
Abstract
Matrix polysaccharides are a diverse group of structurally complex carbohydrates and account for a large portion of the biomass consumed as food or used to produce fuels and materials. Glucuronoxylan and arabinogalactan protein are matrix glycans that have sidechains decorated with 4-O-methyl glucuronosyl residues. Methylation is a key determinant of the physical properties of these wall glycopolymers and consequently affects both their biological function and ability to interact with other wall polymers. Indeed, there is increasing interest in determining the distribution and abundance of methyl-etherified polysaccharides in different plant species, tissues, and developmental stages. There is also a need to understand the mechanisms involved in their biosynthesis. Members of the Domain of Unknown Function (DUF) 579 family have been demonstrated to have a role in the biosynthesis of methyl-etherified glycans. Here we describe methods for the analysis of the 4-O-methyl glucuronic acid moieties that are present in sidechains of arabinogalactan proteins. These methods are then applied toward the analysis of loss-of-function mutants of two DUF579 family members that lack this modification in muro. We also present a procedure to assay DUF579 family members for enzymatic activity in vitro using acceptor oligosaccharides prepared from xylan of loss-of-function mutants. Our approach facilitates the characterization of enzymes that modify glycosyl residues during cell wall synthesis and the structures that they generate.
Introduction
Methyl ethers exist as nonglycosyl modifications of glycosyl residues that are present in several plant polysaccharides. Glucuronoxylan (GX) and glucuronoarabinoxylan (GAX), which are major components of plant secondary cell walls, typically contain a 4-O-methyl ether modification of the α-linked glucuronosyl (MeGlcA) substituents decorating the 1,4-linked β-xylosyl backbone. 1 This modification is a conserved feature of GX and has been shown to co-evolve with the formation of vascular tissues. 1 A reduction in the MeGlcA content of GX can lead to increased xylan release during enzyme-catalyzed saccharification, suggesting that this structural modification may contribute to overall cell wall structure and integrity. 2 Nevertheless, the contribution of polysaccharide O-methylation to cell wall architecture and function remains largely unexplored despite the ubiquity of this modification.
The terminal β-glucuronic acid (β-GlcA) residues of arabinogalactan protein (AGP) often contain 4-O-methyl-etherified glycosyl sidechains. 3 These MeGlcA residues have been implicated in important biological processes, including signaling for proper pollen tube guidance and maturation within plant ovaries.4,5 Further, the GlcA residues on AGPs have been proposed to chelate divalent cations and thereby modulate calcium signaling at the plasma membrane, which has led to the suggestion that methylation of GlcA fine-tunes this process.6,7 AGPs isolated from the avascular moss Physcomitrella patens are known to contain 3-O-methyl rhamnosyl moieties, although this structure appears to be lacking in AGPs isolated from vascular plants. 8 Methyl-etherified xylosyl, fucosyl, and galacturonosyl, and in certain cases rhamnosyl residues, are also present in the complex pectic polysaccharide rhamnogalacturonan II.9–11 The biosynthetic mechanisms leading to these decorations and the consequences of this modification remain largely unknown. Previous studies have demonstrated that methylation of GlcA attenuates the catalytic efficiency of glucuronidases that hydrolyze GlcA sidechains of GX and AGPs. Thus, methyl etherification may be one strategy utilized by plants to defend themselves against microbial enzyme degradation due to increased recalcitrance of plant cell wall material. 12
Methyl-etherified uronic acids present several analytical challenges when assessing cell wall structure. Hexuronosyl residues are typically resistant to acid hydrolysis due to electronic effects rendered by the C6 carboxyl.13,14 The harsh conditions required to cleave the glycosiduronic bond may result in degradation of the carbohydrate. 14 Furthermore, the detection response factor for MeGlcA and GlcA differs in multiple analytical techniques. 15 Oligosaccharides containing methylated glycosyl residues are often isometric with other carbohydrate structures generated from plant cell walls and cannot be distinguished by mass spectrometric-based analysis lacking fragmentation capabilities. For example, MeGlcA linked to a xylose has the same mass as a GalA residue linked to rhamnose. This may confound analysis of complex samples and requires additional techniques such as glycosyl residue composition analysis to establish the structure of the glycan.
Members of the Domain of Unknown Function 579 (DUF579, PF04669) family of proteins have been implicated in the synthesis of methylated GlcA substituents in Arabidopsis.2,16–19 The Arabidopsis DUF579 family contains 10 members (
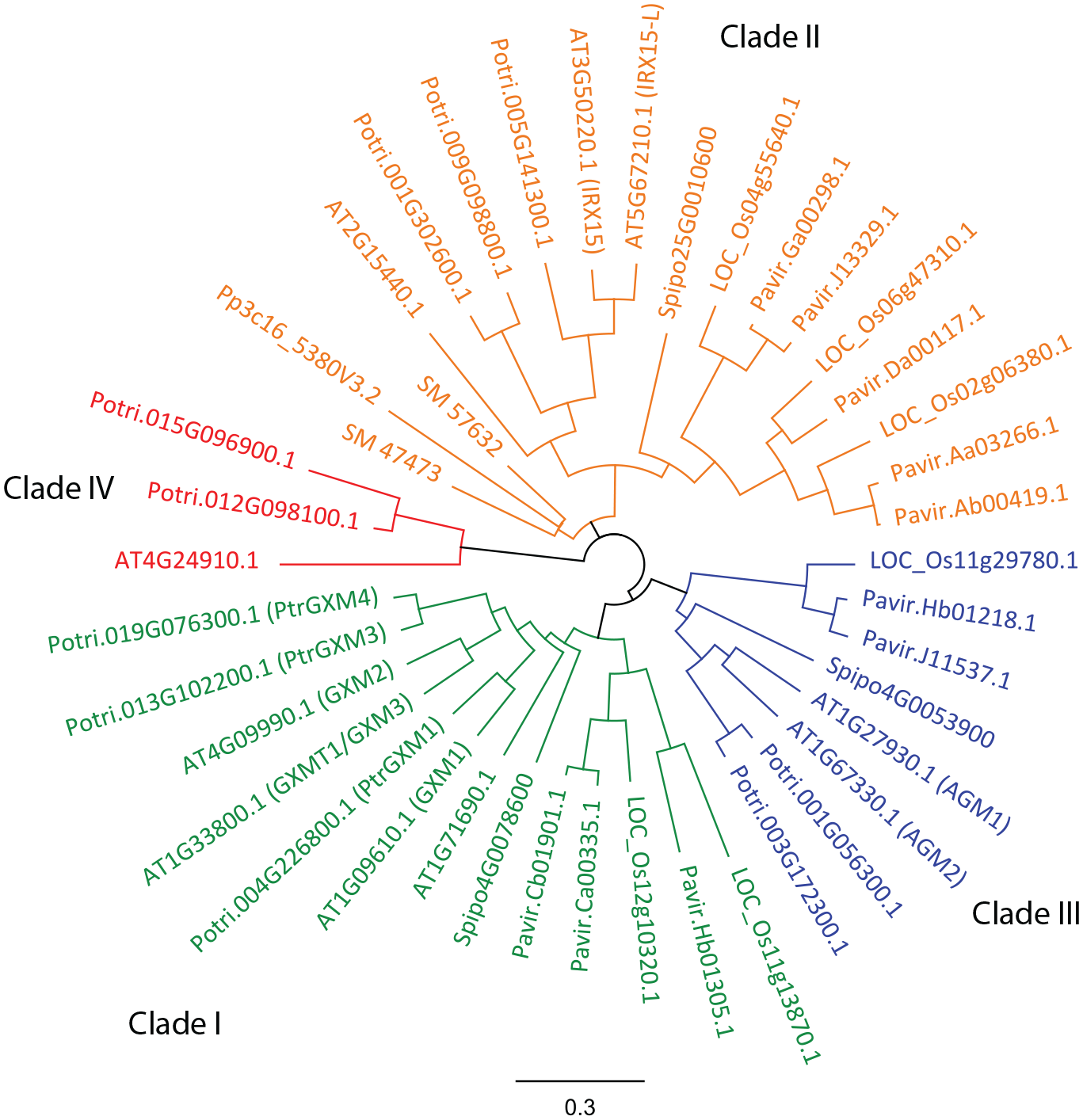
Cladogram depicting protein sequences of DUF579 family members from Arabidopsis, Populus, Panicum, Oryza, Selaginella, Physcomitrella, and Spirodella. The family segregates into four distinct clades. Clade I (depicted in green) contains members characterized as GXMTs. Clade II shown in orange includes IRX15 family members, mutants of which have been shown to affect xylan structure. Recent reports have suggested two members in clade III in Arabidopsis as putative arabinogalactan glucuronosyl 4-O-methyltransferases. Clade IV has not been characterized.
The availability of mutants that lack specific substituent modifications is a useful resource to aid in the development of methods and techniques to detect these methylated structures and their biosynthetic and interacting enzymes. Indeed, there is a growing need for facile methods that demonstrate and delineate the chemotypic effects on altered plant cell wall structures caused by changing or eliminating the expression of DUF579 enzymes. To this end, here we have described methods to functionally characterize the AGP-specific putative methyltransferases in DUF579 clade III, as well as the GX methyltransferases present in DUF579 clade I. Specifically, we detailed analytical methods for detecting the β-linked MeGlcA decorations present on the sidechains of AGPs with the use of loss-of-function Arabidopsis mutants in two members of DUF579 clade III. We also used nuclear magnetic resonance (NMR) spectroscopy to explore the tissue-specific contributions of these enzymes to the synthesis of MeGlcA on AGPs. In addition, we have demonstrated the recombinant expression and characterization of a member of DUF579 clade I from Populus and presented facile mass spectrometry (MS)-based techniques to assay the activity of this enzyme in vitro.
Materials and Methods
Phylogenetic Analysis of DUF579 Protein Family
Protein sequences were obtained via a BLAST analysis using the amino acid sequence of AT1G27930 in the Phytozome database. All protein sequences were downloaded from Phytozome. Sequence alignments were performed with Geneious 8.1.9 gene viewer software (ClustalW, BLOSUM matrix with gap open penalty of 12 and gap extension penalty of 0.1), and the phylogenetic tree was constructed using the Geneious tree builder function (Jukes–Cantor genetic distance model with UPGMA tree build method).
Plant Materials and Growth Conditions
Seeds were obtained from the Arabidopsis Biological Resource Center. Arabidopsis wild type (WT, Col-0) and T-DNA insertional mutants in the genes encoding the two clade III family members AT1G27930 (SALK_000253) and AT1G67330 (GABI_054A04), referred to here as arabinogalactan methylation-deficient 2 (agm2) and 1 (agm2), respectively,23,24 were used for analysis. To generate double mutants, agm1 and agm2 homozygous mutants were crossed and the resultant F2 population was screened by PCR as described below to identify agm1 agm2 double mutants. Populus trichocarpa trees were grown at the University of Georgia greenhouses.
Root and aerial tissue were obtained by germinating and growing Arabidopsis in liquid medium. Briefly, seeds were surface sterilized and one seed was added to each well of a six-well plate containing 5 mL of sterile growth medium (1× Gamborg’s media, 1% sucrose, 0.5 g/L MES, pH 5.8) and wrapped in micropore surgical tape (3M, Saint Paul, MN, USA). Six plates were used for each plant line under investigation. Plates were placed in a 23 °C chamber at 38% relative humidity and gently shaken at 80 rpm under constant light (100 µmol) and grown for 2 weeks. Roots and aerial tissue were separated manually and used to prepare alcohol-insoluble residue (AIR).
Genotyping
Seeds were suspended in water and vernalized for 48 h at 4 °C in the absence of light. Seeds were then sown in soil and grown in a climate-controlled growth room for 8 h in dark (19 °C) and 16 h in light (23 °C). Small sections of leaves from 3-week-old plants were used for genotyping. Leaves of WT, agm1, agm2, and agm1 agm2 were collected from Arabidopsis plants and genomic DNA was prepared. Briefly, two small leaves were placed in 1.5 mL Eppendorf tubes containing 125 µL of extraction buffer (100 mM Tris, pH 8.0, 50 mM EDTA, 500 mM NaCl, 10 mM 2-mercaptoethanol). The tissue was ground in the extraction buffer using a pestle before adding another 125 µL of extraction buffer and 17.4 µL of 20% sodium dodecyl sulfate (SDS). Samples were then kept for 10 min at 65 °C, followed by 10 min on ice. Samples were then pelleted by centrifugation for 10 min at 12,000g. The supernatant was removed and transferred to clean tubes containing 30 µL of 3M sodium acetate (NaOAc) and 320 µL of isopropanol, mixed and kept for 1 h at −20 °C. The tubes were then centrifuged for 20 min at 12,000g. The supernatant was discarded and the pellet suspended in aqueous 70% ethanol and then centrifuged for 5 min. The supernatant was removed, and the pellets were dried for 15 min at 37 °C. Forty microliters of TE buffer was then used to resuspend each sample. Two microliters of genomic DNA was then used in PCRs to genotype the plants. Genomic DNA from WT, agm1, agm2, and agm1 agm2 were subjected to PCR using four different primer pairs (
Preparation of AIRs
AIRs were prepared as described by Tuomivaara et al. 25 Briefly, plant tissue was suspended in cold aqueous 80% ethanol and homogenized using a Polytron tissue disruptor (Ultra-Turrax, Ika-Werke, Staufen, Germany). The suspension was then filtered through 50 µm nylon mesh and the residue suspended in aqueous 75% cold ethanol, stirred, and then filtered through nylon mesh. The residues were suspended in chloroform and methanol (1:1 v/v) and stirred for a minimum of 4 h, before removal of the chloroform and methanol solution by another round of filtration through nylon mesh. The insoluble residue was then washed with acetone and dried for 16 h under vacuum. The dry residue represents the isolated plant cell wall material referred to as AIR herein.
Fractionation of Cell Wall Components
The AIR was treated with shaking for 16 h at 55 °C with 50 mM NaOAc, pH 5 (10 mg/mL), to solubilize and enrich for AGPs. The supernatant was collected by filtration through a Whatman GF/A filter covered by a layer of 50 µM nylon mesh, dialyzed against deionized water, and finally lyophilized to produce the AGP-rich fraction. The NaOAc-insoluble material was then prepared similarly to the description in Zhong et al. 26 and sequentially extracted at room temperature with 50 mM ammonium oxalate (pH 5), 1 N KOH (containing 1 mg/mL NaBH4), and 4 N KOH (containing 1 mg/mL NaBH4). The KOH extracts were neutralized with acetic acid prior to dialysis. One drop of octanol was added prior to neutralization to prevent foaming. Fractions were extensively dialyzed (3.5 kDa molecular weight cutoff [MWCO], Thermo Fischer Scientific, Hampton, NH, USA) against deionized water and lyophilized.
NMR Analysis of AGP-Enriched Fractions
NaOAc extracts of AIR from WT, agm1, agm2, and agm1 agm2 plants and from Arabidopsis roots and stems were suspended in 99.9% D2O and lyophilized. This was then repeated. The samples were then dissolved in 100 µL of D2O containing 0.00026% DMSO as an internal standard. NMR spectra were recorded at 298 K with a Varian Inova NMR spectrometer operating at 600 MHz and equipped with a 5 mm NMR cold probe (Agilent, Santa Clara, CA). Data were analyzed using MRestNova software (Mestrelab Research S.L., Spain). Chemical shifts are reported relative to the methyl proton signal of DMSO at 2.721 ppm. Assignments for MeGlcA and GlcA were made based on previous analysis of similar structures as well as by 2D NMR analysis (HSQC and COSY).27,28
Glycosyl Residue Composition of Cell Wall Extracts
Cell wall fractions were methanolyzed essentially as described by Kelly et al., 29 but with some modifications. The method was modified for a milder hydrolysis to avoid potential degradation of methyl glucuronosyl residues at high temperatures for extended time periods. HCl/methanol (MeOH; 0.5 N) was prepared by diluting methanolic HCl (3 N; Supelco) in anhydrous methanol (Sigma Aldrich). Approximately 200 µg of lyophilized samples was suspended in 300 µL 0.5 N MeOH and heated for 30 min at 60 °C. Samples were concentrated to dryness under a stream of air, washed with 3 mL of MeOH, and dried again. The residue was then suspended in 0.5 mL of 10 mg/mL NaBD4 in water and kept for 3 h at room temperature. The reaction was quenched by the addition of three drops of glacial acetic acid, dried with air, and then washed with 1 mL of 9:1 MeOH/acetic acid, followed by 1 mL of MeOH. The residues were suspended in 3 mL of water and desalted using an OnGuard II H column (Dionex, Sunnyvale, CA, USA) previously washed with 1 mL of 1 N HCl followed by 10 mL of H2O. The eluent was collected and dried under air. Samples were then dissolved in 0.5 mL of 2 M trifluoroacetic acid (TFA) in a glass tube with a Teflon-lined cap and kept for 2 h at 120 °C. Following hydrolysis, samples were dried under air with 3 mL of isopropanol. A second reduction was then performed for 2 h at room temperature with 0.5 mL of 10 mg/mL NaBH4 in 1 M ammonium hydroxide. The reduction was quenched with the dropwise addition of three sequential additions of 1 mL of MeOH/acetic acid (9:1), followed by drying under air. Acetylation was then performed by the addition of 0.5 mL of acetic anhydride and 0.5 mL of pyridine for 10 min at 120 °C. One milliliter of toluene was then added to the samples and they were dried under air. One milliliter of methylene chloride and 1 mL of water were added to the dried sample and mixed. The organic layer was transferred to a clean tube and dried under air. The sample was then resuspended in five drops of dichloromethane and run on an Agilent 7890A gas chromatography–electronic ionization–mass spectrometry (GC-EI-MS, Agilent Technologies, Santa Clara, CA, USA) equipped with a 30 m Supelco SP-2331 bonded phase fused silica capillary column (Sigma Aldrich, St. Louis MO, USA). Experiments were performed in technical triplicates.
Trimethyl Silyl Analysis (TMS) of Cell Wall Extracts
Glycosyl composition analysis was performed by combined GC-MS of the per-O-trimethylsilyl (TMS) derivatives of the monosaccharide methyl glycosides produced from the sample by acidic methanolysis as previously described by Santander et al. 30
Briefly, Arabidopsis buffer extracts of WT and agm1 agm2 double mutants (~100 μg), enriched in AGPs, were heated with methanolic HCl (1 N) in a sealed screw-top glass test tube for 17 h at 80 °C. After cooling and removal of the solvent under a stream of nitrogen, the samples were derivatized with Tri-Sil (Pierce, Thermo Fischer Scientific, Rockville, Il, USA) at 80 °C for 30 min. GC-MS analysis of the TMS methyl glycosides was performed on an Agilent 7890A GC (Agilent Technologies, Santa Clara, CA, USA) interfaced to a 5975C MSD, using an Supelco Equity-1 fused silica capillary column (30 m × 0.25 mm ID; Sigma Aldrich, St. Louis, USA).
Production of gxmt1 Xylooligosaccharides
The acceptor substrate for enzymatic assays of GXMT activity was generated from GX solubilized by 1 M KOH treatment of Arabidopsis gxmt1 null mutant AIR. 2 The lyophilized extract was suspended in water at a concentration of 10 mg/mL, and 1 mL was digested for 12 h at 25 °C with 2.3 U of M1 xylanase (Megazyme International, Bray, Ireland). Undigested polymers were precipitated by the addition of cold ethanol to 70% (v/v). The precipitate was removed by centrifugation for 5 min at 2800g. The supernatant was collected and dried under air. Once dry, it was resuspended in 1 mL of water, frozen, and lyophilized.
Analysis of PtGXMT3 Activity by MALDI-TOF MS
PtGXMT3 (Potri.013G102200.1) was cloned essentially as described. 31 Briefly, to create Gateway entry clones the truncated coding region of PtGXMT3 (amino acids 46–304) was PCR amplified (PtGXMT3_46F, 5′-AACTTGTACTTTCAAGGCAATGATGACCTAACAAACC -3′ and PtGXMT3_304R, 5′-ACAAGAAAGCTGGGTCCTAAGGACAAAAAGGTCTGCC -3′) using cDNA prepared from terminal buds of P. trichocarpa with Phusion High-Fidelity DNA Polymerase (Thermo Scientific, Waltham, MA). The PCR product was cloned into the pDONR221 plasmid vector (Life Technologies, Carlsbad, CA, USA) to create an entry clone, which was then recombined into a Gateway-adapted version of the pGEn2 mammalian expression vector (pGEn2-DEST) 32 using Gateway BP Clonase II Enzyme Mix and Gateway LR Clonase II Enzyme Mix (Life Technologies, Santa Clara, CA), respectively. The final expression construct is designed to express a fusion protein composed of an NH2-terminal signal sequence, an 8xHis tag, an AviTag, the coding region of “superfolder” green fluorescent protein (GFP), and a tobacco etch virus (TEV) protease recognition site, followed by residues 46–304 of PtGXMT3. Recombinant protein expression was performed by transient transfection of suspension-cultured HEK293-F cells (100 mL) as described. 31 Purification of 8xHis-tagged enzyme was performed using a 1 mL HisTrap HP column (GE Healthcare, Uppsala, Sweden) as described.31,33
Purified His-GFP-PtGXMT3 was concentrated to at least 1 mg/mL by diafiltration with a 30 kDa MWCO Amicon Ultra centrifugal filter (Merck Millipore, Cork, Ireland) and then dialyzed (3500 MWCO) at 4 °C into 50 mM HEPES sodium salt/HCl, pH 6.8, containing 5 g/L Chelex-100 resin (Bio-Rad Laboratories, Hercules, CA, USA). Proteins were then used directly for reactions or kept at 4 °C.
Methyltransferase activity was performed in duplicate in Eppendorf tubes in 50 µL of 50 mM HEPES sodium salt/HCl, pH 6.7, containing 10 µM PtGXMT3, 2 mM GX oligosaccharides, 10 mM S-5′-adenosyl methionine p-toluenesulfonate salt (SAM), and 2 mM cobalt chloride. A control reaction lacking SAM was also prepared. The reactions were kept for 3 h at room temperature. A portion (5 µL) of the reaction products was removed and mixed with 5 µL of Dowex 50WX8 hydrogen to form cation exchange resin (Bio-Rad Laboratories, Hercules, CA, USA) and kept for 30 min at room temperature before a brief centrifugation. Two microliters of the supernatant was then mixed with 2 µL of matrix solution composed of 0.1 M 2,5-dihydroxbenzoic acid (DHB, Sigma Aldrich, St. Louis, MO, USA) in aqueous 50% methanol directly on a stainless steel matrix-assisted laser desorption ionization (MALDI) target plate (Bruker Daltonics, Billerica, MA, USA). Data were collected in the positive ion mode by summing at least 200 individual laser shots with a Bruker Microflex LT mass spectrometer and Biospectrometry workstation (Bruker Daltonics, Billerica, MA, USA).
Results
A New Protocol to Identify MeGlcA
Two standard protocols are typically used for cell wall glycosyl residue composition analysis. First is the alditol acetate (AA) method, which requires the hydrolysis of glycosidic bonds, typically by treatment with hot acid, followed by reduction and acetylation of the released monosaccharides to form the acetylated alditols that are separated and analyzed by GC-EI-MS or gas chromatography–flame ionization detection (GC-FID). Second is the TMS method, involving methanolysis and derivatization of the methyl glycoside hydroxyls with TMS groups, followed by GC-EI-MS analysis. The TMS method provides direct analysis of acidic sugars, which are not detected by the standard AA method. However, with TMS each monosaccharide within the sample produces multiple, independently eluting peaks, which can confound data analysis with complex samples. While MS analysis of peaks can determine the presence or absence of a methyl glycoside, unambiguous determination of their identity (e.g., GlcA vs galacturonic acid) requires the use of known glycosyl standards for each monosaccharide or monosaccharide derivative in order to compare retention times for assignment. The use of such standards can present difficulties when assessing rare or modified residues where access to purified standards may be limited, as is often the case with methyl-etherified sugars. Furthermore, TMS analysis of methyl-etherified residues also lacks the capability to determine the position of any methyl-etherified (or otherwise modified) hydroxyl groups present within a sample, information that may have biological relevance.
To minimize the pitfalls of these two methods, we combined portions of the AA and TMS methods (
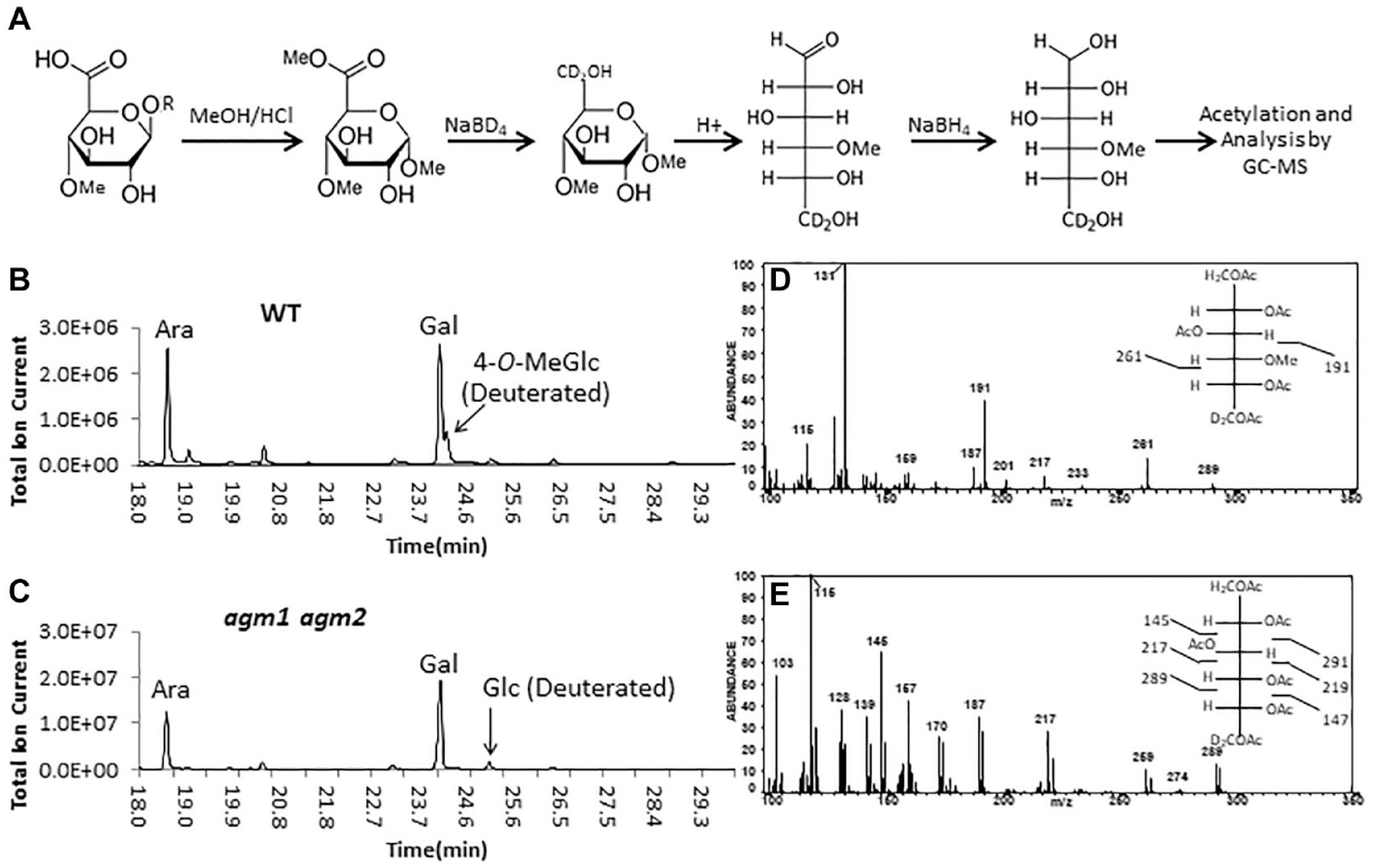
A new method for compositional analysis of methyl uronides. (
When comparing the approach to compositional analysis described in this paper to that of the TMS method for these same samples (
NMR Analysis of AGPs from WT and AGM Mutants
Though methylation of AGPs has been shown to be disrupted in DUF579 clade III knockout mutants, it is not known if the two paralogs of this clade mediate methylation in a tissue-dependent manner. Furthermore, there is a gap in the literature in assignments for the 1H and 13C signals in the NMR spectra of methylated AGPs. As such, we sought to utilize the agm1 agm2 double mutant as a tool to assign signals corresponding to MeGlcA in AGPs, providing the scientific community with a resource with which to nondestructively assess the extent of AGP β-GlcA methylation in plant tissues. We obtained 2D gCOSY and HSQC spectra for AGPs from WT and the agm1 agm2 double mutant (
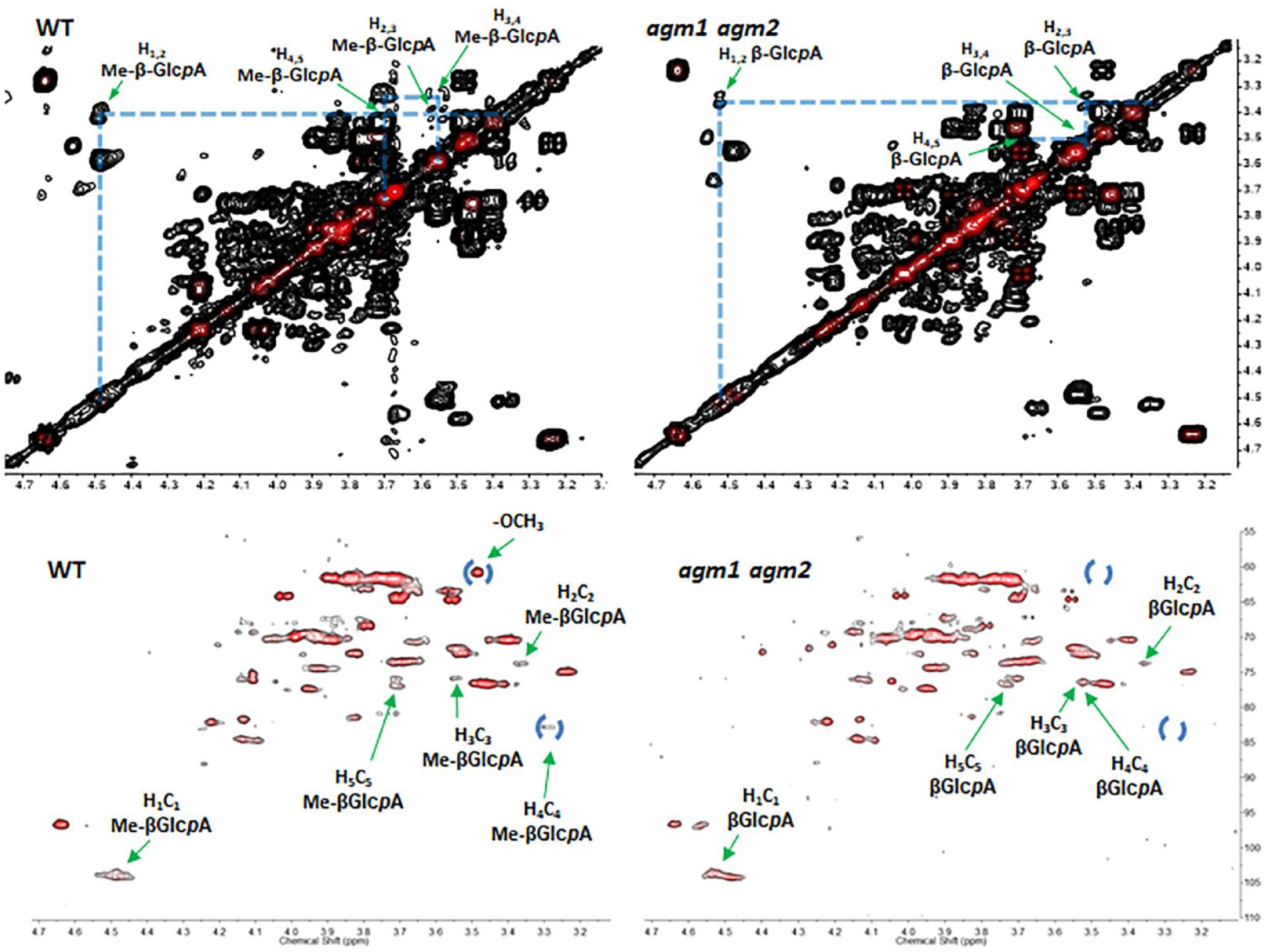
Top: gCOSY spectra of polymeric AGP extracted from WT (left) and agm1 agm2 (right) seedling plants. Labels indicate cross-peaks for terminal β-4-O-MeGlcpA in WT and β-GlcA in agm1 agm2 plants. Bottom: HSQC spectra of polymeric AGP extracted from WT (left) and agm1 agm2 (right). The presence of diagnostic signals (labeled with blue circles) for terminal β-4-O-MeGlcpA in the WT spectrum, which are absent in the agm1 agm2 spectrum, indicated the lack of β-GlcpA methylation in the AGP of the double mutant.
1H and 13C Signal Assignments of β-GlcpA and β-MeGlcpA in the NMR Spectra of Arabinogalactan Sidechains.
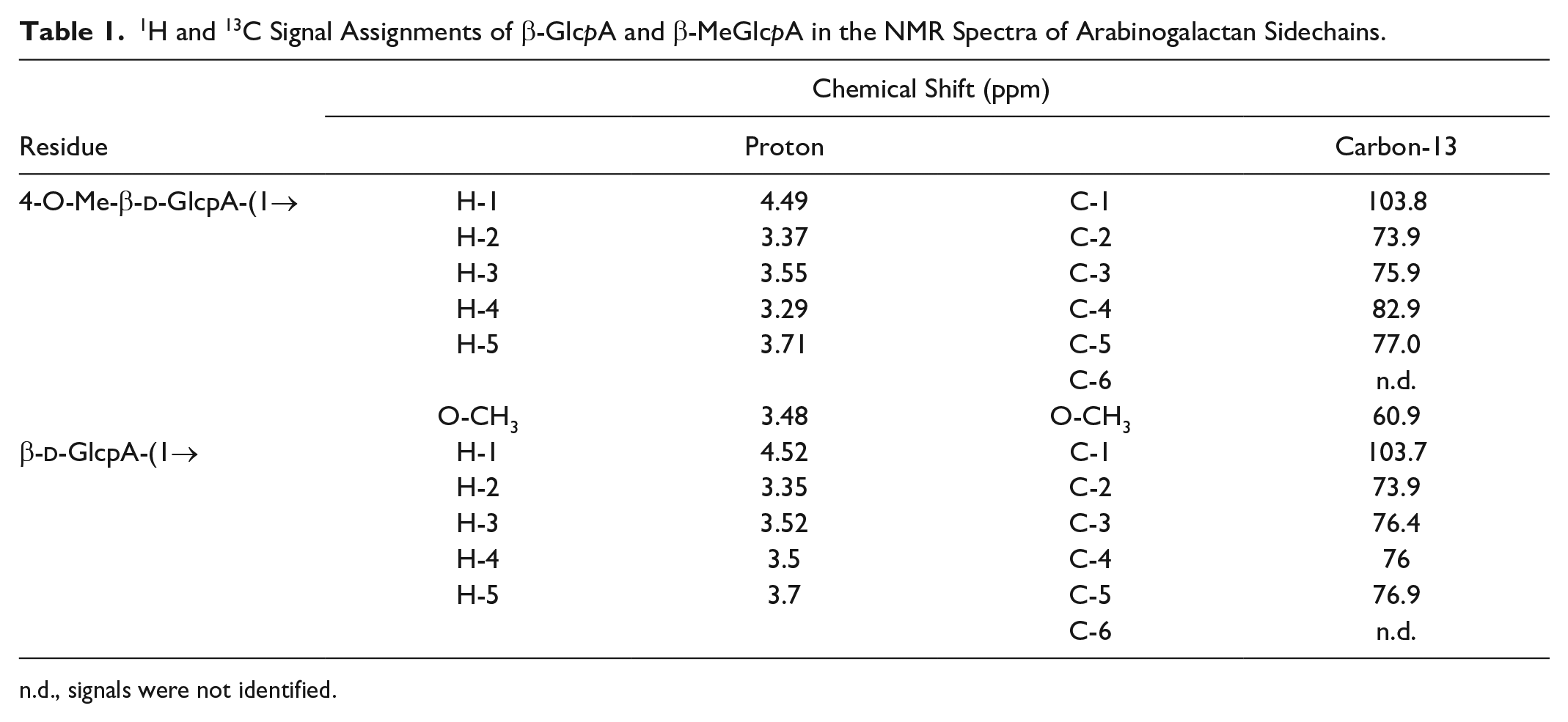
n.d., signals were not identified.
Previously published data suggest that AGM2 is expressed primarily in roots, while AGM1 is expressed fairly ubiquitously throughout the plant (
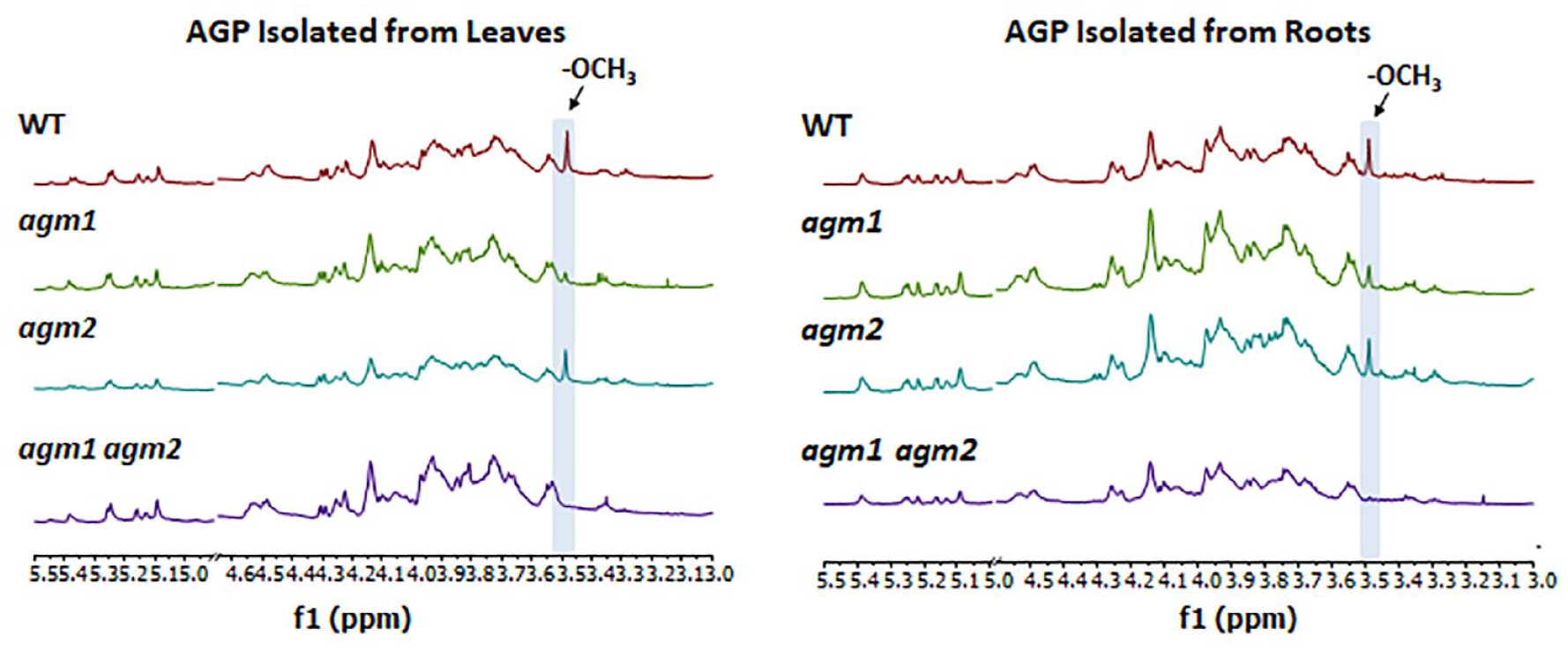
1H NMR analysis of the buffer-soluble fractions from agm1 and agm2 single and double mutants as compared to WT. The diagnostic signal at ~3.49 ppm (indicated by blue bar) that corresponds to the methyl group protons of the terminal β-4-O-MeGlcpA is evident in the WT spectra; however, this signal is reduced or absent in samples originating from single and double mutants.
MS-Based Assays to Evaluate Methyltransferase Activity of a DUF579 Enzyme
Describing the chemotypic effects of DUF579 knockouts on the methyl etherification status of plant cell wall polysaccharides is a useful tool for generating hypotheses regarding the enzymatic activities of proteins. However, given our ability to express functional recombinant versions of proteins involved in plant cell wall biosynthesis,2,31,33 the gold standard remains to directly assay enzyme activity and structurally characterize the reaction products. Thus far, we have been unsuccessful in our attempts to recombinantly express AGM1 or AGM2 from either Populus or Arabidopsis. For this reason, we chose to provide a method for the in vitro activity assay of Populus trichocarpa PtGXMT3, a putative GXM from the DUF579 family. PtGXMT3 groups with clade I in Arabidopsis (
We performed an MS-based assay for methyltransferase activity using recombinant PtGXMT3 and defined GX oligosaccharides. GXMTs have been shown to be cobalt-dependent enzymes; 2 thus, 2 mM cobalt was included in the reaction mixture, along with the methyl donor SAM, as a previous report had demonstrated that cobalt is a cofactor required by Arabidopsis GXMT1 for catalysis. 2 A negative control was performed in the absence of the methyl donor, SAM, included in the reaction mixture.
The MALDI time-of-flight (TOF) mass spectrum (
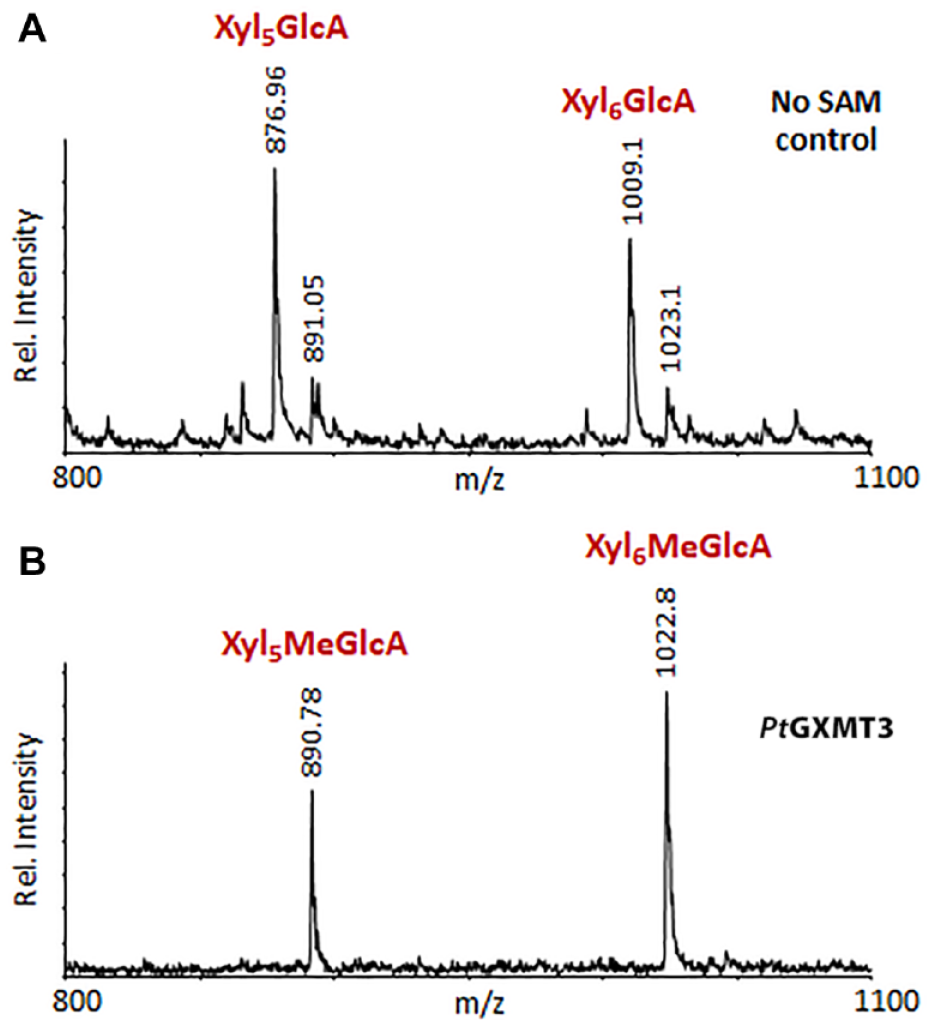
MALDI-TOF-based assay of PtGXMT3 activity. Xylan oligosaccharides with reduced MeGlcA content were prepared from Arabidopsis gxmt1 mutant plants and incubated with PtGXMT3 in the (
Discussion
Plant cell walls are a repository of diverse molecules with a range of structural complexities, which current analytical techniques have only begun to unravel. It is also becoming clear that small and often overlooked structural features such as methylation and acetylation contribute to the overall architecture and biological functions of the wall. Gaining an understanding of the biosynthesis of cell wall polymers and, in turn, allowing for the control of the biochemical mechanisms influencing cell wall structural and biological properties is a crucial step in harnessing the potential of biomass for next-generation fuels, materials, and products. However, as we gain insight into these biochemical mechanisms, we must also continue to develop methods that provide better approaches for the rapid analysis of cell wall chemotypes. Modifying plant cell wall polymers by genetic modification or selective breeding is now a reality as we rapidly advance our understanding of the biochemical and genetic pathways that can be manipulated. However, rapid exploitation of these technologies requires the ability to observe the effects of the structural and architectural changes we have made.
The DUF579 enzyme family is of particular interest for biomass modification given their recently established influence on the fine structural features of several cell wall polymers. Prior work has suggested that Arabidopsis DUF579 enzymes catalyze the methylation of GlcA sidechains present on GX and AGP.2,16 In vitro biochemical evidence for xylan methyltransferase activity has previously been demonstrated for members of clade I in Arabidopsis. However, the analysis of these enzymes and their resultant structures is complicated by the redundancy of paralogous enzymes in many plant tissues, as well as a lack of robust analytical methods to isolate, detect, and quantify methyl-etherified structures in complex mixtures of cell wall carbohydrates.
In this work, we have developed a method involving methanolysis and NaBD4 reduction, which can be used to detect methyl-etherified glucuronosyl residues as their deuterium-labeled AAs. This method presents an advantage over TMS analyses as it generates a single derivative for each methyl-etherified uronic acid. Furthermore, the position of a methyl ether, if present, is readily determined from the electron impact mass spectral fragmentation patterns.
Furthermore, we have provided NMR spectroscopic signal assignments for the detection of MeGlcA-modified AGP polymers and used this tool to demonstrate the contribution of two DUF579 clade III paralogs, AGM1 and AGM2, to the synthesis of methylated AGP sidechains in a tissue-dependent context. This demonstrates how the reported expression patterns of these two proteins correlate with the abundance of MeGlcA in root and leaf tissue AGPs. More specifically, we show that knockout of AGM2, which is expressed primarily in root cells, has little effect on AGP GlcA methylation in leaf or root tissue, potentially due to the compensatory effects of AGM1, which is expressed in nearly all plant tissues. Conversely, knockouts of AGM1 result in a near loss of signals for MeGlcA in leaves, while showing a modest decrease in MeGlcA for AGPs extracted from roots. In addition, agm1 agm2 plants result in a near-complete loss of signals corresponding to AGP MeGlcA in both tissues. Taken together, these data support the robustness of this technique and involve a simple buffer extraction, facilitating scalability. NMR spectroscopy has considerable potential for characterizing cell wall resident methyl ethers and is of great promise as it requires minimal sample preparation and is nondestructive.
We have demonstrated, using a MALDI-TOF MS-based assay, that PtGXMT3 O-methylates the α-GlcA residues of xylooligosaccharides generated from Arabidopsis gxmt1 GX. This illustrates the ease with which MS-based assays can be performed and applied to the characterization of plant cell wall biosynthetic enzymes in vitro. In summary, we have provided a working template and a detailed description of analytical techniques that can be readily applied toward the discovery and analysis of DUF579 protein activities and products and serve as a valuable resource for those who wish to explore the roles of these enzymes in plant biology and biotechnology.
Supplemental Material
Methylether_SLAS_SI-PS_2-11-20_PDF – Supplemental material for Analytical Techniques for Determining the Role of Domain of Unknown Function 579 Proteins in the Synthesis of O-Methylated Plant Polysaccharides
Supplemental material, Methylether_SLAS_SI-PS_2-11-20_PDF for Analytical Techniques for Determining the Role of Domain of Unknown Function 579 Proteins in the Synthesis of O-Methylated Plant Polysaccharides by Peter J. Smith, Malcolm A. O’Neill, Jason Backe, William S. York, Maria J Peña and Breeanna R. Urbanowicz in SLAS Technology
Footnotes
Acknowledgements
We would like to thank Ian Black and Artur Muszynski for their expertise and aid in carbohydrate analysis.
Supplemental material is available online with this article.
Declaration of Conflicting Interests
The authors declared no potential conflicts of interest with respect to the research, authorship, and/or publication of this article.
Funding
The authors received the following financial support for the research, authorship, and/or publication of this article: Funding was provided in part by the Center for Bioenergy Innovation (CBI), a U.S. Department of Energy (DOE) Bioenergy Research Center supported by the Office of Biological and Environmental Research in the DOE Office of Science. This work was also partially supported by the Division of Chemical Sciences, Geosciences, and Biosciences, Office of Basic Energy Sciences of the U.S. Department of Energy.
Supplemental Material
The data that support the findings of this study are present in the paper and/or the Supplemental Materials, and materials are available from B.R.U. upon reasonable request.
References
Supplementary Material
Please find the following supplemental material available below.
For Open Access articles published under a Creative Commons License, all supplemental material carries the same license as the article it is associated with.
For non-Open Access articles published, all supplemental material carries a non-exclusive license, and permission requests for re-use of supplemental material or any part of supplemental material shall be sent directly to the copyright owner as specified in the copyright notice associated with the article.