxidative Stress
The latest addition to the canon of MERC functions was discovered in 2011 through the localization of the NLRP3 inflammasome to MERCs (Zhou et al., 2011) in the presence of oxidative stress signaling (Figure 1, third panel). This key regulator of inflammation depends on the production of ROS within mitochondria and the ER, which triggers association of thioredoxin interacting protein (TXNIP) with the inflammasome (Zhou et al., 2010). While normally acting as an inhibitor of the antioxidant thioredoxins, TXNIP association with the inflammasome further potentiates ROS levels especially if associated with mitochondria (Saxena et al., 2010). This function predicts that the interaction between TXNIP and the NLRP3 inflammasome depends on the formation of MERC redox nanodomains, whose ROS content oxidizes IP3Rs, thus activating MERC Ca2+ signaling (Booth et al., 2016, 2021). Oxidation of cysteines is initiated by their sulfenylation, followed by sulfinylation of responsive cysteine residues, both of which are reversible through the activity of sulforedoxin. Therefore, cysteine oxidation acts as a posttranslational modification not unlike phosphorylation (Paulsen and Carroll, 2013). Upon further oxidation, cysteines can undergo sulfonylation, which is irreversible (Yang et al., 2016). Each of these modifications changes the function of substrates and may achieve their activation through oligomerization (Bassot et al., 2021).
MERCs are a hotspot for such modifications, since ER and mitochondria release ROS through pores like aquaporin-11 (AQP11) (Bestetti et al., 2020). This ROS signaling allows MERCs to respond to stress. This can occur, for instance, from the accumulation of unfolded proteins within the ER (Bassot et al., 2023), a condition long known to increase MERCs lipid and Ca2+ flux through the tightening of the contacts (Csordas et al., 2006; Bravo et al., 2011). While mitochondrial ROS are thought to derive from OXPHOS imbalance (Murphy, 2009), the ER ROS sources with MERC-modulating potential have been identified as NOX4 that seems to promote baseline activity of MERCs (Gutierrez et al., 2020) and the ER oxidoreductase ERO1L that oxidizes the eIF2α kinase 3/protein kinase R like ER kinase (PERK) (Bassot et al., 2023). Thus, PERK acts as a stress-induced tether at MERCs (Verfaillie et al., 2012) and as an activator of MERC Ca2+/lipid flux that switches cell metabolism from glycolysis to OXPHOS (Bassot et al., 2023). Although the cytosolic MERC interface might directly interact with NOX enzymes (Dikalov, 2011), we currently do not know a specific cytosolic MERC redox enzyme that could modulate mitochondria or ER redox release. Such a postulated mechanism is under the control of cytosolic glutathione, thioredoxins, and peroxiredoxins (Jezek et al., 2020).
The link between ROS signaling and MERC lipid metabolism is currently poorly understood. Upon disruption of PS or PC synthesis, ROS levels go up, potentially through the disruption of efficient OXPHOS, and these ROS appear to predominantly affect glial cells, at least in the fly model, leading to a neurodegenerative phenotype (Park et al., 2021). Conversely, several MERC lipid and cholesterol metabolic enzymes (e.g., ACAT1) are targets of ROS-mediated oxidation (Akter et al., 2018). For instance, acyl-coA:acyltransferase 2 (ACAT2) undergoes sulfenylation, which activates and stabilizes this producer of cholesteryl esters (Wang et al., 2017). In contrast, the oxidation of MERC-enriched diacyglycerol acyltransferase 2 (DGAT2) (Stone et al., 2009) inactivates it (Jung et al., 2017), indicating that MERC ROS levels profoundly affect lipid metabolism.
Another topic in this context is the MERC-specific peroxidation of PUFAs during ferroptosis (Dixon et al., 2012; Stockwell, 2022). The MERC marker ACSL4/FACL4 promotes the production of PUFAs through the incorporation of arachidonic acid into phospholipids (Kang et al., 1997). It is therefore not surprising that ACSL4 is a known ferroptosis regulator (Doll et al., 2017). Once peroxidized, lipids can revert to their reduced form through the activity of GPx4 protein, a selenoperoxidase found in the cytoplasm, within mitochondria and the nucleus (Xie et al., 2023). Lipid peroxidation reveals an association of MERC oxidative signaling with mitochondria homeostasis, since mitochondrial ROS production creates MERC redox nanodomains (Booth et al., 2016). Accordingly, the interference with MERC formation also blocks ferroptosis (Zhang et al., 2023). Aside from the ERO1L interactor SELENON that we discuss later, given the largely unknown identity of MERC redox regulators, no rare disease is currently known that is tied to defective MERC redox signaling.
MERCs Mediate Stress-Dependent Metabolic Signaling
The intimate link between mitochondrial and ER redox conditions explains why ER stress is induced upon inhibiting mitochondrial Krebs cycle activity (Kuznetsov et al., 2022). A mechanistic MERC-associated basis for diseases resulting from direct mutations of ER stress and mitochondrial Krebs cycle enzymes might therefore prove difficult to untangle from their other functions. Nevertheless, the now established link between ER stress signaling and MERC metabolic signaling (Simmen and Herrera-Cruz, 2018) explains why gene mutations within PERK are particularly relevant to this review. PERK is best known as a moderator of ER protein synthesis through eIF2α phosphorylation during the induction of the unfolded protein response (UPR) (Ron and Walter, 2007), but also as a MERC-localized interactor with the tethers mitofusin-2 (Munoz et al., 2013) and extended synaptotagmin-1 (E-Syt1) to promote mitochondrial respiration (Sassano et al., 2023). Both E-Syt1 (Chang et al., 2013) and PERK (van Vliet et al., 2017) also localize to ER-plasma membrane junctions, thus implicating them in store-operated Ca2+ entry (SOCE) (Chen et al., 2019; Kang et al., 2019). Mutations in PERK cause Wolcott-Rallison syndrome (MIM#226980) that leads to neonatal diabetes but also epileptic seizures (de Wit et al., 2006; Julier and Nicolino, 2010). Whether this disease spectrum depends more on the role of PERK in the integrated stress response and, thus, the prevention of further ER stress (Pakos-Zebrucka et al., 2016) or on its role in MERC tethering to maintain mitochondria metabolism (van Vliet and Agostinis, 2016) remains unclear. Regardless, Wolcott-Rallison syndrome is reminiscent of Wolfram syndrome (MIM#222300), which leads to diabetes mellitus, optic nerve atrophy, central diabetes insipidus, sensorineural deafness, urinary tract problems, and progressive neurologic difficulties (Urano, 2016). Wolfram syndrome is caused by mutations in two genes, WFS1 and WFS2 (Pallotta et al., 2019). Both proteins encoded by these genes are of interest for this review. WFS1 is a glycosylated transmembrane protein that localizes to the ER, where it not only promotes mitochondrial membrane contact formation but is also a major determinant of the Ca2+-signaling functions at MERCs (Angebault et al., 2018). Due to its interaction with VDAC and the important role of ER-mitochondria Ca2+ flux for cell metabolism, it comes as no surprise that WFS1 maintains mitochondrial ATP production (Zatyka et al., 2023). The WFS2 gene product, CDGSH iron–sulfur domain-containing protein 2 (CISD2), is also an integral ER membrane protein that interacts with and inhibits IP3Rs, as well as autophagy (Chang et al., 2010). Moreover, CISD2 localizes to MERCs and prevents ER stress, possibly through its function as a preserver of mitochondrial bioenergetics (Wiley et al., 2013).
Another connection between ER stress and MERC metabolic signaling is revealed in the syndrome of microcephaly, epilepsy, and permanent neonatal diabetes (MEDS, MIM#614231), which is based on mutations within the immediate early response 3 interacting protein 1 (IER3IP1) gene (Poulton et al., 2011; Yang et al., 2022). IER3IP1 is an ER transmembrane protein (Yiu et al., 2004) that regulates ER protein export towards the Golgi complex and prevents ER stress (Esk et al., 2020). In this case, and for all proteins discussed up to here, the connection of the diseases to MERC dysfunction is currently unclear. However, as we will outline later, some aspects of these pathologies are very reminiscent of the syndrome resulting from mutations in the gene encoding the cytosolic phosphofurin acidic cluster sorting protein 2 (PACS-2) (Simmen et al., 2005).
MERCs and Mitochondrial Dynamics
Mitochondrial dynamics control the fission and fusion of mitochondria, which controls the levels of mitochondrial stress and distribution of mitochondria within the cell (Chan, 2020). This function is tightly linked to MERC tethering (Figure 1, bottom panel). Mitochondrial fusion is mediated by the pairing of the dynamin superfamily GTPases mitofusin 1 and 2 (Mfn1/Mfn2) on neighboring outer mitochondrial membranes (OMM) (Koshiba et al., 2004). Mfn2 and its mutants will be discussed in detail later, since ER- and mitochondria-specific splice variants act as key MERC tethers (Naon et al., 2023). In parallel, the inner mitochondrial membrane (IMM) GTPase Opa1 mediates the second step of fusion (Song et al., 2007). The fusion and elongation of mitochondria is tightly connected to MERCs, as evidenced by PERK activation, which induces stress-dependent mitochondria elongation (Lebeau et al., 2018), accompanied by the accumulation of the CL precursor PA on the OMM (Perea et al., 2023).
Counteracting fusion, mitochondrial fission aims to increase the number of mitochondria. This happens, for instance, during excessive stress that can trigger the phosphorylation (Cribbs and Strack, 2007) and oxidation (Cho et al., 2009) of the GTPase dynamin-related protein 1 (Drp1). Subsequently, Drp1 associates with the OMM proteins Fis1, Mff, MiD49, and MiD51 (Loson et al., 2013). Importantly, the fission machinery is assembled on MERCs (Friedman et al., 2011) with the help of these receptor proteins (Ji et al., 2017). Mutations within the genes encoding the MERC-associated fission machinery share many characteristics with MERC dysfunction-based pathologies, as discussed below.
MERC formation indirectly depends on mitochondria motility along the cytoskeleton, which could be an initial determinant of ER-mitochondria contact formation (Yi et al., 2004). A key factor for mitochondria movement is the small OMM Rho protein 1 (Miro-1), a GTPase enriched in yeast and mammalian MERCs (Modi et al., 2019). Miro-1 is conserved from humans to yeast, where its counterpart Gem1 is a component of the yeast ERMES tethering complex (Kornmann et al., 2011). On the membrane contact site, Miro-1 senses high cytosolic [Ca2+] released from IP3Rs through its EF hand, which arrests mitochondria movement (Saotome et al., 2008; Wang and Schwarz, 2009). Thus, Miro-1 conveys Ca2+-sensitivity to mitochondria positioning (Fransson et al., 2006; Eberhardt et al., 2020). This is achieved through the interaction of Miro-1 with the kinesin and dynein motor proteins (Macaskill et al., 2009; Morlino et al., 2014). The Miro-1 activity is critically important for axonal transport of mitochondria to synapses, firstly shown in Drosophila (Guo et al., 2005). Similar to the presenilins, mutant Miro-1 is observed in Parkinson's disease (Grossmann et al., 2020).
To conclude the introduction, MERC-associated proteins are connected to a variety of diseases, ranging from neurological, muscle, to pancreatic defects. Given their frequent multifunctional aspects, many of these may depend on functions not related to MERCs. In the case of defective mitochondrial dynamics, as related to MERC assembly, such diseases are primarily associated with defects within the central and peripheral nervous system.
Rare Diseases Based on MERC Tethers
As outlined above, MERC functions comprise exchange of lipids, Ca2+, and ROS that then impinge on metabolic functions of mitochondria to a varying extent. Changes in lipid homeostasis appear to act most directly, while Ca2+ signaling acts more subtly and the impact of interfering with MERC redox nanodomains is currently largely unexplored. To gain better assessment of the significance of MERC interference for disease, we will first focus on the physical tethering between the ER and mitochondria. We will further narrow the discussion and exclude proteins are either associated to common diseases (e.g., DJ-1, presenilins) or do not primarily control MERC formation or regulation (e.g., ACSL4, EPT1, PERK, WFS1, CISD2, or IER3IP1). When doing so, we detect highly similar pathologies.
MERC tethering is controlled by a set of proteins that bridge the gap between the two organelles. Typically, tethers are formed by different proteins that are found on the ER or mitochondria, respectively. For example, ER-localized IP3Rs can bridge the two organelles through interaction with mitochondrial VDAC and the chaperone GRP75 (Szabadkai et al., 2006). This function also depends on the mitochondrial protein DJ-1 (Ottolini et al., 2013; Liu et al., 2019), a cytosolic protein that interacts with glycolytic enzymes and prevents ROS formation as a chaperone, but relocates to mitochondrial upon its oxidation (Mencke et al., 2021). One of the best-characterized tethering pair is formed between ER-localized VAPB (VAMP-associated membrane protein B) that connects to mitochondrial protein tyrosine phosphatase interacting protein 51 (PTPIP51), also called regulator of microtubules dynamics 3 (RMDN3) (De Vos et al., 2012; Stoica et al., 2014). ER-localized proteins like gp78/AMFR connect to these and determine the extent of tethering (Wang et al., 2015). Another protein complex is the ARCosome that is formed when ER-localized BAP31 (also known as BCAP31) interacts with mitochondrial Fis1 (Iwasawa et al., 2011) and with the import complex component TOM40 (Namba, 2019). More recently, a protein complex between ER-localized E-Syt1 and mitochondrial SYNJ2BP has been identified (Janer et al., 2024). Given that E-Syt1 is recruited by PERK to MERCs, these two ER tethering regulators may act together (Sassano et al., 2023). There is currently no known association of E-Syt1 or SYNJ2BP with rare diseases.
It is expected that interfering with any of these bona fide MERC tethers directly affects key MERC readouts, as detected by decreased Ca2+ transfer, disrupted mitochondrial lipid content and altered OXPHOS activity. Where known, we will discuss how disease results from mutations in these proteins, as well as their function during homeostatic and stressed situations, where applicable (Figure 1). MERC-associated diseases are indicated in bold.
VAPB and PTPIP51
One of the best characterized MERC tethers is formed between VAPB, an ER-localized tail-anchored protein with an N-terminal major sperm protein (MSP) domain (Dudas et al., 2021), and mitochondrial PTPIP51 (De Vos et al., 2012). Recent findings indicate PTPIP51 contains a tetratricopeptide repeat domain that mediates PA transport and controls mitochondrial CL content, which could singlehandedly explain the function of this complex for MERCs (Yeo et al., 2021). In addition to MERC tethering, VAPB also forms ER-based interorganellar protein complexes with Golgi, plasma membrane, endosome, and peroxisome proteins (Borgese et al., 2021). The VAPB-PTPIP51 heterodimeric interaction increases upon activation of GSK3β (Stoica et al., 2014). At MERCs, VAPB recognizes two motifs characterized by two phenylalanines (FF) in an acidic tract (FFAT) within PTPIP51, which then leads to the interaction of VAPB with a disordered domain within PTPIP51, thus controlling the tethering and lipid transport functions of this complex (Di Mattia et al., 2020; Yeo et al., 2021). Expression of deletion mutants of one FFAT motif within PTPIP51 also reduces the ER Ca2+ signaling towards mitochondria in half in SH-SY5Y cells, compared to wild-type cells (De Vos et al., 2012; Morotz et al., 2022). The VAPB-PTPIP51 tether pair is present at neuronal synapses, where it controls their activity (Gomez-Suaga et al., 2019). MERC-tethering by VAPB-PTPIP51 increases upon synaptic activity and this promotes dendritic spine number, suggesting that neuronal firing correlates with increased MERC tethering and lipid transfer through VAPB-PTPIP51 complexes (Gomez-Suaga et al., 2019).
Different from the PTPIP51/RMDN3 gene for which there are currently no known disease-associated mutations, VAPB mutation is an important and frequent cause of amyotrophic lateral sclerosis (ALS), recently reviewed by us (Chen et al., 2021), leading to its alternate designation as ALS8 (MIM#608627; Nishimura et al., 2004). However, in addition to these relatively frequent ALS-related mutations, there are about 200 cases of VAPB-based genetic disease, characterized by spinal muscular atrophy (Kosac et al., 2013). In this disease, patients suffer from progressive proximal weakness, cramps, and a lack of reflexes. These defects are likely derived from abnormal release of synaptic vesicles and aberrant endosomal trafficking. There appears to be some unclear level of overlap between ALS and this pathology, termed
Mfn2
Amongst the more frequent MERC pathologies are mutations in the Mfn2 tether, which cause
IP3R-VDAC-GRP75 (HSPA9) Tether
A ternary tethering complex forms between the IP3R1 isoform with the voltage-dependent anion channel 1 (VDAC1) and the outer mitochondrial membrane chaperone GRP75 (Szabadkai et al., 2006). It is currently not clear whether IP3R2 and IP3R3 can also do this, however, these forms have been reported to transmit Ca2+ signals from the ER to mitochondria more effectively (Mendes et al., 2005; Bartok et al., 2019). In contrast to VDAC, several GRP75 and IP3R-based rare genetic diseases have been reported to date.
IP3Rs are expressed from three gene loci, ITRP1 (IP3R1), ITRP2 (IP3R2), and ITRP3 (IP3R3). Although these paralogs are overall similar in amino-acid sequences with each other, they exhibit distinct characteristics such as interaction partners and expression patterns. For example, IP3R1 dominates over the other forms in cerebellum and brain (Wojcikiewicz, 1995), while IP3R2 dominates in the liver and heart (Ivanova et al., 2014).
A gain-of-function mutation in the suppressor domain of the IP3R1 (R36C) causes
In contrast to these gain-of-function mutations, dominant-negative IP3R1 mutations cause
Like ITRP1, ITRP3 mutations are known to cause neuronal and developmental disorders. Dominant mutations in ITPR3 (e.g., V615 M) cause
GRP75, also known under the names of mtHsp70, HSPA9, and mortalin, is a chaperone that mainly localizes to mitochondria (Havalova et al., 2021), where it protects from ROS and controls mitochondrial biogenesis, including protein translocation into mitochondria, and the synthesis of Fe–S cluster proteins (Flachbartova and Kovacech, 2013). GRP75 localizes to multiple intracellular compartments. While the main localization of GRP75 is on mitochondria, ∼30% of its amount is found in other compartments (Esfahanian et al., 2023), including endosomes, the cytosol, the ER and the MAM, where it forms the IP3R1-GRP75-VDAC tethering complex (Szabadkai et al., 2006). Several genetic diseases are caused by mutations in the GRP75 gene. One type causes the
BAP31 (BCAP31)
In cells depleted of the MERC regulatory protein PACS-2, the ER protein BAP31 results as cleaved, which no longer allows it to interact with mitochondrial proteins such as Fis1 or Tom40 (Simmen et al., 2005). Mutation of BCAP31 is observed in a rare X-linked recessive disorder termed
PDZD8/VPS13
The mammalian SMP domain-containing protein PDZD8 was first described as an ER-mitochondria tether, mediating efficient Ca2+ flux from the ER to mitochondria in neuronal cells (Hirabayashi et al., 2017). The presence of an SMP domain suggests that PDZD8 may use its localization to ER membrane contact sites to mediate exchange of lipid molecules at these locations (Jeyasimman and Saheki, 2020). This function is particularly evident in yeast, where in addition to the PDZD8 paralog Mmm1 (Hirabayashi et al., 2017; Wideman et al., 2018), several SMP domain-containing proteins (Mdm10, Mdm12, and Mdm34) form the ERMES complex, which mediates lipid exchange between the ER and mitochondria (Kornmann et al., 2009). Premature stop codons in the PDZD8 gene create a shortened version of this tether that is associated with intellectual disability designated as “
Rare Diseases Based on MERC-Mediated Mitochondrial Dynamics
The role of MERCs for mitochondrial dynamics is evident in the case of mitochondrial fission, where ER-associated Drp1 becomes enriched on MERCs and forms a constricting ring with the associated ER membranes around a mitochondrion (Kalia et al., 2018). This mechanism requires ER-associated actin polymerization (Korobova et al., 2013) and mitochondrial CL (Stepanyants et al., 2015), thus highlighting the high significance of MERC formation when disease is associated with mitochondrial hyperfusion from disrupted fission.
Drp1
The ER GTPase Drp1 is mutated in a lethal encelopathy of early childhood (Waterham et al., 2007), a spectrum characterized by epilepsy, and microcephaly (Navaratnarajah et al., 2021). The disease resulting from DRP1 mutations is referred to as
Mff
The assembly of mitochondrial receptors is critical for Drp1 oligomerization. The mitochondrial fission factor (Mff) is the primary mitochondrial receptor for Drp1 (Loson et al., 2013). Its targeting to the ER portion of MERCs greatly increases mitochondrial fission (Ji et al., 2017) Neuronal defects result, for instance, upon mutation of the MFF gene in the
Rare Diseases from Mutated MERC Regulatory or Accessory Proteins
Several proteins that modulate MERC signaling through changes in activity of ER-mitochondria lipid flux, as well as ER Ca2+ pumping and release have been described over the past decade. They include calnexin (Lynes et al., 2012), TMX1 (Raturi et al., 2016), ERdj5 (Ushioda et al., 2016), and GPx8 (Yoboue et al., 2017). Their activity and localization is controlled by cytosolic factors such as PACS-2 (Myhill et al., 2008), as well as by the ER membrane protein complex (EMC) (Christianson et al., 2011). Some of these regulatory proteins and their interactors are listed below, where we discuss their association with MERC-connected rare diseases.
PACS-2
PACS-2, the first identified regulator of MERC tethering, maintains MERC protein complexes by preventing caspase-8-mediated cleavage of BAP31, thus preserving the MERC structure (Simmen et al., 2005). PACS-2 is phosphorylated during homeostatic conditions (Betz et al., 2013) and promotes MERC lipid synthesis as well as Ca2+ signaling during apoptosis (Simmen et al., 2005; Aslan et al., 2009). PACS-2 also controls the MERC localization of important functional and regulatory proteins, including ACSL4 and calnexin (Simmen et al., 2005). Like many proteins that are subject of this review, PACS-2 is multifunctional (Thomas et al., 2017). Its anabolic, pro-survival, and apoptotic functions are controlled by Akt phosphorylation (Aslan et al., 2009). Additionally, PACS-2 can migrate into the nucleus, where it prevents DNA damage through p53 and p21 and controls ATM trafficking toward the cytosol (Barroso-Gonzalez et al., 2016). In the absence of PACS-2, the histone deacetylase sirtuin-1 (SIRT1) is hyperactive, which normally maintains p53-mediated transcription (Atkins et al., 2014). Importantly, however, some of the nuclear functions of PACS-2 may indirectly depend on its MERC functions: disrupted ER-mitochondria Ca2+ flux, followed by mitochondrial fragmentation, as observed upon PACS-2 knockdown, activates SIRT1 (Lovy et al., 2020). Moreover, increased ROS content upon PACS-2 knockdown could migrate into the nucleus and promote DNA damage (Srinivas et al., 2019).
PACS-2 is mutated in PACS-2 syndrome, a subtype of early infantile developmental and epileptic encephalopathy (EIDEE) (Zuberi et al., 2022), referred to as
PACS-2 depletion causes ER stress, reduces PSS1 at MERCs and disrupts Ca2+ signaling (Simmen et al., 2005). This is also observed in DEE66 mutant cells (Thi My Nhung et al., 2023). PACS-2 also controls the pro-apoptotic trafficking of the Bcl2 family protein Bim onto mitochondria and lysosomes (Simmen et al., 2005; Werneburg et al., 2012) and is also found on endosomes, where it can influence major histocompatibility complex class I (MHC-I) downregulation (Atkins et al., 2014). Moreover, PACS-2 also promotes MERC-localized mitophagy in vascular smooth muscle cells (Moulis et al., 2019). Yet, its role for MERCs is likely a main function. As such, PACS-2 interacts with coatomer and retrieves MERC-regulatory proteins such as the transient receptor potential polycystin-2 (TRPP2) and calnexin from the Golgi complex to the ER, where they then get enriched on MERCs (Kottgen et al., 2005; Myhill et al., 2008). Both PACS-2 cargo proteins promote ER-mitochondria Ca2+ communication: TRPP2 likely controls this signaling mechanism through a control of MERC IP3R activity and downregulates Mfn2 (Kuo et al., 2019), while calnexin promotes oxidation of MAM Ca2+ handling proteins such as SERCA and IP3Rs, thus activating their signaling functions (Gutierrez et al., 2020).
In the PACS-2-dependent disease scenario, mutant PACS-2 protein increases apoptosis susceptibility (Zang et al., 2022). While mutant PACS-2 could potentially fail to stabilize ion channels such as TRPP2 (Kottgen et al., 2005), another mechanistic basis of this syndrome could be an increased half-life of mutant PACS-2, leading to its abnormal accumulation that would then abnormally promote MERC formation (Zang et al., 2022). As hypothesized recently, mutant PACS-2 could also change ion channel activity at MERCs or elsewhere (Chou et al., 2023) or modulate MERC-derived autophagy (Hamasaki et al., 2013). This latter function also connects PACS-2 to the prevention of cardiac injury, especially under hypoxic conditions at high altitude, where it helps maintain normal cardiomyocyte metabolism (Yang et al., 2023).
EMC
The EMC is a multiprotein transmembrane complex that promotes the insertion of transmembrane proteins (Guna et al., 2017). This function determines largely the topology of multipass transmembrane proteins (Shurtleff et al., 2018). An important EMC substrate is rhodopsin that is compromised upon EMC3 interference (Taylor et al., 2005). Interestingly, EMC interacts with calnexin (Christianson et al., 2011) that can show high levels of MERC enrichment (Lynes et al., 2013). Consistent with functions of EMC for MERCs, this protein complex also determines MERC formation itself and its knockout compromises PS transfer from the ER to mitochondria at MERCs (Lahiri et al., 2014). At the moment, it is unclear whether this function stems from a direct role of EMC for MERCs, or, more likely, from the insertion of MERC tethers at the level of the ER. Accordingly, genetic diseases associated with EMC mutation are most closely associated with membrane protein insertion, such as EMC3 mutation that is expected to affect vision (Cao et al., 2021). Nevertheless, mutations within the EMC1 and EMC10 genes lead to overlapping disease spectrums, highly reminiscent of MERC disorders (Chung et al., 2022): while EMC1 mutation causes
SEPN1
An interesting pair of ER oxidoreductases is formed by ER oxidoreductin 1 (ERO1) and SELENON, also known as Selenoprotein N1 (SEPN1). ERO1 can oxidize SERCA, which impairs ER Ca2+ filling (Marino et al., 2015), necessary for normal MERC function (Gutierrez et al., 2020). The oxidizing enzyme ERO1 has long been recognized as an important component of MERCs whose enrichment is oxygen and redox-dependent (Gilady et al., 2010). ERO1 oxidizes proteins involved in tethering during the onset of ER stress (Bassot et al., 2023). In addition to tethers, ERO1 also targets IP3Rs and results in their activation through oxidation (Li et al., 2009). Consequently, ERO1 activates mitochondria, especially under ER stress (Bassot et al., 2023).
In contrast, SELENON/SEPN1 is an ER antioxidant protein (Arbogast et al., 2009), whose mutant variants are found in
GDAP1 (CMT4A)
PSS1/PSS2/PISD
As mentioned in the introduction, a main function of MERCs is the transformation of PS into PE, the original function discovered by Jean Vance (Vance, 1990, 1991). Both PSS1 and PSS2 localize to MERCs (Stone and Vance, 2000). A D. melanogaster knockout results in altered mitochondrial morphology and increased ROS production (Park et al., 2021), as is typical when the MERC structure undergoes functional changes, such as the activation of ER-mitochondria Ca2+ flux (Booth et al., 2016). Human patients with a PTDSS1 mutation suffer from
Likewise, PISD activity that converts PS to PE on the IMM in proximity to MERCs is required for mitochondrial OXPHOS at complex I and IV, as well as normal mitochondrial membrane potential (Tasseva et al., 2013). Unlike the viable PSS1 and PSS2 knockout animals (Bergo et al., 2002; Arikketh et al., 2008), the activity of PISD is essential, since it determines embryonic development and mitochondrial integrity (Steenbergen et al., 2005). PISD mutation-associated pathologies have been classified as mitochondrial diseases with skeletal and central nervous system (CNS) dysfunction, characterized by hypomyelination, ataxia, and intellectual disability (Zhao et al., 2019). This rare disease is now referred to as
ACSL4 (FACL4)
Acyl CoA synthetase 4 (ACSL4) is a reliable marker of MERCs (Lewin et al., 2002). This enzyme promotes the production of PUFAs through the incorporation of arachidonic acid into phospholipids (Kang et al., 1997; Golej et al., 2011). Mutations in the ACSL4 gene are found in patients with
Concluding Remarks and Potential Therapeutic Avenues
The past decade has seen increasing insight into rare diseases stemming from MERC disruption. An overall characteristic of these diseases emerges as predominantly resulting in neurological defects, with less frequent skeletal muscle deficiency (Figure 2). In many ways, PACS-2-linked DEE66 is the prototype of these pathologies with a developmental delay, intellectual disability, facial dysmorphism, and ophthalmologic defects. Additional symptoms include seizures, epilepsy, deafness, ataxia, or peripheral neuropathy. The key example of muscle defects is seen with SEPN1-RM that results in rigid spine muscular dystrophy. Of course, the insight that MERC defects lead to common characteristics in rare diseases opens up the possibility to find common or similar treatment avenues. At the moment, however, it is difficult to foresee whether intervention at the level of lipid, Ca2+, or ROS functions of MERCs will be most successful. Therefore, current treatment options include vitamin B6 in the case of DEE6 (Chou et al., 2023). This is based on the role of this vitamin for OXPHOS through its control of iron–sulfur and NAD+ biosynthesis (Ciapaite et al., 2023). Another example is Mfn2 agonists such as MiM111 that aim to preserve mitochondrial bioenergetics (Franco et al., 2020). Similarly, Drp1 inhibition with the cell-permeable quinazolinone Mdivi-1 could address some of the diseases described here (Xie et al., 2016).
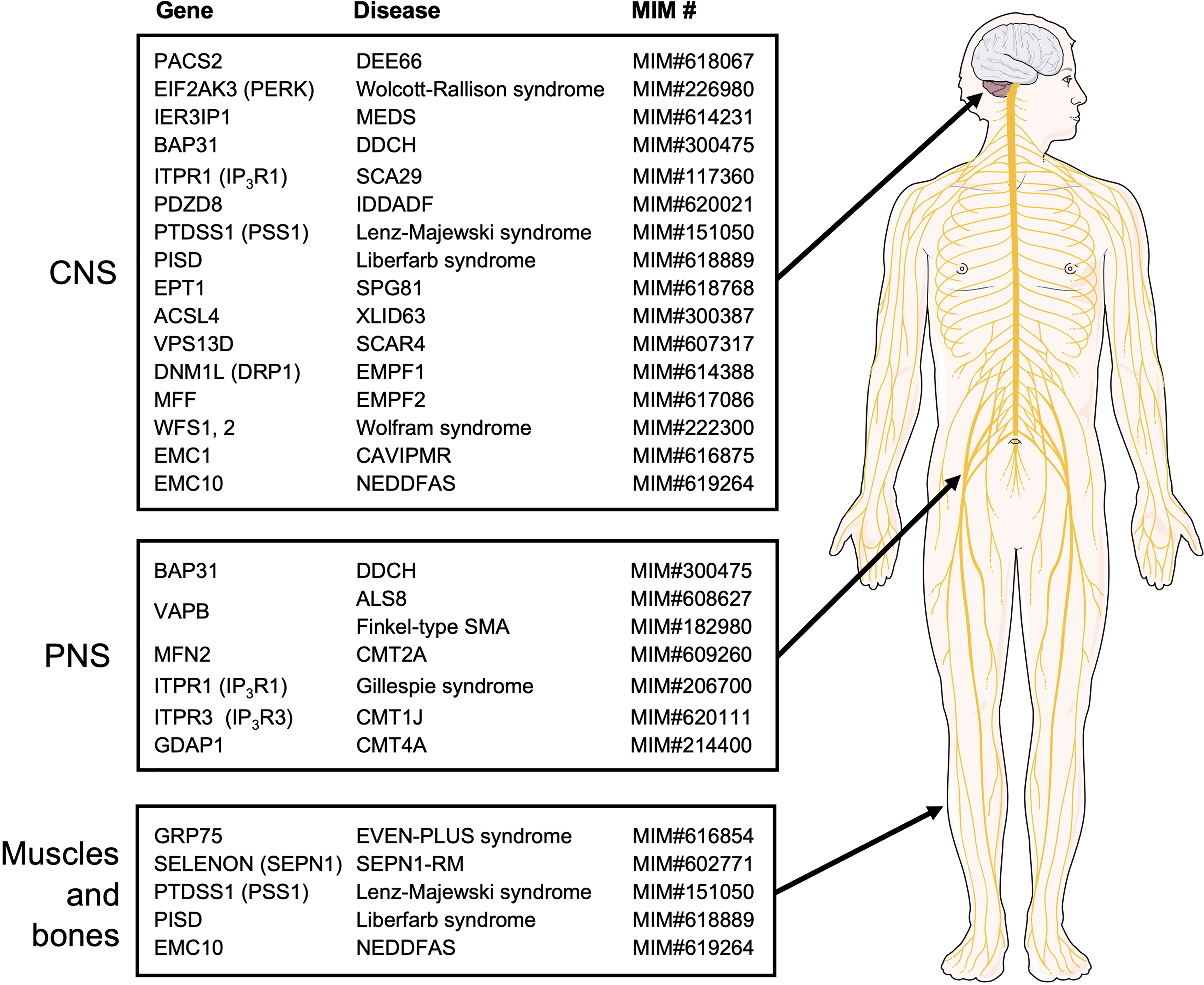
Rare diseases caused by mutations in the MERC-related genes. The MERC-related genes whose mutations cause rare diseases are summarized. They are categorized by the systems mostly affected by their respective diseases. The MIM numbers of the genetic diseases given by OMIM (https://www.omim.org/) are also shown. Please note that additional systems may be affected by a disease. The illustration is adapted from Servier Medical Art (https://smart.servier.com/).
MERC dysfunction could respond to increased or decreased SERCA activity, as well as altered IP3R Ca2+ release. While experimental cancer therapeutics make use of this idea through inhibition of SERCA and presumably MERCs, the activation of SERCA could also be promising (Tadini-Buoninsegni et al., 2018). This latter drug group, for instance the allosteric activator CDN1163 (Kang et al., 2016), could be of particular interest in the case of SEPN1-RM. Another approach in this context is the use of IP3R-modulating agonists, like adenophostins (Gambardella et al., 2021), and antagonists, like the macrocyclic bis-1-oxaquinolizidine xestospongin, which induce relaxation of blood vessels in vivo (Nakagawa et al., 1984). However, IP3R-modulatory compounds currently seem to suffer from a lack of specificity. Future research will have to determine whether compounds or proteins based on the endogenous IP3R antagonist IRBIT (IP3R-binding protein released with IP3R) could be more specific (Ando et al., 2003, 2006).
Additionally, ROS intervention appears an obvious approach, due to the wide availability of pro- and antioxidants, which, however, would have to be targeted to the mitochondria of the tissue affected most by any rare disease (Jiang et al., 2020). Such approaches are considered, for instance, in the case of SEPN1-RM (Arbogast et al., 2009). However, ROS may increase or decrease upon MERC interference, therefore lipid and Ca2+ flux changes currently seem more straightforward. Given that PE defects are most closely associated with MERC and mitochondrial dysfunction, in vitro supplementation with ethanolamine and lyso-PE appears a promising approach (St Germain et al., 2022). However, in the case of PISD-mutant in vitro studies, this approach required the additional modulation of ROS (Girisha et al., 2019) and lyso-PE supplementation can cause liver disease (Yamamoto et al., 2022). An indirect approach could be the use of peroxisome proliferators that increase, ER, mitochondria, and peroxisome function in the liver to modulate PE metabolism (Mizuguchi et al., 1999).
Not all MERC proteins fit into our proposed common MERC-disease pattern. For instance, the PERK-connected Wolcott Rallison syndrome has a predominant diabetic consequence. At this point, we hypothesize that such discrepancies stem from the predominant function of the respective protein for functions other than MERC formation or regulation, in the case of PERK as a regulator of the UPR. Similarly, problems with vision upon EMC protein mutation are likely connected to the role of EMC for rhodopsin, while the weakness, cramps, and a lack of reflexes in the case of VAPB could derive from the functions of this tether in ER MCS with organelles other than mitochondria. The further use of animal models, further insight about the global functions of MERC proteins and the advent of new compounds will provide the necessary insight required for testing any of these approaches based on MERC ROS, lipid, and Ca2+ signaling.
Footnotes
Acknowledgments
We thank Junsheng Chen and Megan Yap for many helpful discussions.
Declaration of Conflicting Interests
The authors declared no potential conflicts of interest with respect to the research, authorship, and/or publication of this article.
Funding
The authors disclosed receipt of the following financial support for the research, authorship, and/or publication of this article. This work was supported by the Institute of Genetics, Natural Sciences, and Engineering Research Council of Canada [grant number CIHR (PG 162449), RGPIN-2021-02765].