
Abstract
The inverse relationship between the cost of drug development and the successful integration of drugs into the market has resulted in the need for innovative solutions to overcome this burgeoning problem. This problem could be attributed to several factors, including the premature termination of clinical trials, regulatory factors, or decisions made in the earlier drug development processes. The introduction of artificial intelligence (AI) to accelerate and assist drug development has resulted in cheaper and more efficient processes, ultimately improving the success rates of clinical trials. This review aims to showcase and compare the different applications of AI technology that aid automation and improve success in drug development, particularly in novel drug target identification and design, drug repositioning, biomarker identification, and effective patient stratification, through exploration of different disease landscapes. In addition, it will also highlight how these technologies are translated into the clinic. This paradigm shift will lead to even greater advancements in the integration of AI in automating processes within drug development and discovery, enabling the probability and reality of attaining future precision and personalized medicine.
Introduction
Drug development is a time-consuming and expensive process, with a duration ranging from 12 to 15 y 1 and an average cost of about 2.6 billion. 1 At the same time, failure rates of clinical trials exceed 90% after preclinical studies, and only 14% of compounds clear clinical trials. Because of the high attrition rate across the entire drug development pipeline, there is a very low probability that a newly identified molecule will result in an approved medicine, with only 10% of small molecules within the industry transitioning to candidate status, failing at multiple stages. 2 This could be due to several factors, including, but not limited to, no developable hits from high-throughput screening (HTS), the inability to configure a reliable assay, toxicity in preclinical studies, off-target effects, and the inability to obtain a good pharmacodynamics or pharmacokinetics profile.1,2 Coupled with these factors, the high cost of failure rates has resulted in alternative approaches that make the processes at each step of the drug development more efficient and accelerate the entire pipeline. This has paved the way toward the integration of artificial intelligence (AI) within these processes. AI in drug development aims to effectively analyze huge amounts of data and project better solutions based on these learned data. In this review, we look into the different permutations of AI used specifically in the drug discovery stage (from target to hit and lead optimization) and preclinical research, compare and contrast the AI used in the different stages, highlight platforms that are at the forefront of these AI applications, and present current hurdles of AI implementation in drug discovery and development.
Types of AI in Drug Development
Historically, computational approaches have been used in drug development, particularly for rational drug discovery. Virtual screening (VS) has been widely used to guide rational drug discovery. 3 The quantitative structure-activity relationship (QSAR) was developed for VS to generate models based on the hypothesis that similar compounds have similar activities. Hence, the models generated were based on molecular structures and target activities (e.g., physicochemical properties, therapeutic activities, and pharmacokinetic properties). 4 However, with the expansion of the search space and more HTS assays, the traditional computational approaches for QSAR are not suitable for big data.
Generally, AI is the ability of a machine to perform tasks in response to a learned range of environments, aiming to replicate human intelligence. The term was first coined in 1956 by John McCarthy to describe the integration between engineering and science to generate intelligent machines. 5 Even though the concept dates back to many decades ago, there have been booms and busts in the development of AI since then. In the peak of the 1980s, advancements in AI led to mathematical models such as the multilayer feed-forward neural network and the back-propagation algorithm. This led to IBM’s Deep Blue victory against chess legend Garry Kasparov, which paved the way toward other advances in AI.6,7
The most common permutations within AI are primarily machine learning 8 and deep learning (DL), with the latter built from the foundations of the former. Briefly defined, ML involves algorithms that have been programmed to learn and improve future decision making based on past input data, without reprogramming or human intervention. The algorithms become more robust with increasing and higher quality data. There are 2 types of ML: supervised and unsupervised. Supervised learning involves developing predictive models based on continuous data, in which the input and output variables are known. Unsupervised learning involves identifying intrinsic patterns within data and clustering them in a meaningful manner. Examples of ML include support vector machines, 9 decision trees, k-nearest neighbors, naïve Bayesian methods, and artificial neural networks (NN), summarized in Table 1 . DL is the next generation of artificial NN. In artificial NN, input data are fed into an input layer, which is subsequently transformed within the hidden layers, eventually generating the predictions in the output layer. DL is more complex than its predecessor, integrating multiple layers of learning from massive data sets, with each layer comprising a refined classification to improve the accuracy of the model. DL carries out large-scale data extraction by processing multilayered deep NN, building on its foundation on artificial NN. Some examples include convolutional NN, stacked autoencoders, deep belief networks, recurrent NN, and Boltzmann machines, summarized in Table 2 . DL has been largely applied in image recognition and video and sound analyses, but there has been growing interest in the application of DL in drug development.
Permutations of Machine Learning used.
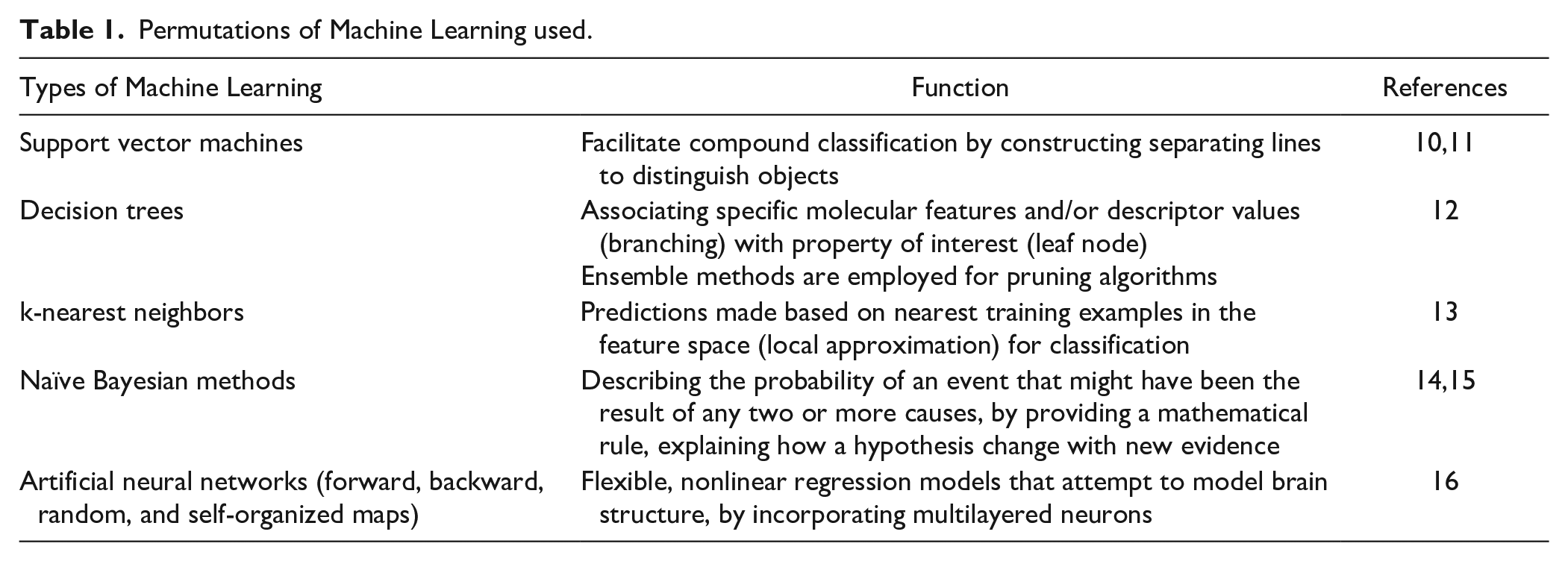
Permutations of Deep Learning Used.

NN, neural network.
The drug development pipeline encompasses drug discovery (where target identification and drug lead discovery occurs), preclinical development (where the efficacy of the drug is interrogated at in vitro and in vivo phases and assessment of drug toxicity properties), and clinical phases (in which the safety of drugs in humans is investigated; Figure 1 ). Using AI at the different stages allows for the processing of massive amounts of data by identifying patterns of functional properties and projecting a response based on new situations. The following sections will explore the different interventional AI approaches that have been used to aid some drug developmental processes.
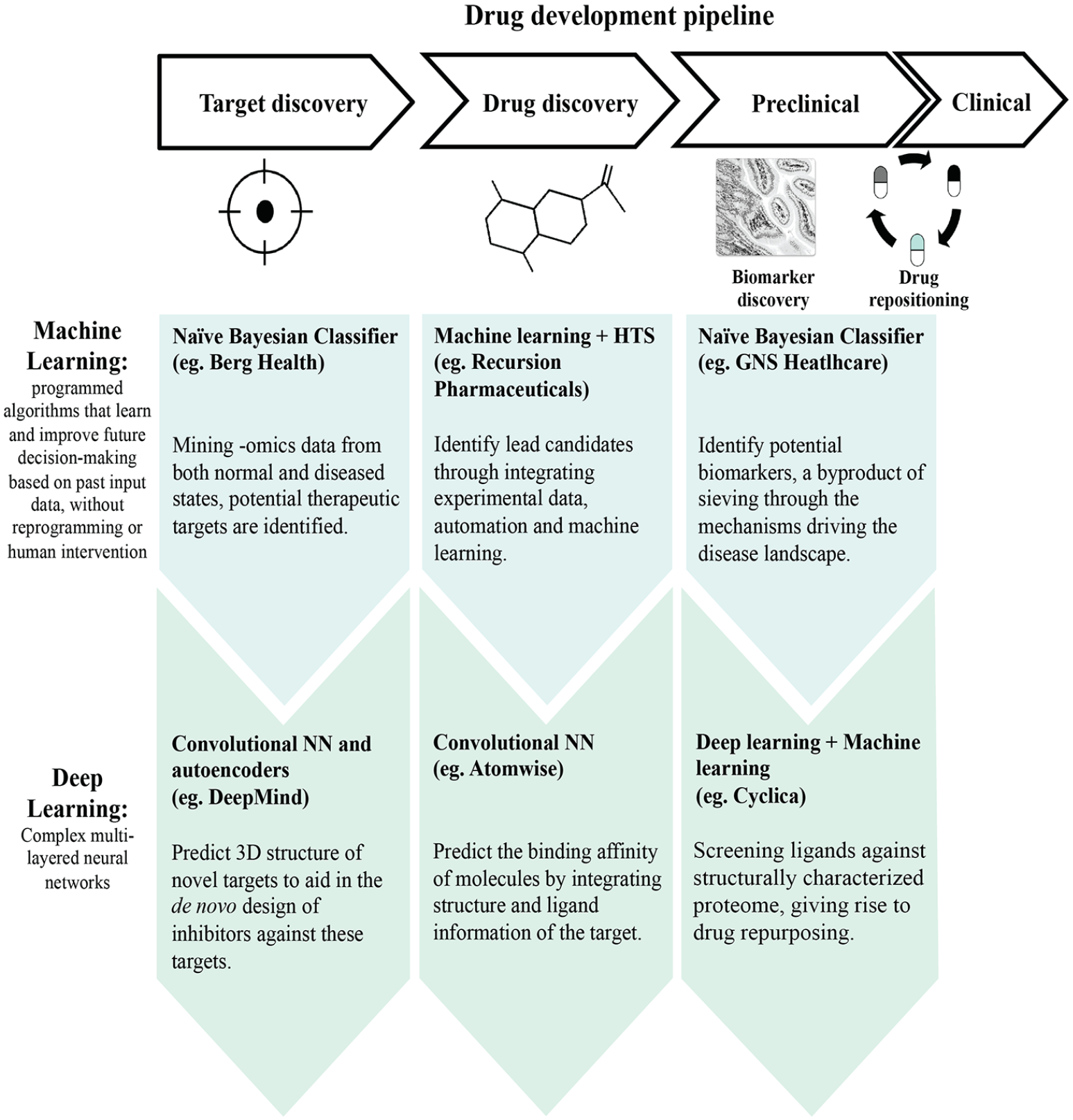
Integration of artificial intelligence within the drug development pipeline, as exemplified by companies within the industry.
Application of AI in Drug Discovery
VS has been an integral component in chemoinformatics, in which computational tools are used to plough through huge databases for new leads, with a higher probability of strong binding affinity to the target protein. VS can be classified into structure or ligand based, where the former is used when there is enough information on the three-dimensional (3D) structure, whereas the latter is used when there is little 3D information. 4 Machine learning approaches ( Table 1 ) have been integrated into VS, particularly for ligand-based VS. The objective of applying machine learning is to generate models, based on training sets, which are then used to predict compound class labels and rank these compounds according to their probable activites.28,29 The first application of machine learning was structural analysis (SSA), a tool for the automated analysis of biological screening data, as described by Cramer et al. 30 This involves deriving a weight for each substructural fragment, independent of others within a molecule, to evaluate the estimated activity of the fragment-containing molecules. This has released prior restrictions that only allowed optimization of a previously recognized lead structure, hence allowing prediction of active compounds beyond the structural class of established biological interest. SSA was used in the US government’s anticancer program, 31 and it was later “reidentified” as naïve Bayesian classifier. 32 Hence, AI has been historically integrated into drug discovery to identify potential new therapeutic targets or to generate new lead molecules. But this is only a small fraction of the capabilities of AI in effecting changes in drug discovery.
With the increasing amount of available data, the hurdle for drug design is ploughing through the vast space of medicinal chemistry data related to multiple targets and finding an optimal solution from these. The following examples illustrate the application of machine learning in drug discovery. The method to be employed depends on the prediction problem, data source, and prediction performance, for instance, the Berg Health harnesses Bayesian method in their AI platform, bAIcis, for target identification (
In the drug design space, Recursion Pharmaceuticals uses a combination of experimental data, automation, and machine learning to help facilitate drug compound design (
Harnessing DL in target identification and drug design has also been extensively explored. Moreover, DL has been demonstrated to have greater predictive capabilities over machine learning, as evident from Merck Kaggle 38 and the NIH Tox21 39 challenge, in which such approaches have triumphed for compound and toxicity prediction, respectively. In the QSAR competition sponsored by Merck, using the same descriptors and training data sets, teams were challenged to predict the biological activities of different molecules based on the numerical descriptors generated from the chemical structures by leveraging and comparing the different machine learning approaches. The challenge was composed of 15 targets, 164,024 compounds, and 11,081 features, with both descriptors and activities provided for the training set but only the latter for the test set. The winning team (submitted by one of the authors, George Dahl), which used multitask deep NN, achieved an improved accuracy of approximately 15%, as compared with Merck’s. The mean R2 averaged from 0.42 to 0.49, as compared with random forests. Even though a small number of targets interrogated is not reflective of the volume processed in pharmaceutical companies, it attracted the attention for future advancements in the QSAR field, 38 especially with the ability of deep NN to process thousand number of predictors. In addition, another study by Ma et al. 40 also demonstrated the superiority of deep NN over random forests for QSAR. Using the same data sets used in the Merck kaggle challenge and adapting the DNN algorithms derived from George Dahl’s team, they have shown deep NN outperforming random forests based on the mean R2 values. This study highlighted the predictive capability of deep NN in which having a single set of values for all the deep NN algorithmic parameters is applicable for most large volumes of QSAR data sets in industrial drug discovery 40 and that it is not necessary to optimize for the individual data sets.
In the Tox21 challenge, DeepTox was demonstrated to be more superior in toxicity prediction (which included 12 stress response and nuclear receptor effects) when juxtaposed against other computational approaches, such as SVM and random forests. DeepTox pipeline consists of deep NN representing the different toxicophores. With a training set of 11,764 compounds, leaderboard set of 296, and test set of 647 compounds, the computational models had to predict the outcome of the HTS assay. All information regarding the compound structures and assay measurements for the 12 different toxic effects were made available for the training set, whereas the assay results have been withheld for the leaderboard set to evaluate the performance of the models and thereafter released so that participants could further refine the models. DeepTox demonstrated high performance, winning 9 of 15 challenges, using the area under the ROC curve (AUC) as a performance criterion. DeepTox achieved the best AUC for both toxicity panels. 39 These aforementioned examples illustrate the superiority of DL over machine learning, including the following: ability to process and analyze large-scale data, mining relationships between input and output features, flexibility of the NN architectures resulting in its efficacy, and the automated extraction of features from raw data representations, without any predefined structure descriptor. However, these hand-engineered features are limited in predictive power to some extent, as these approaches cannot fully encode the structure information, and these predefined features are not data driven. The analysis underlying DL still involves predefined input predictors, limiting the full potential of direct raw data extraction. In addition, the currently available architectures are not suited for irregular structured data, such as molecules.
To minimize bias and improve the robustness of existing approaches, alternative methods such as graph convolution networks (GCN) aim to address these limitations. GCN, an extension of convolutional NN, is driven by the aggregation of information from neighboring nodes to represent a particular node. This neighborhood information is represented as graph substructures, and these are projected onto similar or different spaces. 41 GCN is thus able to encode the structural information of a molecular graph. A pioneering example was carried out by Duvenaud et al., 42 in which GCN was leveraged to generate data-driven fingerprints, representing the substructures within molecules. This was carried out by graphically representing the atoms as nodes and the interatom chemical bonds as the edges. These graphs are then used as input to learn molecule representations. The fingerprints were then used to evaluate drug properties, including solubility and drug efficacy, and were demonstrated to be superior when juxtaposed against circular fingerprints. 43
Another important aspect of drug discovery is the evaluation of molecular dynamics simulation. Gilmer et al. reformulated available models42,44,45 and proposed a common network framework termed message-passing NN within the GCN. 46 This NN extracts features from molecular graphs. Bond types and interatomic distances are translated into neighborhood messages. These messages are subsequently fed into the center atom via a set2set model. This approach was evaluated on the QM9 data set, comprising approximately 130,000 molecules with 13 properties and various types of molecular energies. This study demonstrated the high predictive capability of the platform, by accurately predicting DFT (a quantum mechanical simulation method) on 11 of 13 targets. 46 It has also highlighted the importance of stretching and effectively generalizing the model toward larger molecular sizes.
Based on these studies, pharmaceutical companies have also leveraged GCN. As exemplified in Chemi-Net, this approach has been used for absorption, distribution, metabolism, and excretion (ADME) prediction endpoints, which encompass human microsomal clearance, human CYP450 inhibition, and aqueous equilibrium solubility. 47 This analysis was carried out by first evaluating and reducing the neighboring information around each atom. The center atom is then defined by congregating the aforementioned information. This condensed information is then combined with an atom input feature. This study, which involved 250,000 data points, was demonstrated to perform better than Cubist, a machine learning approach adopted by Amgen. 47
Other permutations of DL within drug discovery include the Google-owned AI company DeepMind, which has leveraged deep NN (namely, convolutional NN and autoencoders) to predict the properties of protein from its primary sequence. Named AlphaFold, it successfully predicted 25 of 43 structures, which includes the distances between pairs of amino acids and the ϕ-ψ angles between neighboring peptide bonds.48,49 This advancement is useful in predicting the 3D structure of the target, so as to allow the de novo design of inhibitors against these targets (
De novo molecule design is challenging because of the vast search space and especially so for novel targets without extensive prior data and knowledge. Predictive models were initially generated using Simplified Molecular-Input Line-Entry System (SMILES), a linear string notation used in chemistry to describe molecular structures. This approach, however, does not capture molecular similarities succinctly, and certain chemical properties are better projected on a graph. Hence, the evolution and introduction of deep generative models for de novo molecule design, in which graphical representations serve as subunits for molecular structures. Building and improving upon these advances, Jin et al. 51 has demonstrated a new generative model of molecular graph, the junction tree variational autoencoder. This approach places importance on evaluating structures as a whole, instead of using the node by node approach, where the likelihood of generating chemically invalid intermediates is high. To circumvent this, junction tree variational autoencoder breaks down molecules into subgraphs, serving as units both when encoding a molecule into vector representation and when decoding vectors back into molecular structures. However, this involves an indirect method of optimizing the molecular properties. You et al. 52 demonstrated an approach that is capable of directly optimizing the molecular characteristics of the molecular graphs through a process termed graph convolutional policy network. 52 Another approach, GraphAF, combines autoregressive and flow-based principles for molecular graph generation. 53 GraphAF is highly efficient, generating a high percentage of 68% of valid molecules. These examples illustrate the role of deep generative models, such as generative adversarial networks (GAN), 25 and variational autoencoders, 54 in aiding de novo molecule design. These models are trained based on real examples and learned to generate similar, novel synthetic counterparts beyond the boundaries of the defined data set.
In silico medicine has leveraged the power of GAN, coupled with reinforcement learning, giving rise to generative tensorial reinforcement learning (GENTRL) technology. 55 GAN trains models using a generator and discriminator, where these components compete. The generator produces artificial data, whereas the discriminator distinguishes it from real data. This process is repeated until the discriminator is unable to discern artificial from real data.26,27 The integration of reinforcement learning allows active exploration and optimization of the space beyond samples defined within the data set. Shedding off a significant amount of time, they have generated, validated, and designed a novel small-molecule targeting discoidin domain receptor 1 (DDR1) within 2 mo. DDR1 is a proinflammatory receptor tyrosine kinase involved in fibrosis. Six data sets were used to generate a robust model: molecules obtained from the ZINC data set, known DDR1 inhibitors, common kinase inhibitors, nonkinases inhibitors, patent data for biologically active compounds, and 3D structures of DDR1 inhibitors. The identified six lead candidates were validated both in vitro and in vivo. This approach demonstrated the capabilities of GAN in generating molecular design in a rapid and efficient fashion through optimizing biological activity and synthetic feasibility.
Mendez-Lucio et al. of Bayer combined GAN with transcriptomic data and demonstrated its capabilities in proposing hit molecules based on a gene expression signature of the target knockout. 56 This approach was based on the concept that a knocked-out protein would generate a gene expression signature that is analogous to the pharmacological inhibition of the same target. This platform could be applied to any target, as no prior background information on it or its active molecules are required. It comprises the training phase, in which approximately 20,000 compounds from the L1000 data set are trained via GAN; the prediction phase, in which new molecules are generated based on the desired gene expression signature; and finally the validation phase, in which molecules are checked against inhibitors beyond the training set. Each signature produced approximately 10% valid molecules, where the generated molecules share functional groups with the active molecule. This platform paves the way in reconciling chemistry and biology in drug molecular design, without having any prior information on the intended target.
For drug design, both Atomwise and Benevolent.AI have used convolutional NN for their big data analyses (
List of Pharmaceutical Companies That Leverage AI to Enhance Drug Development Processes.
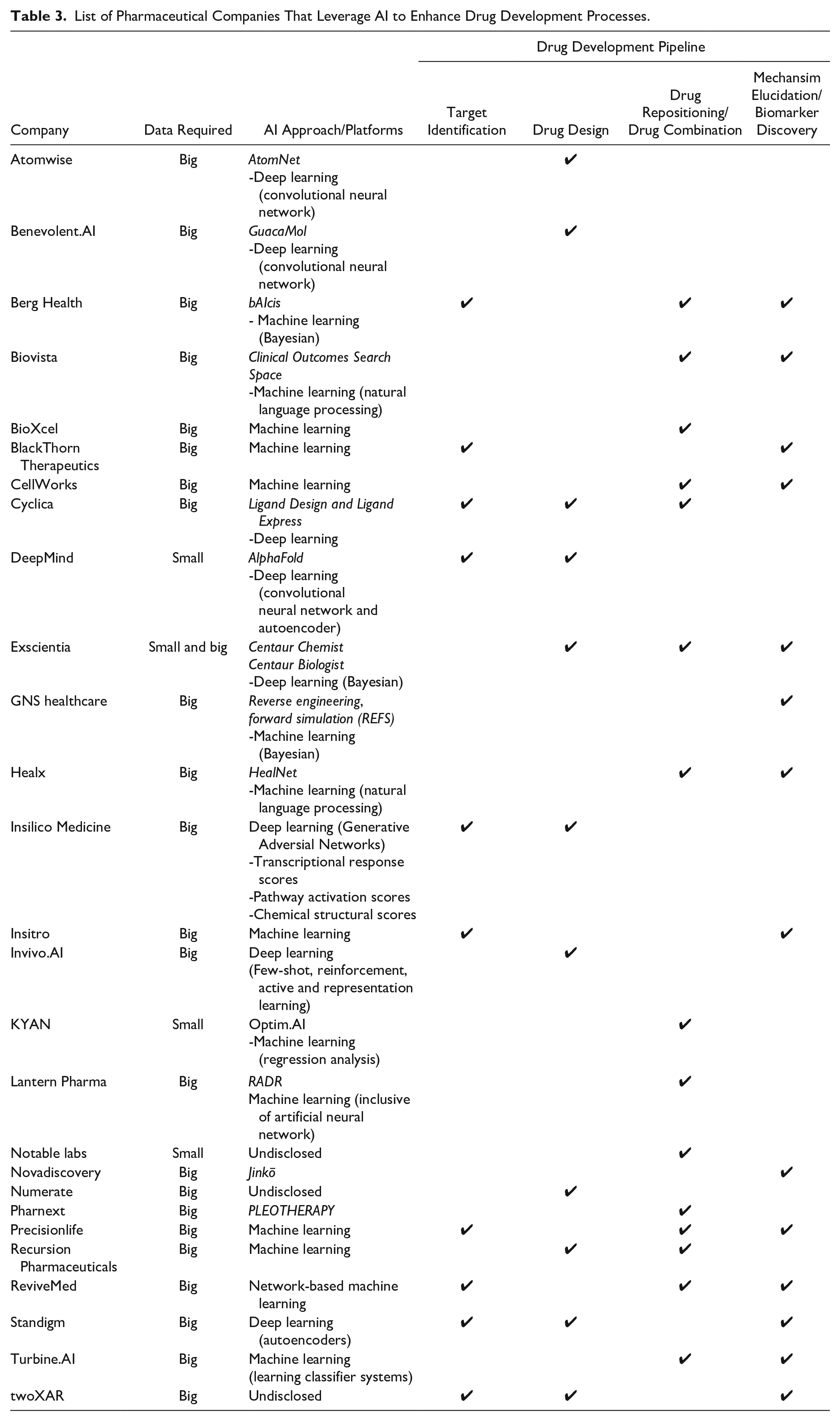
Another company that harnesses DL, Exscientia, has recently developed an obsessive compulsive disorder drug, DSP-1181, within 12 mo, as juxtaposed with the conventional norm of 4 y to reach phase 1 of clinical trials.59,60 DSP-1181 is a long-acting potent serotonin 5-HT1A receptor agonist. This molecule was developed in partnership with Japan’s Sumitomo Dainippon Pharma, which has expertise in G-protein–coupled receptor drug discovery. With their platforms, Centaur Chemist and Centaur Biologist, Exscientia is able to optimize multiple SARs in a systemic method. This entails mining data from ChEMBL, a public database that contains compound and activity data from decades of published medicinal chemistry literature. In the aforementioned OCD drug, data from ChEMBL were extracted to construct Bayesian models of ligand activity across 784 human protein targets, including G-protein–coupled receptors.61,62 These models aim to identify compounds that exert multitarget effects while minimizing off-target effects. This approach also provides an edge over other approaches that focus on only a single molecular target.63,64 Exscientia differentiates itself from the others by being able to drive drug discovery using very limited data points, especially useful for novel targets. Using donepezil, an acetylcholinesterase inhibitor to improve cognitive enhancement, as a starting point, this approach evolved its structure with additional improvements in D2 dopamine receptor activity and penetration of the blood-brain barrier (BBB). 62 Based on these defined dimensions with their corresponding Bayesian scores, including an additional combined score representing the ADME properties suitable for BBB penetration, alternative chemical structures were generated. In addition to evolving donepezil’s negligible D2 activity, inhibitors in the benzolactom series were also generated. This class was not present in the initial database, providing further confidence in the capabilities of the algorithm in generating novel structures. 62 As illustrated, with limited literature information and starting from a single point, Exscientia is able to expand alternative options within neuropsychiatric conditions.
The other companies that are within the same DL space include Insilico Medicine, Cyclica, Standigm, and DeepMind ( Figure 1 ; Table 3 ).
Application of AI in Drug Repositioning
There has been interest in drug repositioning, or drug repurposing, due to the challenges in designing new molecules. This is especially useful when these drugs have known mechanisms of action and ADME and toxicity (ADMET) data, thus saving resources with repurposed drugs approved sooner (3–12 y)
65
and at reduced cost (50–60%).66,67 As mentioned in the previous section, Cyclica uses DL for target identification and drug molecule design. In addition, Cyclica is also able to execute drug repositioning through their Ligand Express platform. This platform provides insights into the polypharmacology of small-molecule ligands by identifying the on- and off-target interactions. Leveraging DL, the platform screens ligands against structurally characterized proteome (
Centered on rare diseases, Healx agglomerates their proprietary data, both high-quality structured data and public unstructured data, as processed using natural language processing (NLP), and analyzes them to repurpose drugs, sieving out potential biomarkers and combinatorial treatments in tandem. NLP, which relies on machine learning techniques, is the ability of software to decipher and make sense of human languages. Leveraging NLP, Healx have demonstrated the capability of their platform to predict 22.2% synergistic antimalarial combinations from 1,540 combinations. 69 The novel combinatorial therapy includes efflux or transporter inhibitors with compounds possessing antimalarial activity. Healx has also identified repurposed therapeutics for Fragile X syndrome, a genetic condition that results in learning disabilities. 70 Spanning more than 15 mo from inception to readiness for clinical trial, Healx harnessed their AI analytics as the basis of its in silico Disease-Gene Expression Matching (DGEM) pipeline. This project identified eight potential candidates, which were also validated in mice. Sulindac, a nonsteroidal anti-inflammatory drug, and metformin, a hepatic glucose production inhibitor, have been identified as promising repurposing candidates for Fragile X. 70 Following this, the most promising ones will be chosen to progress through phase IIa trials. The efficiency in repurposing therapeutics puts forth AI technologies such as Healx as viable options for indications that lack readily available therapeutics.
Even though AI has been synonymous with big data analysis, generating predictive models from small data is particularly useful, as demonstrated earlier by Exscientia for novel targets. Another approach that uses small, experimentally derived data sets is able to highlight optimal drug combinations using existing available drugs via regression analysis. These data could be derived experimentally at the in vitro, in vivo, and ex vivo stages to rationally identify the most optimal drug combination. This platform has been leveraged on many indications including multiple myeloma 71 and T-cell lymphoma, 72 for which epigenetic drugs have been repurposed for these hematological disorders. Recently, the platform has also repositioned antiviral drugs to inhibit SARS-CoV-2. With a 12-drug search space spanning more than 530,000 possible drug combinations, the platform identified remdesivir, ritonavir, and lopinavir as the most optimal regimen to inhibit SARS-CoV-2 live virus. This regimen demonstrated a 6.5-fold improvement in efficacy juxtaposed to remdesivir alone. On the other hand, combinatorial hydroxychloroquine and azithromycin were shown to be relatively ineffective. 73 Completed within 2 wk, these studies also demonstrate the application and translational capabilities of AI to identify optimal therapeutics from the existing pool of drugs to address future outbreaks.
Application of AI in Biomarker Discovery
Identifying a robust biomarker is powerful in identifying potential responders, thus enabling precision medicine and improving clinical trial success rates.74,75 Under the master protocol, both basket and umbrella trials aim to extend the reach of targeted therapies to potential patients. Basket trials translate this by enrolling patients harboring the same mutation or biomarker, regardless of indication, to the same treatment.76,77 Umbrella trials, on the other hand, focus on a particular indication but enroll patients to the respective treatments based on the different molecular alterations or biomarker. 76 The search space to identify a suitable biomarker is huge, having to plough through extensive amounts of data.
By leveraging the Bayesian approach to sieve through mechanisms driving the disease landscape, a by-product of the GNS platform is the identification of potential biomarkers (
To increase the sensitivity of this process, a combination of features was also used to facilitate patient stratification. This involves the agglomeration and analysis of genomic, epigenetic, epidemiological, and other patient data to match the patients to the right treatments. Precisionlife uses this machine learning clustering approach to aid the process of therapy selection. Mining through genotype data of 547,197 single-nucleotide polymorphisms (SNPs) from 11,088 breast cancer cases and 22,176 controls, this platform was able to highlight high-order combinatorial genomic signatures within 1 h. Clustering of the SNPs provided insights into the complex disease population, where 175 risk-associated genes were found to be relevant to different patient subpopulations. From these, P4HA2 and TGM2 were demonstrated to have high repurposing potential in breast cancer, with accompanying validation. 80 P4HA is involved in collagen synthesis, whereas TGM2 translates into an enzyme (transglutaminase 2) involved in posttranslational modification of proteins to facilitate crosslinking. Collagen deposition has been proven to hasten cell growth and development, 81 thus making it a good biomarker to recruit potential patients. Transglutaminase 2 has been shown to be upregulated in breast cancer, and its interaction with interleukin-6 mediates metastasis. 82 Without taking these mechanisms into account prior to the analysis, the robustness of the platform is able to highlight these genes that would have high patient stratification potential. This analysis provides multiple insights into the disease, identifying both signature biomarkers and their corresponding viable drug treatments. Precisionlife has also leveraged their analytics platform on other non-oncology indications such as amyotrophic lateral sclerosis, Alzheimer’s disease, and asthma.
Besides analysing extensive and huge data sets, an ex vivo analysis on patient cells is able to highlight the most optimal, actionable treatments for the patient within a short turnaround time of 5 to 7 days. As demonstrated on a rare subtype of peripheral T-cell lymphoma (PTCL), hepatosplenic T-cell lymphoma, this machine learning–driven platform by KYAN has a relatively high concordance to previous clinical trials. 72 Before ex vivo analysis, the patient was administered the standard-of-care combinations (HyperCVAD, pembrolizumab, GVD [gemcitabine, vinorelbine, and liposomal doxorubicin], and pralatrexate), but the disease was refractory to them. Upon carrying out the ex vivo analysis as harnessed through KYAN’s platform, panobinostat and bortezomib emerged as the optimal combination for the same patient. After eight cycles on the regimen, the patient experienced complete metabolic remission. Subsequently, the patient underwent an autologous stem cell transplant and reported wellness 1 y later. Following this patient, 20% of subsequent patients with similar clinical manifestations responded to the same regimen, reflective of a previous phase II clinical trial on relapsed or refractory PTCL. 83 Following up, this ex vivo analytical platform could recruit patients who are sensitive to the desired treatment of interest, whereas those who are not will be administered the standard of care, akin to the basket trials. Paving the way toward personalized medicine, this platform could potentially be a viable clinical decision support system in recruiting and matching the right patients to the best therapeutic options.
The companies exemplified above highlight the importance of selecting the right biomarkers to facilitate patient recruitment, hence increasing the success of clinical trials.
We have demonstrated the role of AI in expediting and increasing efficiency across various stages along the drug development pipeline. Even though there is higher density and greater development of pharmaceutical companies in the drug discovery space, we have also showcased the application of AI for drug repositioning, biomarker discovery, and patient recruitment for clinical trials (
With the pervasive use of AI, especially in drug development, some factors should be taken into consideration. Algorithm transparency still remains an issue, in which platforms are akin to black boxes, making it difficult to sufficiently explain how the results are derived. Before implementation, complementary and follow-up experiments are important for validating the results that have been obtained via AI. This gap in interpretation would also result in troubleshooting difficulty when failure occurs.
Because AI is synonymous with big data aggregation and analysis, another issue that comes with big data is the availability of quality and accurate data. AI is based on the premise of predictive model generation, so any noisy data may skew the results and future predictions using this training set. One solution is for pharmaceutical companies to enforce high standards for data quality and management. Another is collaborative effort, in which high-quality shared data resources can be made more publicly available. These should encompass both successful and failed drug development efforts to allow more accurate prediction.
In addition, these large data sets can be integrated for training, validation, and feature data for the related algorithms. Integrating multifaceted data improves the accuracy and robustness of these algorithms. There are many ongoing efforts including ChEMBL, a large-scale chemical bioactivity database, 84 which contains and is not limited to compounds in clinical development, phenotypic data associated with similar compounds, and data from patents. Probe miner, a web resource that enables the objective, large-scale analysis of chemical probes, provides valuable information that assists in the identification of potential chemical probes. Approximately 300,000 (out of 1.8 million) published bioactive compounds were objectively explored and quantified against at least 2,000 human targets. 85 This database also highlights the gaps in knowledge, including human targets that lack chemical tools to probe their function. CanSAR is another platform that brings together multidisciplinary data across biology, chemistry, pharmacology, structural biology, cellular networks, and clinical annotations. 86 Machine learning can also be applied for predictions in drug discovery. In addition, these open-source data sets aid the accuracy of AI prediction in drug discovery.
With the burgeoning number of AI approaches and companies sprouting within the industry, the presence of publicly available data also serves as a standardized data set that allows the evaluation of these newly developed algorithms. For instance, MoleculeNet (built upon the open-source package DeepChem) provides a suite of software encompassing many known molecule representations and DL algortihms.87,88 Built on multiple public databases, it covers approximately 700,000 compounds that have been tested across different properties. For drug property prediction, the integration of attention and gate mechanisms improved the performance of graph convolutional networks. 89 This open access code was evident from the approach of Ryu et al, which is able to distinguish two separated molecular regions related to charge-transfer excitations for highly efficient photovoltaic molecules without any electronic structure information. These were proposed with the availability of publicly available codes. These efforts are pertinent particularly because the eventual goal for AI is the successful implementation and integration into patient care.
With the growing number of companies occupying the AI space, there is a need to use AI intelligently to make drug development processes more efficient. There are many companies offered at each stage along the entire drug development pipeline. Every problem is unique; hence, a different approach is needed to answer the intended question elegantly. This highlights the importance of selecting the most appropriate company, and their accompanying AI platforms, to solve the issue. In addition, with the burgeoning of AI companies, there is a worry that the drug development space would be saturated. To prevent this saturation, it is essential to keep these companies in check and to remind potential startups to employ an approach that has not been explored before, providing alternative options and promoting diversity as well.
From the examples illustrated in this review, we are just scratching the surface in unveiling the capabilities of AI in transforming drug development processes. Upon addressing the issues raised earlier, models of high predictive validity holds the key to increased efficiency in drug development. With the availability of more effective therapeutics coupled with accurate biomarker identification and patient stratification, there is great potential in AI to advance precision and personalized medicine at unprecedented rates.
Footnotes
Declaration of Conflicting Interests
The authors declared no potential conflicts of interest with respect to the research, authorship, and/or publication of this article.
Funding
The authors received no financial support for the research, authorship, and/or publication of this article.