
Abstract
An effective oligonucleotide preparation approach for the thermodynamically balanced, inside-out (TBIO) PCR-based assembly of long synthetic DNA molecules (synthons) is described in the current work. We replaced the necessity to purify individual oligonucleotides with just one purification procedure per approximately 500 base pairs (bp) of duplex DNA. So for an enhanced green fluorescent protein (EGFP) gene of 717 bp, we synthesized 24 oligonucleotides with a length of 50 bases and performed just two solid-phase extraction (SPE) purification procedures. It was found that the capacity of ZipTip microextractors, usually used for sample desalting in proteomics, perfectly corresponds to the gene synthesis scale (40–60 pmol). The robustness of the approach was validated with a 65-mer oligonucleotide design of the same gene. The modification of the oligonucleotide concentration gradient from the original TBIO scheme substantially increased the purity of the PCR product. We proposed a mechanism for the formation of supramolecular structures, which often occur during TBIO assembly. By using the proposed workflow, any laboratory with a standard facility for molecular biology manipulation, a 16-channel oligonucleotide synthesizer, and a conventional thermocycler has the ability to prepare one gene with a length of about 700 bp per day.
Introduction
The de novo synthesis of various DNA sequences or artificial genes has become a cornerstone of modern biotechnology and molecular biology. The ability to synthesize long duplex DNA served as the basis for a new field of science—synthetic biology. The modern laboratory gene synthesis pathway starts from synthesis design and usually finishes at the plasmid vector with a target insert. The multistage procedure is usually laborious and takes a lot of time. Although there are many techniques that can be used for every stage of gene synthesis, current gene synthesis workflows rely on important factors: oligonucleotide synthesis yield, oligonucleotide preparation (cleavage, deprotection, and purification) time, quality/purity of intermediate products, synthesis scale, and possibility of automation ( Fig. 1 ). The time spent on the process of gene synthesis consists of the following consecutive steps: solid-state oligonucleotide synthesis time, cleavage and deprotection, purification time, time for the enzymatic assembly step, target sequence reamplification or purification, time for the cloning step if it is required, quality control, and error correction. 1
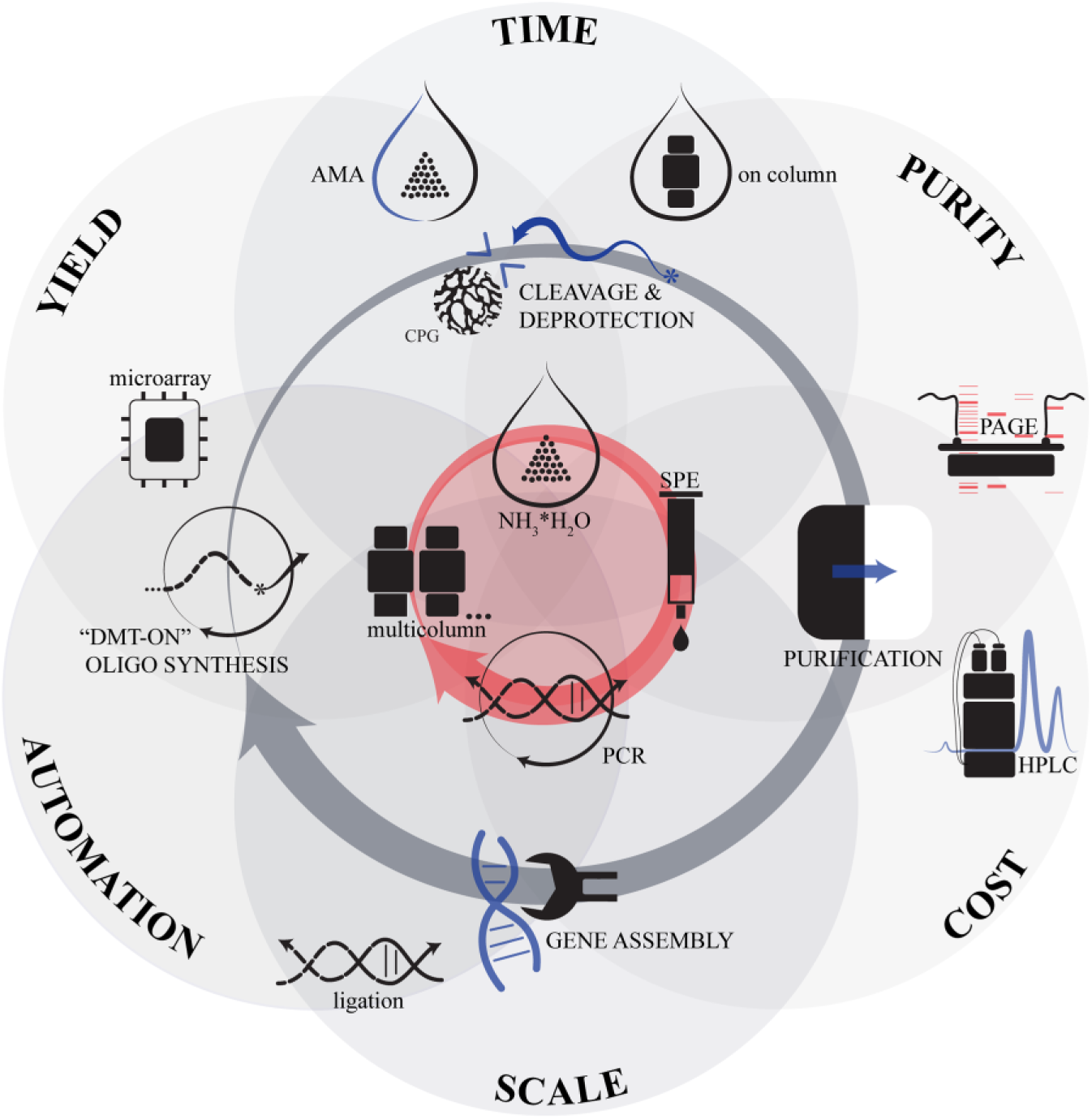
Optimal techniques for gene synthesis. The typical problems and factors of the different techniques are presented as six intersecting circles. A central red circle is situated at the intersection of all six factors and represents their balanced state. The general stages of gene synthesis workflow are shown in an inner gray loop. Variants of the techniques for each stage are arranged around a stage icon. A red loop that frames the central red circle indicates the optimal choice of techniques, which were used in the current work.
The typical oligonucleotide length for gene synthesis starts at 40 bases. 2 Oligonucleotides with sequences of more than 80 bases may contain errors with high probability. 3 So the most commonly used oligonucleotides for gene synthesis contain 50–70 bases. A typical cycle in β-cyanoethylphosphoramidite chemistry takes from 5 to 10 min, so the synthesis of one oligonucleotide takes about 8 h. 4 Multicolumn or array synthesizers allow us to perform multiple syntheses simultaneously, which allows us to run synthetic processes in parallel and prepare oligonucleotides for a list of genes.5–7 Although microchip-based oligonucleotide synthesis is very promising for reducing waste and the cost of the process, it is not widely used for gene synthesis today because of its high complexity and total synthesis scale, which is very low for most gene synthesis applications.8,9
Classical cleavage from controlled pore glass (CPG) support and deprotection with a concentrated ammonia solution take a lot of time (8–16 h), which is comparable to the duration of the synthesis procedure. 10
The AMA method (1:1 mixture [v/v] of aqueous ammonium hydroxide and aqueous methylamine) speeds up the deprotection process substantially (from 8–16 h to 5–15 min) but requires dCAc instead of the commonly used dCBz phosphoramidite. 11
The next stage of gene synthesis is oligonucleotide purification. The quality and purity of oligonucleotides have a significant influence on the number of errors in the target sequence. 12 Many gene synthesis protocols rely on the purification of full-length oligonucleotides from capped, truncated variants by polyacrylamide gel electrophoresis (PAGE); this provides a simple and relatively inexpensive method for synthesizing long, accurate DNA sequences, 13 but the procedure takes approximately 1 week. PAGE purification can be characterized as a low-throughput, labor-intensive process that is difficult to automate. It should also be noted that PAGE cannot separate full-length oligonucleotides from single-deletion variants, which are the main cause of errors in any gene assembly process. 8
High-performance liquid chromatography (HPLC)-based oligonucleotide purification (ion-pairing, reverse-phase [RP] or ion-exchange variants) is possibly the most effective method to obtain oligonucleotides of the highest purity, but this approach requires expensive equipment and materials and takes a lot of time, since each oligonucleotide should be purified individually.14–18
“Trityl ON” purification is an alternative method for target oligonucleotide purification. 12 This technique utilizes the hydrophobicity of the dimethoxytrityl (DMT)-protecting group, which protects the 5′-hydroxyl group during coupling steps. 12 DMT is not removed after the last synthetic cycle, which allows us to differentiate full-length oligonucleotides from truncated by-products capped with acetylation during the synthesis. 12 This hydrophobicity differentiation of the target sequence from impurities may be used for HPLC separation or solid-phase extraction (SPE). 12 SPE is a fast and simple method that usually gives a yield of about 95% for a sequence length of about 20 bases. 12
The need to purify each oligonucleotide turns gene synthesis into a very laborious procedure.
The thermodynamically balanced, inside-out (TBIO) PCR-based gene synthesis approach was introduced in 2003 19 and proved to be one of the most high-fidelity gene synthesis methods available. The error rate for the TBIO synthesis method ranges from 0% to 0.3%, which is substantially lower than that obtained by the thermodynamically balanced conventional (TBC) method.20–22 The TBIO approach has a unique synthesis design and assembly scheme: half of the oligonucleotides for synthesis represents a sense strand and the other an antisense strand. 19 So during PCR cycles oligonucleotide fragments assemble in pairs from the center to the edges (inside-out) with every additional cycle. In order to reduce the formation of shortened assembly products, fragments are added in a concentration gradient with the center pair having the lowest concentration and the terminal oligonucleotide pair having the highest concentration. 19
The exact concentration for each pair of oligonucleotides is very important for the correct assembly and purity of the synthesis product. The need to thoroughly prepare the concentration gradient for oligonucleotides creates certain difficulties for this assembly scheme: every oligonucleotide should be purified separately, and the concentration must be measured carefully based on the extinction coefficient for each oligonucleotide.
The aim of this work was to develop the optimum strategy for oligonucleotide preparation in the TBIO gene assembly approach, which could minimize time and material consumption.
We tested a number of silica-based RP-SPE sorbents for their ability to purify long oligonucleotides with the “DMT-ON” method. This allowed us to determine an important criterion—the pore size of the sorbent—to select silica-based RP-SPE sorbents that allowed for the best quality of long oligonucleotides. In our approach we used the SPE sorbent with the best performance in minimizing time and material consumption and demonstrating excellent results in TBIO assembly. We also showed that ZipTip microextractors with a pore size of 200 Å, which are commonly used for protein and peptide desalting in proteomics experiments, have a capacity that perfectly matches the oligonucleotide amount required for gene assembly (40–60 pmol). The proposed approach is suitable for error-free gene assembly from oligonucleotides with lengths up to 65-mer, as confirmed by our experiments.
Materials and Methods
Materials
DMT-dABz, DMT-dGiBu, DMT-dCBz, and DMT-dT phosphoramidites; dichloromethane; acetic anhydride; lutidine; 1-methylimidazole; and trifluoroacetic acid (TFA) were from Sigma-Aldrich (St. Louis, MO). HPLC-grade acetonitrile (MeCN) with a low water content (0.002%) was from Fisher Chemical (Waltham, MA). 5-(Ethylthio)-1H-tetrazole and oxidizer reagent for phosphoramidite synthesis were from Emp Biotech (Berlin, Germany). DreamTaq DNA polymerase and dNTP mix were from Thermo Scientific (Waltham, MA). Thirty-two percent ammonia solution was from Roth (Karlsruhe, Germany). Triethylamine was from Alpha Aesar (Kandel, Germany). 5′-DMT-dABz-, 5′-DMT-dT-, 5′-DMT-dCAc-, and 5′-DMT-dGiBu-CPG with a pore size of 1000 Å and loading capacity of 42–45 μmol/g were from BioAutomation (Irving, TX). The BigDye Terminator v3.1 cycle sequencing kit from Applied Biosystems (Waltham, MA) was used for sequencing.
General Methods
A260 and electronic spectra for oligonucleotide solutions were recorded with a Carry 5000 UV-Vis-NIR spectrophotometer from Agilent (Santa Clara, CA), and NanoDrop UV-Vis spectrophotometer from Thermo Scientific (Waltham, MA). Confirmation of the synthesized product structure was performed with electrospray ionization (ESI) mass spectrometry. Oligonucleotide mass spectra were registered with an LCQ-Fleet ion-trap mass spectrometer (Thermo Scientific) in the negative ESI mode (ESI–) with a mixture of 80% 5 mM NH4OAc, pH 7.5, and 20% MeCN as a mobile phase. High-resolution mass spectra were registered with a Q-TOF 6550 high-resolution mass spectrometer (Agilent) in the ESI– mode with a mixture of 80% 5 mM NH4OAc, pH 7.5, and 20% MeOH as a mobile phase. Analytical HPLC analysis with a diode array detector (DAD) and an MS detector was performed on an Agilent 1290 HPLC system according to the application note “Characterization of Synthetic Oligonucleotides Using Agilent LC/MS Systems” from Agilent. Mass spectra of oligonucleotides were analyzed in the presence of target sequences, truncated sequences, and not fully deblocked sequences with software developed by the authors. Denaturing urea PAGE of oligonucleotides was performed with Mini-PROTEAN II Electrophoresis Cell from Bio-Rad (Hercules, CA).
Design of Oligonucleotides for the TBIO Method of PCR-Based Gene Synthesis
In order to design oligonucleotides for gene synthesis, we used DNAWorks software from https://hpcwebapps.cit.nih.gov/dnaworks.23,24
The input shown is from the EGFP amino acid sequence:
The EGFP nucleotide sequence was altered to obtain optimized codon usage percentages for K12 protein expression strains of Escherichia coli for 50-mer and 65-mer oligonucleotide designs as shown below.
The EGFP nucleotide sequence obtained for the 50-mer design is as follows:
The EGFP nucleotide sequence obtained for the 65-mer design is as follows:
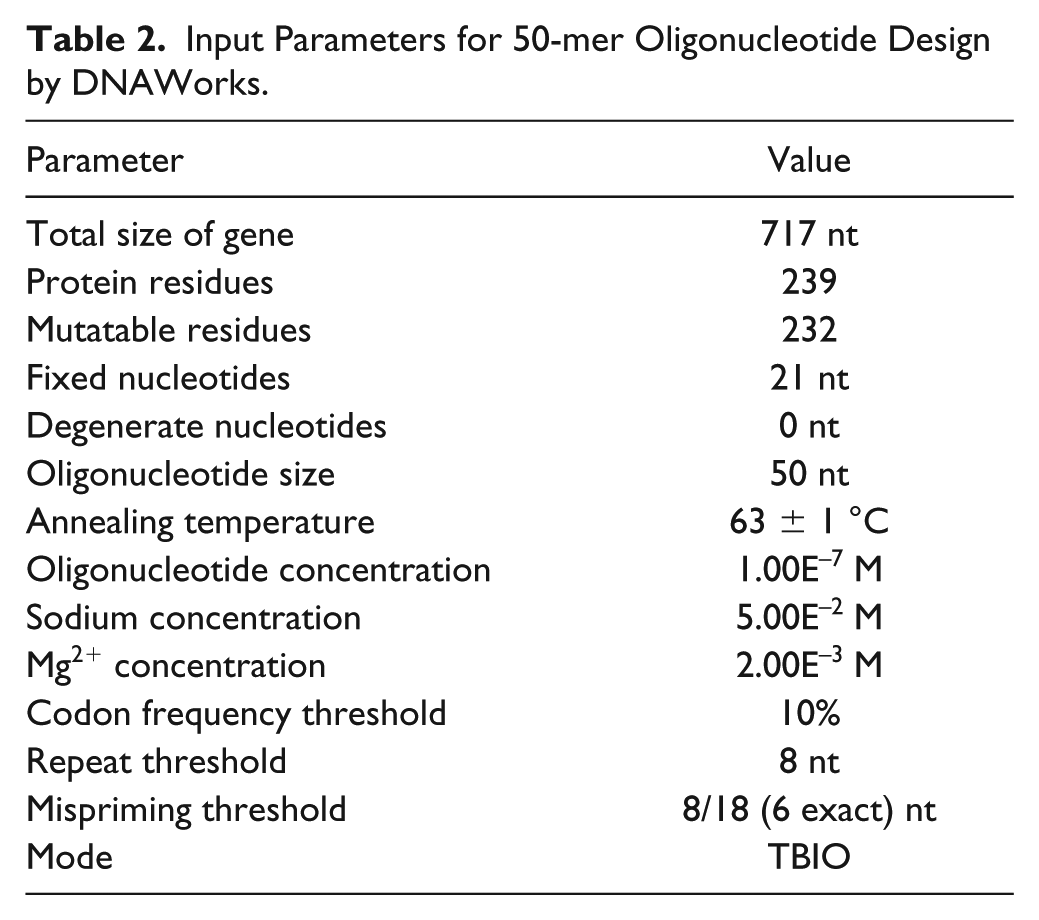
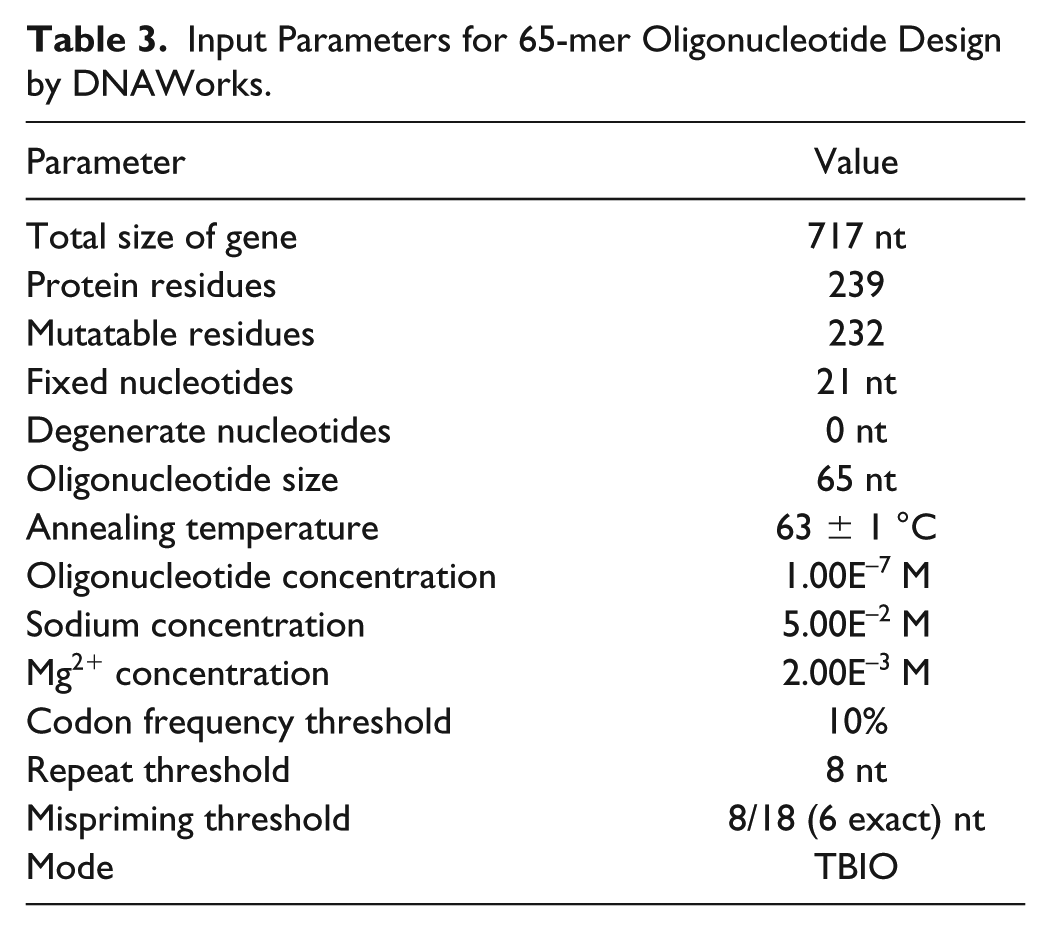
The input parameters for DNAWorks are listed later for 50-mer and 65-mer building blocks (see Tables 2 and 3 ).
All work on the optimization of gene assembly conditions, as well as SPE procedures, was performed with the 50-mer design. The 65-mer design was used to confirm the performance of the proposed approach for longer oligonucleotides, as well as to prove correct assembly by Sanger sequencing.
Oligonucleotide Synthesis
Oligonucleotides were synthesized on a DNA synthesizer H32 (K&A, Germany) on a 100 nm scale with a 1000 Å wide pore CPG. The concentration of phosphoramidite was 0.05 M. The oxidation reagent was 0.05 M I2 in a pyridine/water (90:10) mixture. The detritylation reagent was 2% trichloroacetic acid in dichloromethane. The capping reagents used were acetic anhydride/lutidine/tetrahydrofuran (THF) and 1-methylimidazole in THF.
Deprotection and Cleavage
CPG with frits was extruded from the column to a 1.5 mL chromatographic vial, cooled in a freezer (–20 °C), and 0.5 mL of 32% aqueous ammonia was added to each vial, which was immediately closed tightly with a polytetrafluoroethylene (PTFE)-coated silicon septum. Vials were transferred to a conventional gas-air thermostat set at 55 °C. After 16 h the vials were cooled in a freezer for 15 min, and after that the vials were opened and aqueous ammonia was evaporated with a vacuum concentrator ScanVac (LaboGene, Denmark).
Preparation of Individually Purified Oligonucleotides for TBIO Gene Synthesis
To each vial with evaporated crude synthesis product 0.5 mL of triethylamine acetate (TEAAc) (pH 8.0) buffer was added and remained for 5 min with mixing. After that, each oligonucleotide was purified by SPE on a Supelclean ENVI-18 Supelclean 1cc (100 mg) column according to the DMT-ON procedure described in Gilar and Bouvier, 12 with a minor modification. Briefly, the column was conditioned with three bed volumes of MeCN and three bed volumes of 100 mM TEAAc buffer (pH 8.0), and 0.5 mL of crude oligonucleotide solution in 100 mM TEAAc buffer (pH 8.0) was slowly pulled through the column. Afterward, the column was washed with three bed volumes of 11% MeCN in 100 mM TEAAc buffer (pH 8.0). Detritylation was performed with three bed volumes of 2% TFA, with subsequent washing with three bed volumes of 100 mM TEAAc buffer (pH 8.0). The column was dried and oligonucleotides were eluted with three bed volumes of 20% MeCN in water. The solution obtained was diluted with water 10 times and A260 was measured by a NanoDrop spectrophotometer. The concentration was calculated with an extinction coefficient obtained by the nearest-neighbor method.
Proposed Method of Oligonucleotide Preparation for TBIO Gene Synthesis
To each vial with evaporated crude synthesis product 0.5 mL of 100 mM TEAAc buffer (pH 8.0) was added and remained for 5 min with mixing. Afterward, the nonsoluble material was allowed to settle for 5 min and solution absorbance at 260 nm was detected with a NanoDrop spectrophotometer using the previous 10-fold dilution with buffer. In order to calculate extinction coefficients for oligonucleotides, we used the nearest-neighbor method. 25 The target oligonucleotide concentration was detected under the assumption that the synthesis yield for every stage has the same value:
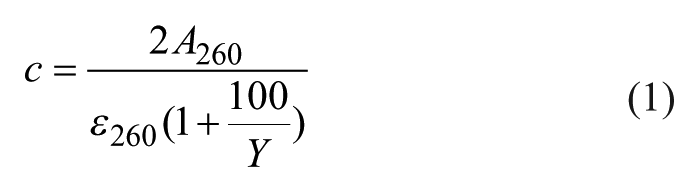
where A260 is the solution absorption at 260 nm corrected to a 1 cm optical pathway; ε260 is the extinction coefficient for the oligonucleotide, calculated by the nearest-neighbor method (mM–1cm–1); and Y is the total synthesis yield as detected by online or offline trityl monitoring during parallel synthesis (%).
After that, we calculated the volumes of each crude synthesis product solution in order to perform one PCR in a 50 µL volume and multiplied these volumes by the volume correction coefficient, which takes into account the need to minimize volume errors and the expected number of PCR repeats. In this work, we used a volume correction coefficient of 100. Oligonucleotide crude solutions in TEAAc buffer were mixed according to the script output and SPE DMT-ON purification was performed as described earlier.
ZipTip Oligonucleotide Purification for TBIO Gene Synthesis
A mixture of crude DMN-ON oligonucleotides was prepared as described earlier. Using a 10 µL pipette with an extraction tip, we completed the following procedures:
Fill in and discard on a filter paper 10 µL of MeCN (repeat three times).
Fill in and discard on a filter paper 10 µL of 100 mM TEAAc buffer with pH 8.0 (repeat three times).
Fill in and discard on the same tube a crude mixture of oligonucleotides (repeat five times).
Fill in and discard on a filter paper 10 µL of 100 mM TEAAc buffer with pH 8.0 (repeat three times).
Fill in and discard on a filter paper 10 µL of 5% MeCN solution in 100 mM TEAAc buffer with pH 8.0 (repeat three times).
Fill in and discard on a filter paper 10 µL of 2% TFA solution in water (repeat three times).
Fill in and discard on a filter paper 10 µL of 100 mM TEAAc buffer with pH 8.0 (repeat three times).
Fill in and discard to a PCR tube 2 µL of 20% MeCN solution.
After that procedure, the PCR master mix, containing water, buffer, dNTPs, and DreamTaq polymerase, was transferred to the same tube, mixed with purified oligonucleotide mixture, and placed in a thermocycler. Under the described conditions, the MeCN concentration does not exceed 1% and does not reduce polymerase activity.
PCR-Based Gene Assembly and Target Sequence Amplification
Enzyme reactions were carried out in 200 μL of sterile thin-walled PCR tubes with a Tercyc multiblock thermocycler from DNA-Technology (Russia). The reaction conditions were almost the same (except for annealing temperature) for both gene assembly and subsequent amplification: incubation at 95 °C for 2 min, 25 cycles (94 °C for 15 s, 58 °C [62 °C for amplification of target sequence] for 30 s, 68 °C for 45 s), 68 °C for 5 min, and 10 °C for storage. In order to increase the specificity of the PCR during bidirectional elongation of the central fragment, the annealing temperature was readjusted from 58 to 62 °C. Each 50 µL of PCR mix contained 5 µL of 10× DreamTaq PCR buffer (proprietary formulation that includes MgCl2 at a concentration of 20 mM), 0.4 µL of 50× dNTP mix (25 mM each), 0.25 µL of DreamTaq DNA polymerase (5 U/µL), the calculated amount of oligonucleotide mix, and water to 50 µL. To minimize the possibility of pipetting errors, the PCR master mix was prepared by mixing water, buffer, dNTP, and enzyme. The concentration of external primers S12–A12 during amplification of the gene assembly product was 0.2 µM each. Reaction products were analyzed by horizontal electrophoresis in 2% agarose gel.
Cloning and Sequencing
A blunt-ended EGFP gene assembled from 65-mer oligonucleotides was cloned into the pJET1.2/blunt cloning vector with the commercially available CloneJET PCR Cloning Kit (Thermo Scientific) according to the manufacturer’s sticky-end cloning protocol without electrophoretic purification. The product of the ligation was transformed into DH5α competent cells (E. coli), prepared according to Chung and Miller. 26 Sixteen colonies from the Petri dish were analyzed by colony PCR in order to screen colonies with an insert of appropriate length. Plasmid DNA was further sequenced with the pJET1.2 forward and reverse sequencing primers supplied with the kit.
Results and Discussion
Selection of SPE Sorbent for DMT-ON Oligonucleotide Purification
We tested a number of silica-based RP-SPE sorbents ( Table 1 ) with different modifiers from different suppliers (Macherey-Nagel, Agilent, and Supelco Analytical). The extraction procedure was the same for every sorbent.
RP-SPE Sorbents Tested for Their Ability to Purify Long Oligonucleotides.
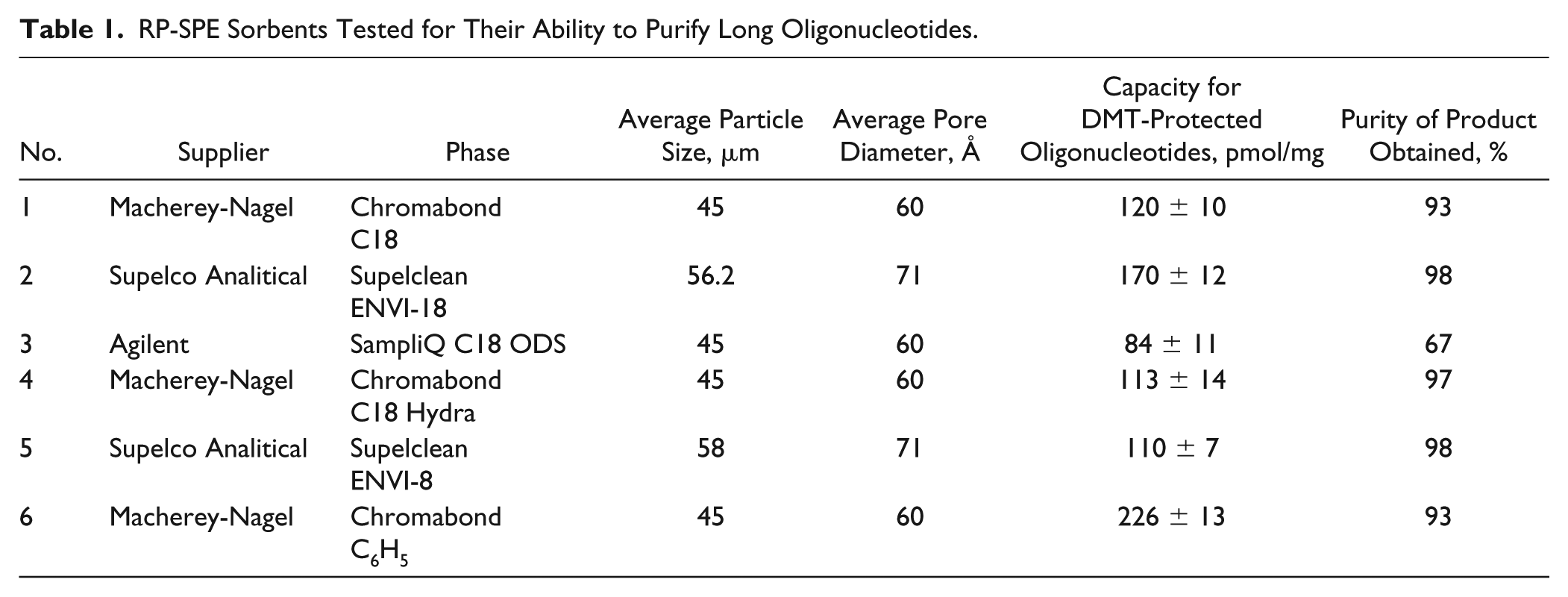
We expected to obtain good results for phenyl-modified silica, concerning the aromatic nature of the DMT protection group, and limited selectivity of the phenyl-modified silica to aromatic compounds. However, experimentation showed that although this sorbent type has a high binding capacity, the quality of the final product was worse than that observed for other sorbents. Octadecyl-modified silica from a number of Agilent SPE columns practically does not bind DMT-ON oligonucleotides, as shown in Figure 2 , and we could not find an explanation for this. A high-purity final product was obtained using a C18 Hydra from Chromobond, but that sorbent has a low binding capacity. Two C18-SPE sorbents from Chromobond and Supelco Analytical showed various results, and the main difference among them was pore size: 60 μm for Chromobond and 71 μm for Supelclean ENVI-18.
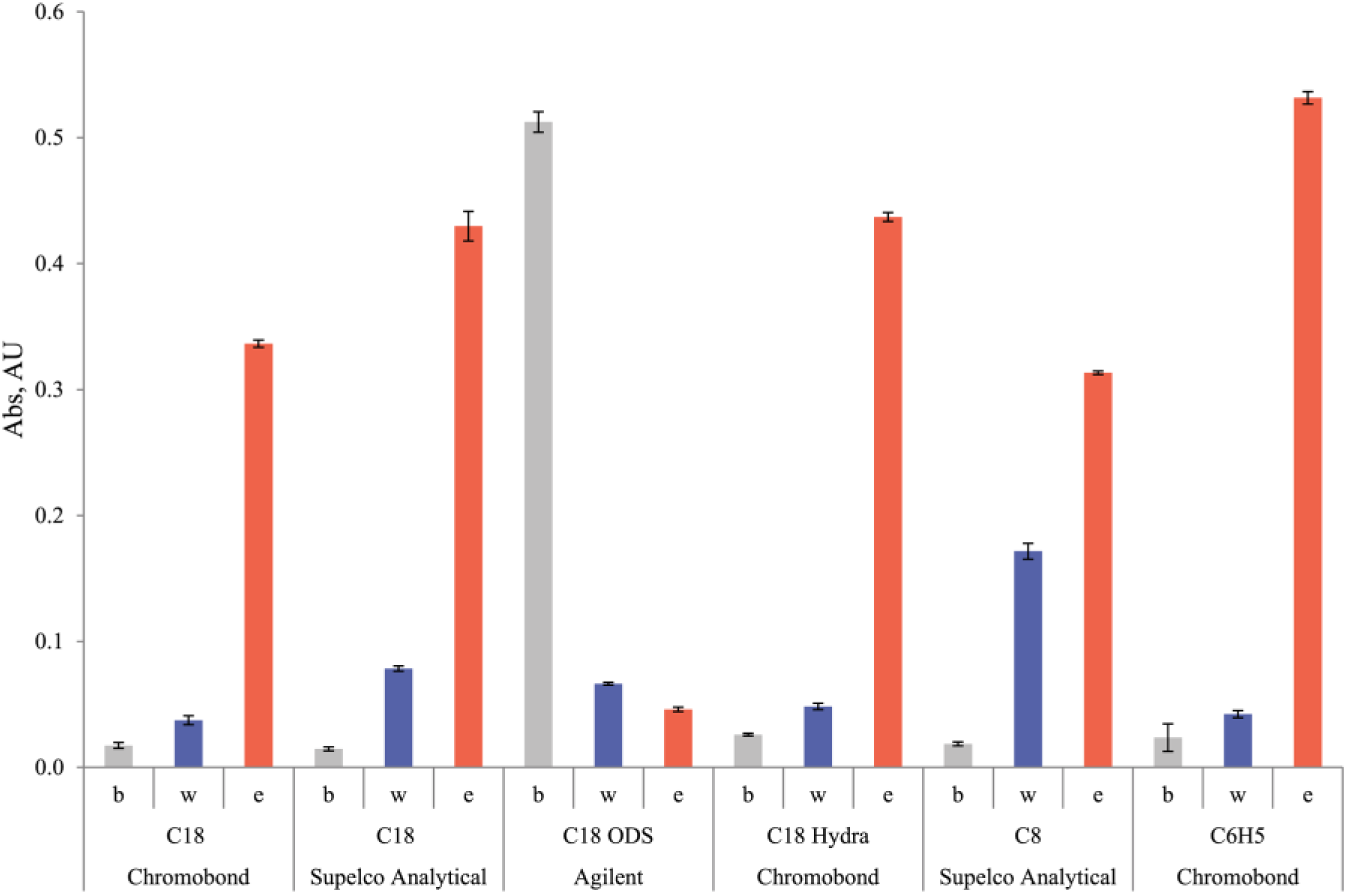
Binding of 45-mer DMT-ON oligonucleotides to silica-based RP-SPE sorbents: A260 at different stages of DMT-ON oligonucleotide purification, illustrating the different binding capacities of the sorbent and phases. The A260 of the initial solution was 0.8 (optical length 0.1 cm). b, absorption of oligonucleotide solution in 100 mM TEAAc buffer (pH 8.0) that was passed through the SPE column; w, washing with 11% MeCN in 100 mM TEAAc buffer (pH 8.0); e, elution with 20% MeCN in water.
The properties of the sorbents and their manufacturer, capacity, and purity of product obtained are listed in Table 1 .
The best results were observed for Supelclean ENVI-18 cartridges. We tested this cartridge not only on individual oligonucleotides 40–65 bases in length, but also on oligonucleotide mixtures prepared for the TBIO assembly procedure. The possible formation of hydrogen bounds between complementary oligonucleotides in the mixture may result in loss of binding ability, but in TBIO oligonucleotide mixtures such possibility is minimized—duplex formation can occur only between two oligonucleotides in the gene center. It should be mentioned, however, that if some possibility of unspecific complex formation between DMT-OFF-shortened oligonucleotides and full-length DMT-ON oligonucleotides exists, then extraction is desirable to carry out under elevated temperatures, which can be easily achieved by using a solid-state thermostat.
Since we used a silica-based SPE sorbent in this work, it was not possible to use the oligonucleotide solution immediately after deprotection, and therefore an evaporation stage was required. In a future work, we will try to adapt fast deprotection techniques to the current approach.
Oligonucleotide Purification with ZipTip Microextraction
We tested the capacity of ZipTip microextractors with respect to 40-mer to 60-mer oligonucleotides and found that the value obtained perfectly corresponds to the amount required for one to three PCR assembly reactions.
The high-resolution ESI– mass spectra and results of PAGE of S12 and A06 oligonucleotides are provided in Supplemental Material ( Suppl. Fig. S1–S3 ).
Oligonucleotide Design
The oligonucleotide design for the EGFP gene that was performed with DNAWorks (
Table 2
) is shown in
Figure 3
, and a list of oligonucleotides is given in
Input Parameters for 50-mer Oligonucleotide Design by DNAWorks.
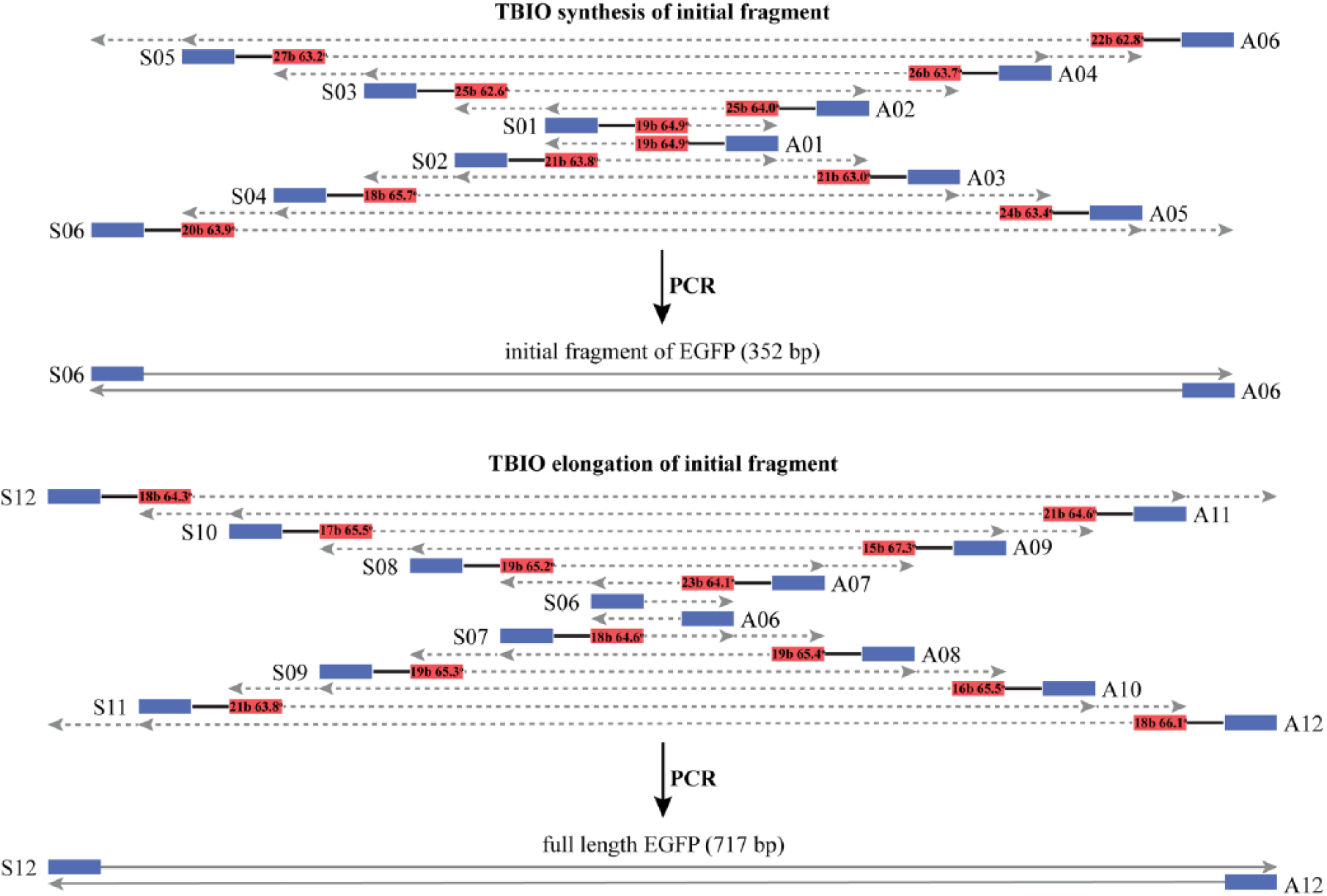
Oligonucleotide design for EGFP gene synthesis. Overlapping segment length and Tm shown on corresponding sites.
The minimum Tm for overlapping segments was 62.6 °C (S02–S03 overlap) and the maximum Tm was 67.3 °C (A08–A09 overlap). The proposed scheme allowed us to perform PCR with an annealing temperature of 58 °C, and it works well for the assembly of central fragments from six pairs of oligonucleotides (SA01–06). Regarding a bidirectional elongation at 58 °C, the annealing temperature gives several nonspecific products, so we had to raise it to 62 °C in order to improve the clarity of the strip on the electrophoretic gel (see Fig. 6 ).
It is known that usually the number of errors in synthetic gene sequences increases with increasing oligonucleotide sequence length. 1 So in order to demonstrate the effectiveness of the proposed approach, we tested the assembly from the 65-mer design with DNAWorks ( Table 3 and Suppl. Table S2 ) that was targeted for assembly from 65-mer oligonucleotides.
Input Parameters for 65-mer Oligonucleotide Design by DNAWorks.
Proposed Approach
In order to avoid time- and material-consuming work on purification, concentration, determination of concentration, and other processes for oligonucleotide preparation for the TBIO assembly, we developed a workflow from crude oligonucleotide synthesis product to PCR assembly that substantially shortens the procedure and could be easily automated ( Fig. 4 ). For accurate work, the DMT-ON synthesis mode and online trityl monitoring are required. If the latter is not available, the approximate step yield may be measured for current reagents by offline trityl assay before and after the synthesis of model sequences. The obtained values can be used for calculation of the theoretical yield for sequences with known lengths.
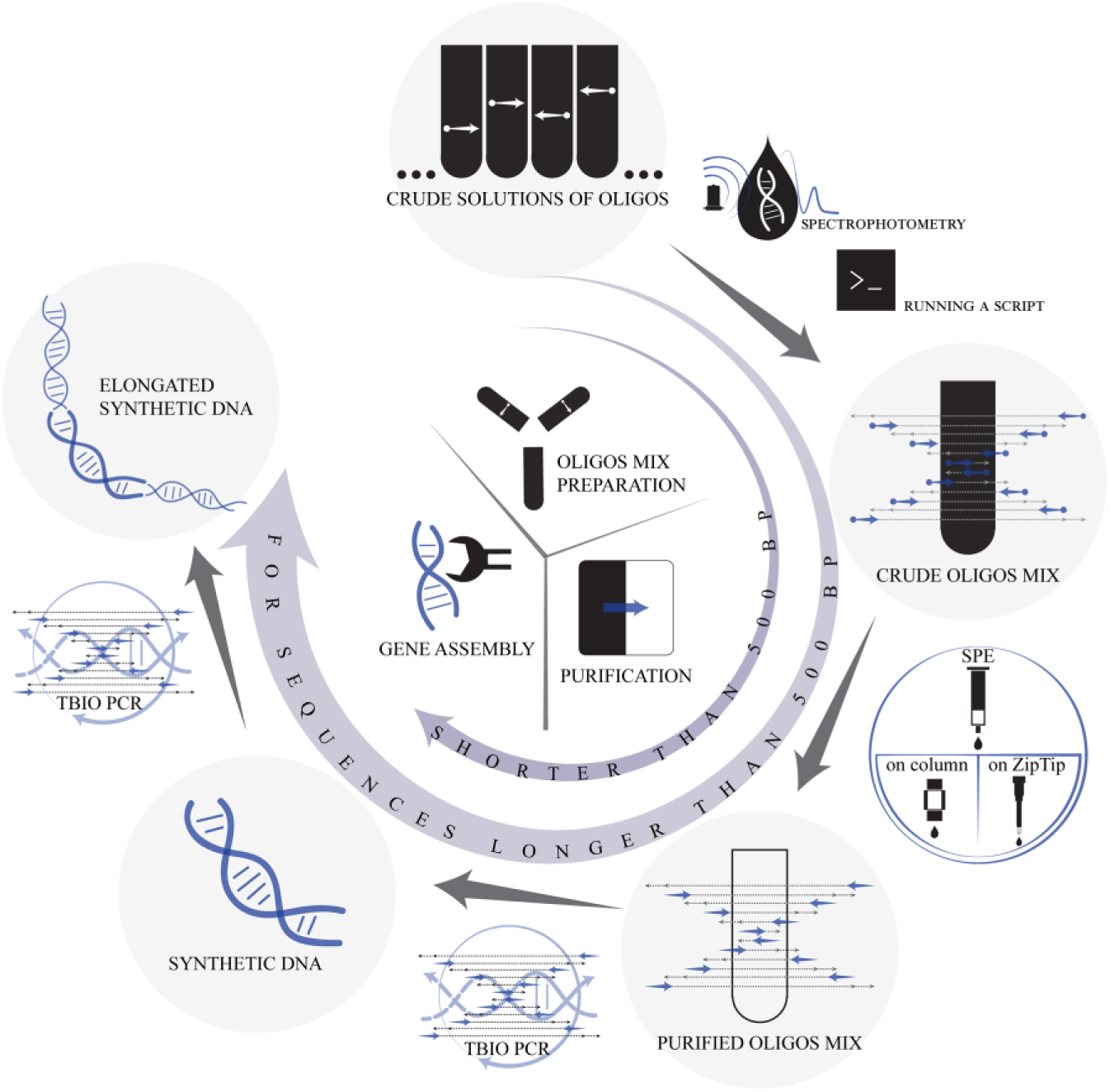
Proposed workflow for oligonucleotide preparation for the TBIO scheme.
Our workflow includes the following steps:
Dissolution of crude oligonucleotides in 100 mM TEAAc buffer (pH 8.0) (~0.5 min for 1 sample, 5–15 min for 12–24 oligonucleotides)
Measurement of A260 for crude oligonucleotide solution on NanoDrop spectrophotometer (0.5–1 min per 1 sample, 10–20 min for 24 oligonucleotides)
Calculation of TBIO scheme with algorithm realized on Python script (0.5–1 min)
Mixing of crude solutions according to calculated output data (5 min)
DMT-ON RP-SPE purification of oligonucleotide mix (1 extraction per approximately 500 base pairs [bp] of target sequence, 15 min for 1 extraction)
PCR assembly with SPE eluate from the previous stage (2 h for standard PCR procedure)
The procedure for manual oligonucleotide preparation takes approximately 1 h, and if PCR time is added, the total time required to obtain the amplified target gene from deprotected oligonucleotides will be 3–5 h.
Since we used silica-based SPE sorbent in this work, it was not possible to use the oligonucleotide solution immediately after deprotection, so an evaporation stage was required. Although a concentrated ammonia solution evaporates quickly, in a future work we will try to adapt this approach to fast deprotection techniques (AMA) and to polymeric sorbents that have stable high pH values.
Currently we are not sure about the applicability of the proposed oligonucleotide preparation approach in other TBC-based gene synthesis methods, because of the possible tendency to form large duplex molecules, which may create spatial limitations for interaction of the DMT group with the sorbent.
The significance of the approach proposed in this work is that we replaced the need to purify individual oligonucleotides with just one purification procedure per approximately 500 bp of duplex DNA (depends on the oligonucleotide length). So in the model example for the EGFP gene of 717 bp we synthesized 22 overlapping fragments with a length of 50 bases and 2 with lengths of 39 and 49 bases and performed just two SPE purification procedures. In order to calculate the TBIO scheme, we developed Python script that uses a text file with input data (oligonucleotide name, oligonucleotide sequence, total synthesis yield, and A260 for crude synthesis product solution) and input parameters (PCR volume and number of reactions). The output of the script is a table with volumes of crude oligonucleotide solutions, which need to be mixed to perform copurification and further TBIO PCR assembly.
In order to eliminate the evaporation stage after the elution step, we minimized the SPE column and elution volume. We used empty DNA synthesis columns (LGC BioSearch Technologies) with polyethylene frits (1/8 inch × 0.163) that can hold 10–40 mg of silica-based sorbent.
The presence of MeCN in the PCR mixture at a concentration of 2% does not interfere with the enzyme reaction, so the total volume of the oligonucleotide mix solution should be about 5 µL. This implies that the oligonucleotide mix can be used immediately after elution from the SPE column without evaporation.
Results of Gene Assembly and Modification of Original TBIO Scheme
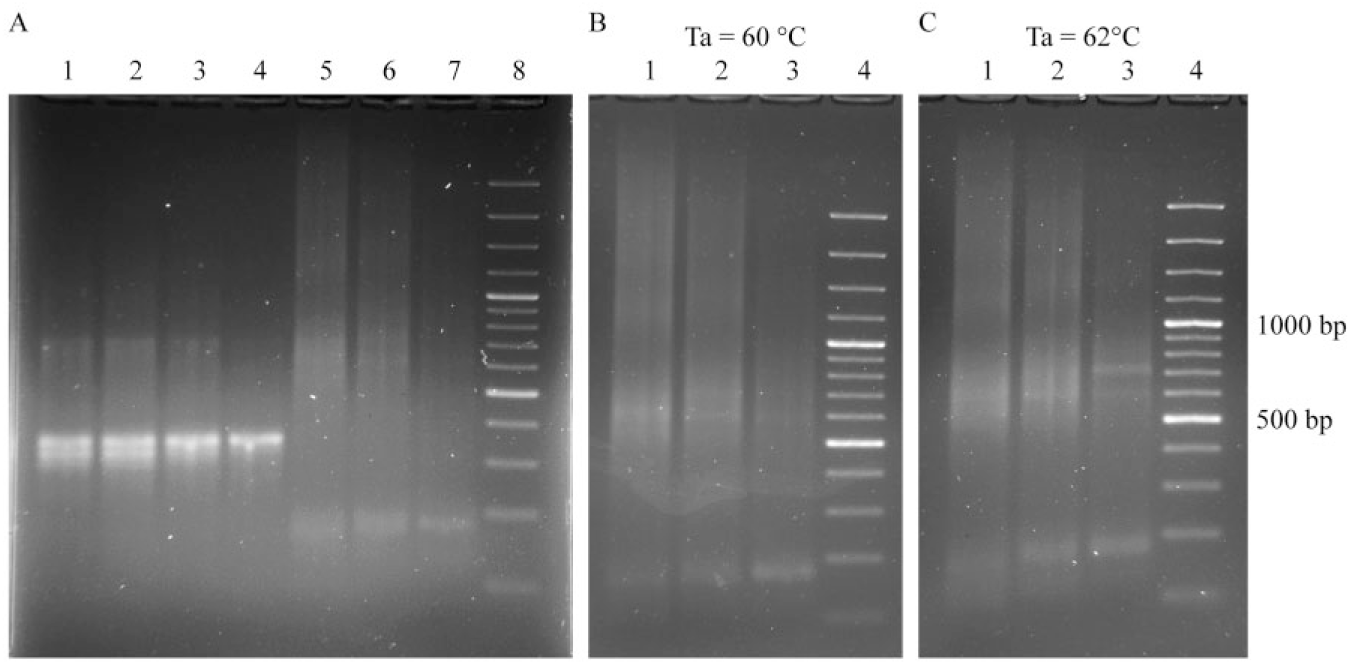
As we can conclude from Figure 5A , there is no difference between PCR product obtained from individually purified oligonucleotides (lane 1) and that obtained from copurified oligonucleotide mix according to the proposed scheme (lane 2). In order to achieve a clearer PCR product, we tried to modify the oligonucleotide concentration gradient compared with the original TBIO scheme. 19
(
Leaving the concentration of external oligonucleotides unchanged, we reduced the concentration of inner oligonucleotides twofold ( Fig. 5A , lane 3) and fivefold ( Fig. 5A , lane 4). The tendency of the PCR product to form a narrow band from the original to the last scheme confirms that modification of the original gradient, which consists of a fivefold decrease of inner oligonucleotide concentration, can yield a nearly pure PCR product, which corresponds to DNA with a length of 352 bp. An attempt to elongate the obtained gene fragment with the next six pairs of oligonucleotides under the same conditions led to unspecific PCR product formation ( Fig. 5 , lanes 5–7). So we performed 2° step optimization of the annealing temperature for PCR until a narrow band for the full-length gene was achieved in our scheme with a fivefold decrease of inner oligonucleotides ( Fig. 5B , 5C ).
Along with the target product, the originally proposed TBIO scheme with gradient 40, 60, 80, 100, 120, and 200 nM gives discrete blurred bands that correspond to the molecular weight of the dimer, but we did not observe bands corresponding to trimers. It was reported earlier that high-molecular-weight species result from equilibrium mixtures of noncovalent cross-hybridization between monomers that form multimers. 19 We proposed a model ( Fig. 6 ) according to which high-molecular-weight species are the result of heteroduplex formation from a target sequence and unfinished sequences. Such heteroduplexes have long sticky ends that contribute to dimer formation.
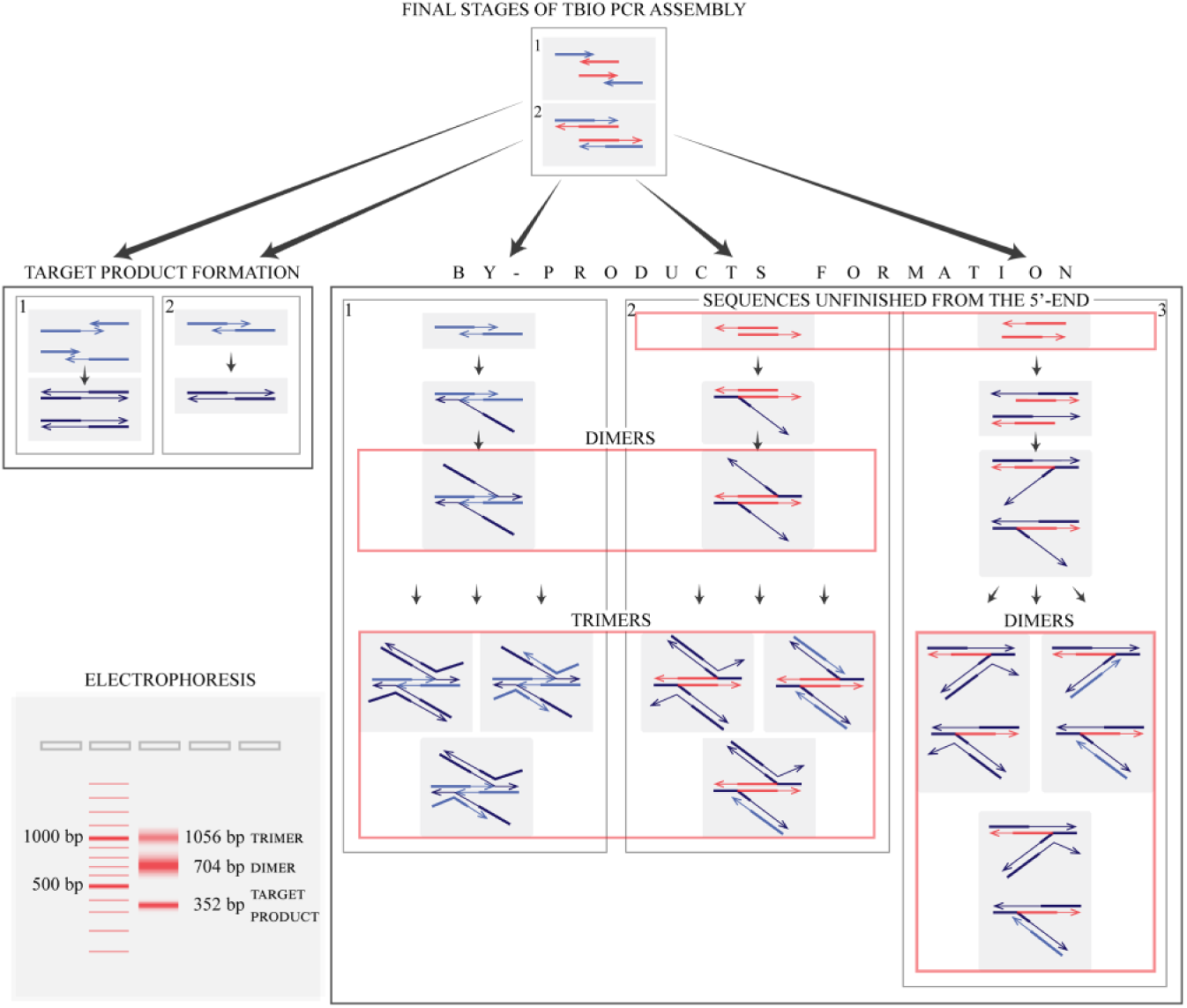
Proposed mechanism, which explains the formation of electrophoretic bands corresponding to supramolecular structures formed from the target sequence and shortened by-products.
Primers (light blue arrows) anneal to both strands of the gene fragment (red arrows) in the first stage. Then DNA polymerase elongates gene fragment strands as well as primers in the second stage. As a result, there are two strands unfinished from the 5′-end (red longer arrows) and from the 3′-end (light blue arrows). The former can form only by-products as DNA polymerase cannot elongate 5′-ends. The latter can give rise to target product (dark blue arrows) or by-products in the case of a high amount of unfinished 3′-end strands versus a low amount of primers.
Target product is synthesized in two ways. First, primers remaining from the first stage can anneal to 3′ unfinished sequences. According to the original concentration scheme (40, 60, 80, 100, 120, and 200 nM), 120 nM of 3′ unfinished strands is formed from 120 nM of gene fragment and 120 nM of primers (whose concentration is 200 nM). So 80 nM (200 nM – 120 nM) of primers remains to elongate the 120 nM of 3′ unfinished fragments with a formation of target DNA. Thus, 40 nM (120 nM – 80 nM) of 3′ unfinished sequences cannot be elongated in such a way. The excess 3′ unfinished ones can take part in two types of events: in a second type of target product formation and in by-product formation. In the former case, 3′-ends of sense and antisense strands of 3′ unfinished fragments anneal to each other and DNA polymerase elongates both strands. On the contrary, there are two sticky ends of the joint structure in the latter case. That is why already formed target sequences can anneal with complementary ends of that structure, which leads to dimer formation. Different trimers can be formed by interactions between complementary strands of target sequences as part of a dimer and free target or 3′ unfinished strands.
It is possibly to get rid of by-products that come from 3′ unfinished sequences by correction of the concentration scheme. Even though the concentration of the outer primers is two times higher than the previous primers’ concentration, there will be no excess of 3′ unfinished DNA that can form such dimers and trimers. This assumption is in accordance with our experimental data ( Fig. 5A ), which show that a five times decrease of inner oligonucleotide concentration allows us to get rid of dimers.
By-products can be formed from 5′ unfinished strands. The sticky ends of two joined 5′ unfinished strands can also interact with two strands of the target sequence (the second way of by-product formation). In addition, individual 5′ unfinished ones can anneal to a target product strand and then another strand of the latter joins one sticky end of that structure (the third way). The mechanism of trimer formation is the same as that described above. Tetramers, pentamers, and so on, cannot be formed because of steric reasons.
It is impossible to dispose of formation by-products from the 5′ unfinished fragments as they appear in the same amounts as the 3′ unfinished ones that are the only source of target product. But we can partially decrease contamination of the PCR product by such structures using the same modification of the original scheme as it reduces the amounts of 3′ unfinished DNA.
Advantages of Oligonucleotide Extraction with ZipTip
We paid attention to ZipTip microextractors when trying to find an RP sorbent with a large pore size in order to purify long oligonucleotides. Although the main application of these microextractors is the desalting of peptides and proteins during sample preparation for matrix-assisted laser desorption ionization time-of-flight (MALDI-TOF) mass spectrometry, we tried to use them for other purposes.
We found that the capacity of ZipTip perfectly fits to scale which is required for TBIO gene synthesis (40–70 pmol per reaction). So usage of a concentrated crude oligonucleotide mixture for SPE with ZipTip extractors does not require correction of oligonucleotide concentrations after extraction. Also, large pore sizes of ZipTip allow the use of specifically long oligonucleotides in this approach.
We also discovered that a fivefold decrease in the internal oligonucleotide concentration did not work when ZipTip extraction was used ( Fig. 7A , lane 2; Fig. 7B , lane 2). The reason for this may be that the saturation concentration of the oligonucleotide mix is used. Thus, internal oligonucleotides with low concentrations are being replaced by highly concentrated outside primers. So it is necessary to use an oligonucleotide quantity that is lower than the SPE column capacity in order to apply the modified TBIO scheme with a decreased concentration of internal oligonucleotides.
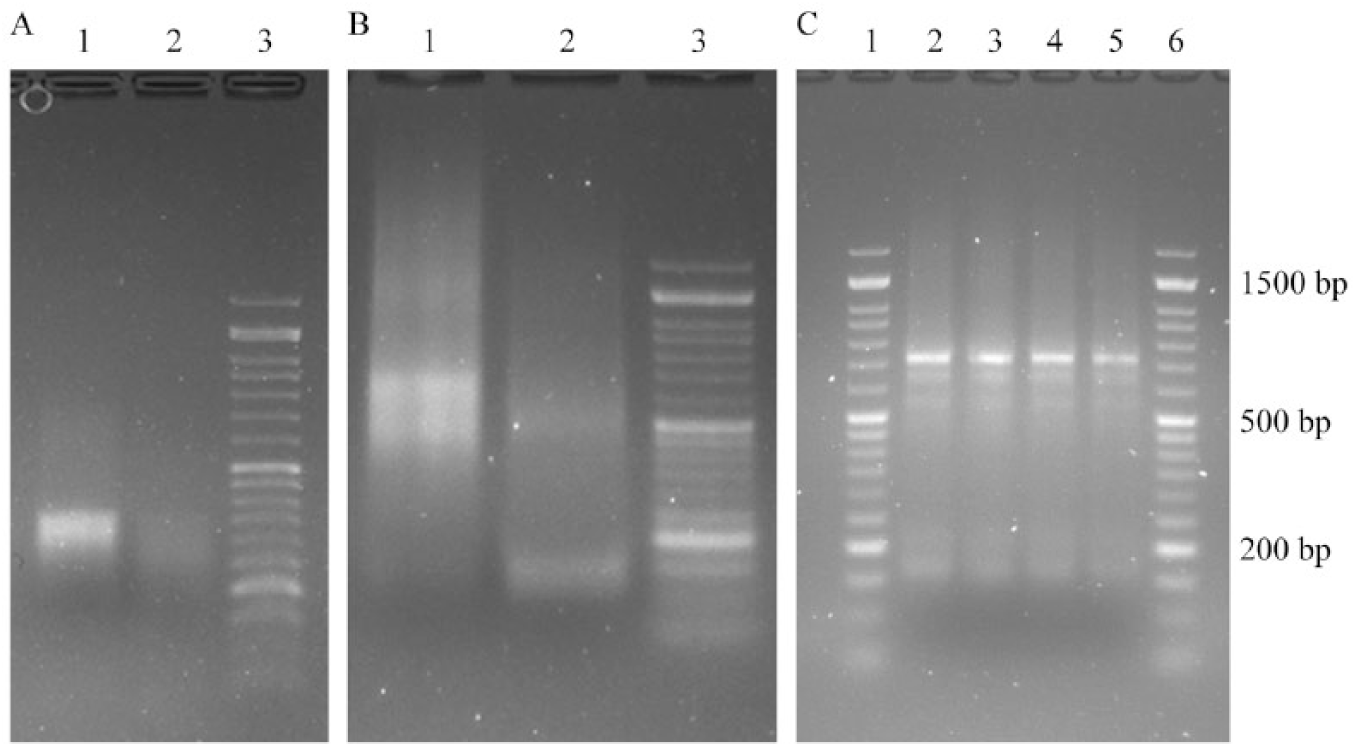
(
Products of PCR after EGFP gene assembly from individually purified oligonucleotides (original scheme), copurified by column SPE (original and modified scheme), and ZipTip mixtures (original scheme) of oligonucleotides were reamplified with oligonucleotides S12 and A12 and showed the same length fragment ( Fig. 7C ). Sequencing of the EGFP gene assembled from 65-mer oligonucleotides and cloned in the pJet2.1 vector confirmed correct gene assembly under the described conditions.
The TBIO gene assembly method developed earlier demonstrated good results in a number of works: it is characterized by a low error rate and gives a relatively pure product after one-pot PCR compared with other TBC methods. In order to avoid time- and material-consuming work on purification, concentration, determination of concentration, and other processes for oligonucleotide preparation for the TBIO method we developed a workflow from the crude oligonucleotide synthesis product to PCR assembly that substantially shortens the procedure and could easily be automated. For accurate work, the DMT-ON synthesis mode and online trityl monitoring are required, but if the latter is not available an approximate step yield may be measured for current reagents before and after synthesis for model sequences by offline trityl assay, and the values obtained can be used for the calculation of the theoretical yield for sequences with known lengths. Our workflow includes the following steps:
Dissolution of crude oligonucleotide synthesis in 100 mM TEAAc buffer (pH 8.0) (~0.5 min for 1 sample, 5–15 min for 12–24 oligos)
Measurement of A260 for crude oligonucleotide solution NanoDrop spectrophotometer (0.5–1 min per 1 sample, 10–20 min for 24 oligonucleotides)
Calculation of TBIO scheme with algorithm realized in Python script (0.5–1 min)
Mixing of crude solutions according to calculated output data (5 min)
DMT-ON RP-SPE purification of oligonucleotide mix (1 extraction per approximately 500 bp of target sequence, 15 min for 1 extraction)
PCR assembly with SPE eluate from previous stage (2 h for standard PCR procedure)
The procedure for manual oligonucleotide preparation takes approximately 1 h, and if PCR time is added, the total time from deprotected oligonucleotides to assembled DNA is approximately 3–5 h.
We proposed usage of the ZipTip microextractor with this scheme and found that the capacity of the ZipTip perfectly fits to scale that required for TBIO gene synthesis (40–70 pmol per reaction). So use of a concentrated crude oligonucleotide mixture for SPE extraction with ZipTip extractors helps us to avoid correction of the oligonucleotide concentration after extraction. Also, the large pore size of ZipTip provides us with the ability to use especially long oligonucleotides in this approach. Modification of the original TBIO oligonucleotide gradient allows us to obtain pure product, which is very important for TBIO bidirectional elongation because of elimination of the gel purification step from the procedure.
Currently, we are not sure about the applicability of the proposed oligonucleotide preparation approach in other TBC-based gene synthesis methods, because of the possible tendency to form large duplex molecules, which may create spatial limitations for interaction of the DMT group with the sorbent.
Supplemental Material
DNA_synthons_assembly_Supplementary_File – Supplemental material for Oligonucleotide Preparation Approach for Assembly of DNA Synthons
Supplemental material, DNA_synthons_assembly_Supplementary_File for Oligonucleotide Preparation Approach for Assembly of DNA Synthons by Aleksei V. Yantsevich, Veronika V. Shchur and Sergey A. Usanov in SLAS Technology
Footnotes
Acknowledgements
The authors acknowledge A. Ivanchik, O. Bokut, A. Karpenko, and N. Bokshits for their excellent technical assistance.
Supplemental material is available online with this article.
Authors’ Note
The raw data that support the findings of this study are available from the corresponding author upon request.
Declaration of Conflicting Interests
The authors declared no potential conflicts of interest with respect to the research, authorship, and/or publication of this article.
Funding
The authors disclosed receipt of the following financial support for the research, authorship, and/or publication of this article: It is funded by a separate project from the National Academy of Sciences of Belarus.
References
Supplementary Material
Please find the following supplemental material available below.
For Open Access articles published under a Creative Commons License, all supplemental material carries the same license as the article it is associated with.
For non-Open Access articles published, all supplemental material carries a non-exclusive license, and permission requests for re-use of supplemental material or any part of supplemental material shall be sent directly to the copyright owner as specified in the copyright notice associated with the article.