
Abstract
Ovarian cancer is the fifth-most lethal cancer among women due to a lack of early detection and late-stage treatment options, and it is responsible for more than 14,000 deaths each year in the United States. Recently, there have been advances in RNA interference therapy, specifically with small interfering RNA (siRNA), to reduce tumor burden for ovarian cancer via gene down-regulation. However, delivery of siRNA poses its own challenges, as siRNA is unstable in circulation, is unable to be effectively internalized by cells, and may cause toxicity in off-target sites. To address such challenges, nanoparticle carriers have emerged as delivery platforms for the biocompatible, targeted delivery of siRNA-based therapies. Several preclinical studies have shown the promising effects of siRNA therapy to reduce chemotherapy resistance and proliferation of ovarian cancer cells. This review evaluates the recent advances, clinical applications, and future potential of nanoparticle-mediated delivery of siRNA therapeutics to target genes implicated in ovarian cancer.
Introduction
Ovarian cancer was responsible for more than 14,000 female deaths in the United States in 2017 and is the most lethal type of gynecological malignancy. 1 In 2017 alone, an estimated 22,440 new cases of ovarian cancer were diagnosed in the United States. 1 Although it is the eighth most common cancer, ovarian cancer is among the five most lethal cancers in women due to a high rate of metastasis and insufficient early detection methods.1,2 The 5-y overall survival rate is relatively promising (>70%) when detected at an early stage (I-II); however, survival drops to about 30% when detected at stages III-IV. 3 Because of a lack of effective early detection strategies, more than 60% of ovarian cancers are not diagnosed until advanced stages, resulting in high rates of mortality. 2 Approximately 70% of the deaths due to ovarian cancer are caused by the advanced, high-grade serous subtype, a particularly lethal and fast-spreading cancer of epithelial origin. 4
Although survival rates for other cancers have drastically improved since the introduction of platinum-based chemotherapy, the prognosis for women with epithelial ovarian cancer has not changed significantly over the past 30 years. 2 Today’s standard of care consists of surgery followed by cytotoxic chemotherapy. 5 Even after treatment, however, platinum-resistant cancer recurs in 25% of patients, potentially leading to uncontrolled tumor proliferation and malignant bowel obstruction, which is the most common cause of death in ovarian cancer patients. 6
The Genetic Basis of Ovarian Cancer
A lack of understanding about the origin and pathogenesis of ovarian cancer contributes to its high rate of morbidity and mortality in affected indivuals. 7 Rather than a single disease, ovarian cancer is better classified as a heterogeneous group of tumors that share a common anatomical location. 8 An improvement in our understanding of the exact mechanisms behind the development of ovarian cancer could lead to strategies for early detection and treatment. Given the heterogeneity of tumors, researchers have predicted that the most effective therapies will rely on molecular characterization of ovarian cancer subtypes to allow for both specific targeting and general treatment with the identification of markers unique to ovarian cancer. 8 Thus, recent investigations have focused on characterizing the genetic indicators of ovarian cancer.7,9,10 A 2011 genomic analysis revealed that more than 96% of lethal ovarian cancers contain mutations in TP53 and BRCA. 10 Moreover, another study identified 54 genes essential for the survival and proliferation of ovarian cancer that were differentially expressed or amplified in tumor cells. 9 Among others, these gene targets have been under further investigation as potential avenues for treatment through gene therapy. 11
RNA Interference
One method of gene therapy that holds promise for the treatment of ovarian cancer is RNA interference (RNAi). In RNAi, strands of native or synthetic RNA interfere with the expression of genes with which they share a homologous sequence. 12 The gene-silencing potential of RNAi was first observed in plants and subsequently demonstrated in the roundworm Caenorhabditis elegans. 13 MicroRNA (miRNA) and small interfering RNA (siRNA) are two small RNA molecules that play an important role in RNAi by binding to messenger RNA and regulating the activity of the transcript—often inhibiting the expression of a gene. 14 For the purpose of this review, we will focus on siRNA molecules, which are 21 to 23 nucleotides in length and produced by the breakdown of long double-stranded RNA in the cytoplasm via the enzyme Dicer. 12 siRNA is then incorporated into an RNA-induced silencing complex (RISC) and unwound into single-stranded RNA (ssRNA). 15 This ssRNA acts as an antisense guide for the RISC complex and binds to complementary mRNA molecules, which are degraded by Argonaute proteins ( Fig. 1 ).16,17
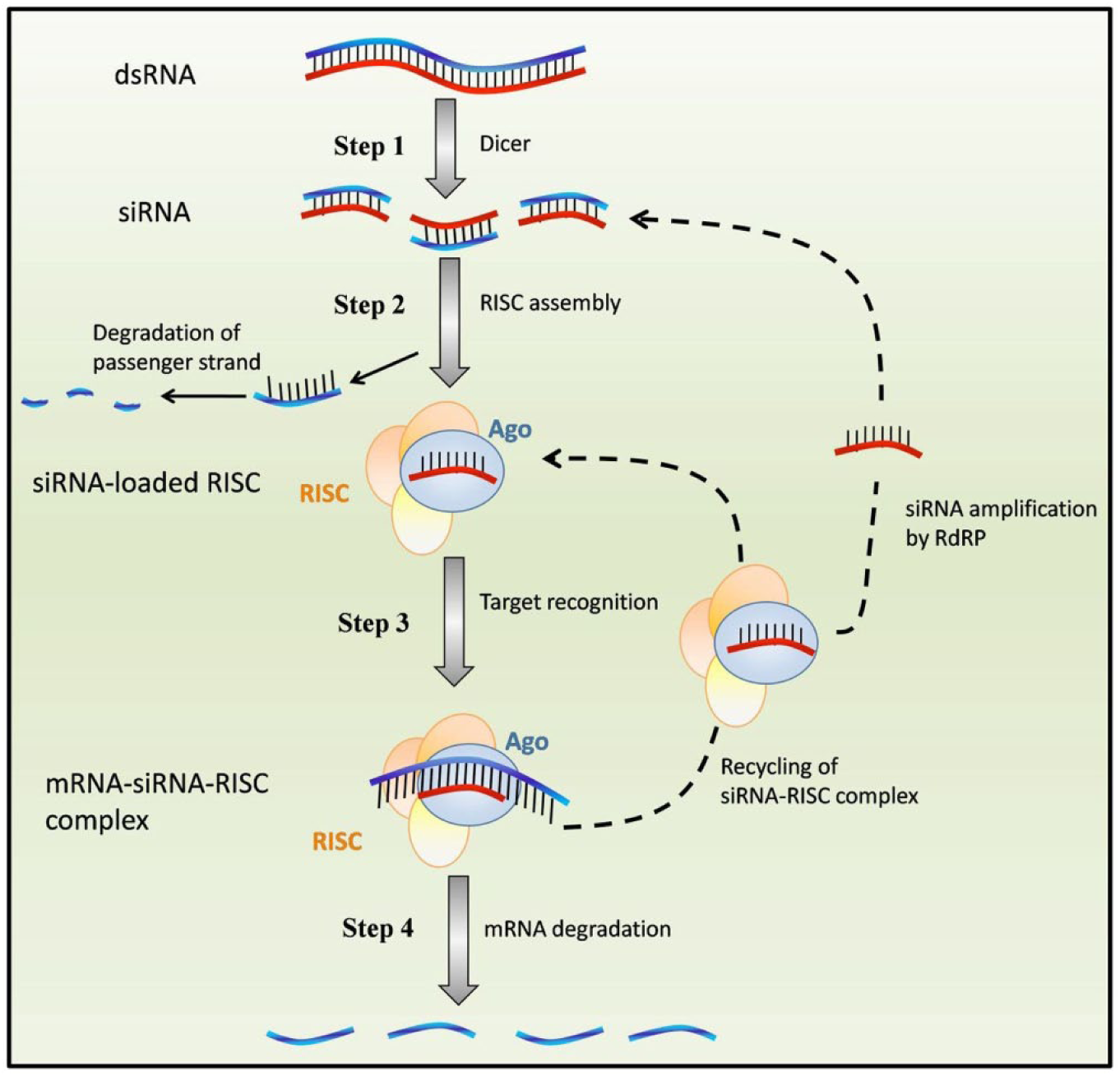
Small interfering RNA (siRNA)–based gene silencing. First, a precursor long double-stranded RNA (dsRNA) is broken down to 21 to 23 base-pair siRNA in the cytoplasm by the enzyme Dicer. The siRNA is incorporated into an RNA-induced silencing complex (RISC) and unwound into single-stranded RNA (ssRNA). The passenger strand is degraded. This ssRNA guides the siRNA-RISC complex and binds to complementary mRNA molecules. Finally, the Argonaute protein component of RISC activates and degrades the mRNA molecules, thus silencing expression of the transcript. Reprinted from Cuccato et al. 17 BMC Systems Biology 5(1): 19. © 2011 Cuccato et al; licensee BioMed Central Ltd. Open Access.
The potential of synthetic siRNA for gene silencing was first demonstrated by Elbashir et al. in 2001 through sequence-specific knockdown of genes in mammalian cells. 18 Later, McCaffrey et al. showed the therapeutic potential of siRNA by knocking down a sequence from the hepatitis C virus in transgenic mice. 19 Since 2004, siRNA has been employed in clinical trials to knockdown gene targets implicated in the life cycle of HIV, macular degeneration, respiratory disease, cancer, and other maladies. 12 These nucleic acids have also been the focus of efforts in computational biology to design algorithms for the synthesis of siRNA with greater gene-knockdown efficiency and fewer off-target side effects. 20
The selective gene-silencing capability of siRNA provides an opportunity to treat cancer and other diseases that are caused by abnormal expression of one or several genes. A total of 291 oncogenes have been characterized, 21 and many have been employed as targets for siRNA-based therapy. 12 In addition, siRNA has been used to knock down gene targets involved in tumor resistance to chemotherapy, resulting in decreased proliferation and reduced cancer cell survival. 22 Breast, lung, ovarian, prostate, and cervical cancer are among the cancers currently under investigation for potential siRNA therapy. 23
Challenges in siRNA Therapy
Although siRNA has shown potential for therapeutic applications, several intracellular and extracellular barriers must be overcome before the full potential of siRNA can be realized. Among the most pronounced challenges in siRNA therapy are off-target effects, instability in circulation, immune stimulation, and difficulties involving cellular uptake. 24 Ten percent of siRNAs may produce significant off-target effects, which involve the knockdown of a gene that is not intended to be targeted by the siRNA. 20 Toxicity may also result from stimulation of the innate immune system and the release of inflammatory cytokines caused by the introduction of foreign RNAs. 25 Synthetic siRNA may further cause unintended side effects by saturating cellular RNAi machinery. As a result, this may disrupt the gene silencing performed by endogenous miRNA and cause unpredictable downstream effects, such as the overexpression of certain proteins. 26 Finally, siRNA is hydrophilic and anionic and cannot readily bypass the hydrophobic cellular membrane to be internalized by cells via passive diffusion. Currently, most siRNA delivery in clinical trials is accomplished through local administration via intravitreal or intranasal routes to minimize time spent in circulation, thus avoiding targeting issues and plasma degradation of siRNA. 12
Nanoparticle Delivery of siRNA
Nanoparticles as carriers present a platform for targeted delivery of siRNA therapeutics. An ideal delivery method for siRNA would be biocompatible, nonimmunogenic, and biodegradable and provide targeted delivery with efficient and selective uptake only at the target tissue. siRNA-conjugated nanoparticle delivery methods are advantageous as they allow for evasion of the innate immune system through modification with polyethylene glycol (PEG), 27 protection from serum inactivation and enzymatic degradation, efficient entry into the cell, 28 and avoidance of intracellular degradation (i.e., endosomal escape). 29 To function as an effective carrier, cationic lipids and polymers are often used to complex with the negatively-charged siRNA. 30 Although the potential for nanoparticle delivery of siRNA has been widely investigated, most siRNA-nanoparticle drug delivery systems are still only in the preclinical stage of development. 31 However, patisiran, an siRNA drug delivered via a lipid nanoparticle, recently became the first siRNA-based treatment to gain Food and Drug Administration approval as a drug. 32 Although patisiran is used to treat hereditary transthyretin-mediated amyloidosis rather than cancer, its regulatory approval has general clinical implications for the efficacy of siRNA-nanoparticle–based treatment to target underlying diseases rather than their symptoms alone. 33
Ovarian cancer is a good candidate for RNAi therapy as it has many well-established gene targets and is anatomically accessible for localized delivery.7,9,10 Codelivery of siRNA therapeutics with chemotherapy agents could reduce drug resistance and allow for more effective therapy. 34 In this review, we will discuss recent advances, clinical applications, and future potential of nanoparticle-mediated delivery of siRNA therapeutics to target genes implicated in ovarian cancer. We will first focus on the applications of siRNA-conjugated nanoparticles to knock down oncogenes implicated in the proliferation of ovarian cancer, to reduce metastasis, and to induce apoptosis. Then, we will discuss the use of siRNA nanoparticles to down-regulate genes that contribute to the drug resistance of ovarian cancer cells. Finally, we will examine the potential of combination treatments that use nanoparticles to co-deliver siRNA with chemotherapeutic agents for synergistic, antitumor effects.
Knocking Down Ovarian Cancer Oncogenes
Late-stage diagnosis of ovarian cancer presents a limiting factor for the use of traditional therapeutics, as treating the cancer in a metastatic, advanced state often requires aggressive treatment methods, including chemotherapy. 35 The contemporary approach to addressing ovarian cancer includes surgical excision of primary tumors followed by chemotherapy. However, the majority of patients who followed such a treatment plan saw recurrent tumors that developed a resistance against chemotherapy.6,36 Given that tumors are heterogeneous, with some cells harboring secondary mutations that confer drug resistance, the use of traditional chemotherapeutics may lead to the selection of resistant cells, resulting in the development of drug-resistant tumors. It is therefore beneficial to prevent the formation of such tumors by finding molecular-based alternative therapies before the tumors become drug resistant. 35
The use of siRNA in cancer therapy has the potential to down-regulate ovarian cancer oncogenes that can lessen the tumor burden and induce selective apoptosis of the cancer cells. Although siRNA-conjugated nanoparticles for treating ovarian cancer are not yet in human clinical trials, preclinical studies have shown the applicability of nanoparticles to lessen the tumor burden in mouse xenograft ovarian cancer models. 37
Identifying Oncogene Targets
As mentioned, in 2011, Cheung et al. 9 identified 54 oncogene targets that were essential to ovarian cancer growth and survival using short-hairpin RNA (shRNA) screening in 25 human ovarian cancer cell lines. Microarray hybridization was first used to measure the abundance of shRNA sequences, and their effects on ovarian cancer cells were then correlated to the shRNA abundance data. 9 The genetic targets identified here served as a baseline for future in vivo studies discussed in this review. For example, expanding on the Cheung study, Ren et al. 38 conducted shRNA screenings across human cell lines to take into account inherent tumor heterogeneity in identifying clinically relevant oncogene targets. With these databases of genetic targets in place, the studies described below were able to use siRNA to silence these oncogenes to restrict tumor metastasis or reduce the primary tumor burden.
Restricting Metastasis
Ma et al. 39 used a generation-6 poly(amidoamine) (G6 PAMAM) dendrimer as a nanovector to deliver siRNA targeting p70S6K. p70S6K is an effector of the phosphatidylinositol 3-kinase (PI-3K)/Akt pathway that is up-regulated in ovarian cancer and associated with an aggressive malignant phenotype. This target plays a role in cell adhesion, migration, and invasion to promote the metastatic growth of ovarian tumor cells.40–42 To study the effectiveness of p70S6K as a therapeutic target, SKOV-3 ovarian cancer stem cells (CSCs)—ovarian cancer cells with a phenotype responsible for initiating the metastatic cascade—were isolated from tumor tissues based on their spherical morphology and the presence of stem cell biomarkers Oct4, c-Kit, Nanog, and Bmi-1. Nanoparticle delivery of siRNA treatment resulted in 90% gene silencing of p70S6K, and a subsequent MTT assay showed that the treatment inhibited more than 50% of CSC proliferation whereas normal ovarian surface epithelial cell growth was unaffected. Furthermore, the role of p70S6K in the stem cell–like state of CSCs was confirmed, as reverse transcriptase polymerase chain reaction revealed that the expression of Oct4, c-Kit, Nanog, and Bmi-1 stem cell biomarkers was reduced by 60% to 80% in SKOV-3 cells treated with p70S6K siRNA/G6 dendriplexes compared with treatment with nonspecific siRNA dendriplexes. To test for the role of p70S6K in the metastatic cascade, CSC adhesion was investigated with a co-culture assay in which transfected CSCs were fluorescently labeled and cultured onto a monolayer of human mesothelial cells. The cells were then analyzed based on their ability to adhere to the mesothelial layer. Migration was also investigated by adding Matrigel to transwell plates to mimic the extracellular matrix. It was found that the sip70S6K/G6 dendriplexes were able to significantly inhibit CSC adhesion, migration, and invasion compared with cells treated with nonspecific nanoparticles, free siRNA, or free dendriplexes. In CSC xenograft mouse models, nanoparticle treatment led to accumulation and retention of the sip70S6K in tumors for more than 24 h, a period during which tumor growth was inhibited without acute toxicity. Importantly, unlike other treatments, all mice with the sip70S6K nanoparticle treatment survived, further supporting the use of sip70S6K/G6 dendriplexes to inhibit tumor growth and metastasis. 39 Such a strategy, although effective in reducing metastasis, remains susceptible to the challenges aligned with surgical tumor resection followed by chemotherapy. Although the metastatic progression of ovarian tumors was limited, this approach failed to induce apoptotic antitumor effects for the primary tumor.
Reducing the Primary Tumor Burden
To directly treat the primary tumor with siRNA-conjugated nanoparticles, researchers have attempted to induce apoptosis in primary tumor cells in addition to curbing metastasis. Extending from their previous work in characterizing ovarian cancer oncogene targets in human cancer cell lines, Ren et al. 38 evaluated the transcriptional regulator inhibitor of DNA binding 4 (ID4) as a novel oncogene amplified in 32% of primary ovarian tumors. However, as ID4 is not overexpressed in normal ovarian tissues, it was identified as a prime candidate for RNAi therapy. To investigate the potential of this oncogene as a target, the researchers used a tumor-penetrating nanocomplex (TPN) as a carrier for siRNA that would target ID4 (siID4). In addition, the TPN was conjugated to two peptide sequences, one for membrane translocation, transportan, and another for tumor-penetration, cyclic nanopeptide lysine-specific permease 1 (LyP1). LyP1 contained domains for both siID4 binding and p32 targeting, whereas the transportan enabled tumor parenchyma penetration.43,44 The design allowed for specific targeting of p32 on OVCAR-4 and OVCAR-8 human ovarian cancer cell lines and subsequent accumulation in cancer cells, rather than relying on passive targeting via the enhanced permeability and retention effect. In vivo studies in mice expressing OVCAR-4 human ovarian tumor xenograft models treated with siID4-TPN revealed an 80% to 90% decrease in ID4 levels and a 375% increase in apoptotic rate accompanied by suppressed growth of primary tumors and 80% of the mice surviving 60 days after treatment cessation compared with controls treated with saline, TPN-LyP1 carrier without siRNA, or TPN/siGFP carrying siRNA targeting green fluorescent protein (GFP). ( Fig. 2 ). Intravenous delivery of the siID4-TPN was found to be more effective for treating subcutaneous ovarian tumor xenograft models, whereas intraperitoneal delivery was more effective for orthotopic ovarian tumor xenograft models, as given by suppression in tumor growths by 82% and 87%, respectively. Notably, compared with the saline control, treatment with silD4-TPN resulted in a 550% increase in expression of CDKN1A, an ID4 target that mediates cell cycle arrest and contributes to apoptosis. Moreover, HOXA9, another downstream regulator for differentiation, proliferation, and self-renewal, was down-regulated in cells treated with siID4-TPN. As a result of this study, ID4 was confirmed as a regulator of ovarian cancer cell proliferation essential for cancer cell survival and tumorigenic properties, as well as a genetic target for the treatment of ovarian cancer.37,38
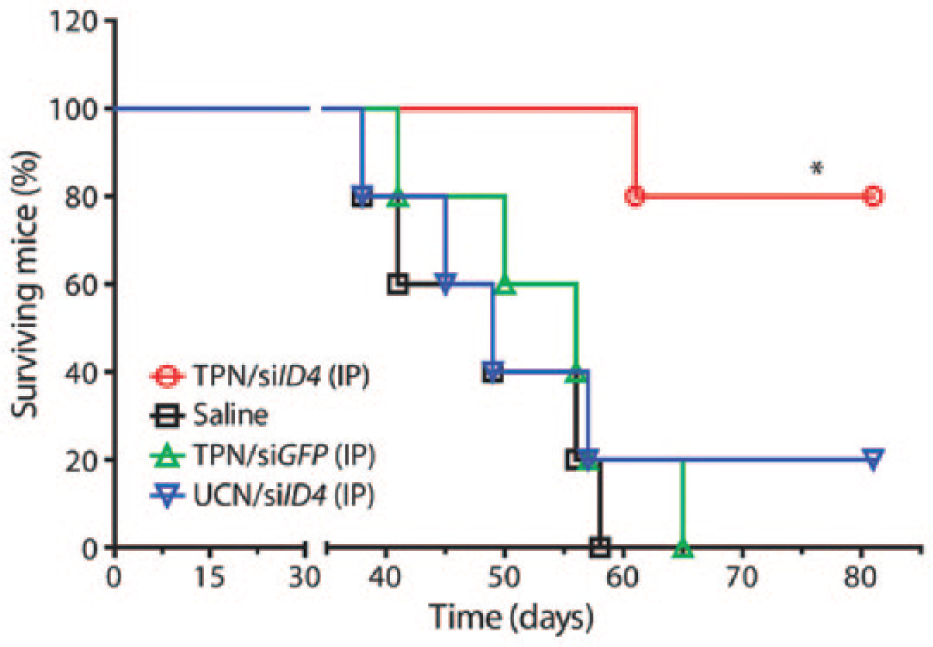
Kaplan-Meier survival curve for mice with orthotopic OVCAR-8 tumors treated with a tumor-penetrating nanocomplex that carried small interfering RNA targeting ID4, an ovarian cancer oncogene. Reprinted with permission from Ren et al. 38 Science translational medicine 4 (147): 147ra112. © 2012 American Association for the Advancement of Science.
Wang and coworkers identified Grb2-associated binder 2 (GAB2) as an oncogene that contributes to the development of ovarian cancer.45–50 GAB2 is a regulator for migratory behaviors and E-cadherin expression via the PI-3K pathway. 51 Whereas Ren et al. used a peptide-based approach, Wang and coworkers used a chitosan-polyethylenimine (PEI) nanoparticle, due to the biocompatible nature of chitosan and its ability to transport charged nucleic acids across epithelial surfaces. The polycationic PEI was used for its high transfection efficiency and ability for endosomal escape via the proton sponge effect. In vitro studies with SKOV-3 human ovarian cancer cells showed that the chitosan-PEI-siGAB2 nanoparticles had the highest rate of transfection efficiency compared with other siRNA/chitosan controls. In addition, siGAB2 demonstrated endosomal escape, and MTT assays revealed that the nanoparticles also resulted in antitumor activity compared with the siGFP control. Furthermore, treated cells had only 5.9% GAB2 RNA expression, illustrating a significant down-regulation of GAB2. A 6.84% early apoptosis rate and 24.2% late apoptosis rate demonstrated that GAB2 siRNA was able to induce apoptosis, specifically by down-regulating the expression of Akt, an effector protein that inhibits drug-induced apoptosis. Although no in vivo studies were performed, the silencing effects and 90% down-regulation of Akt protein expression observed in nanoparticle-treated cells, along with low toxicity in healthy cells, demonstrated that chitosan-PEI-siGAB2 nanoparticles may be a promising strategy to treat primary tumors that can modulate Akt2 activity. 29
Goldberg et al. 52 took an alternative approach by relying on the concept of synthetic lethality, a process by which the loss of two protein functions confers cellular lethality but the loss of a single protein function alone is not lethal. In this case, BRCA1-deficient ovarian cancer cells, which constitute 10% of all ovarian cancers, were treated with NC100 lipidoid nanoparticles. These nanoparticles carried siRNA specific to the DNA repair enzyme poly(ADP-ribose) polymerase (PARP1) to induce apoptosis by interfering with DNA repair pathways.53–58 BRCA1-deficient cells have been previously shown to be particularly sensitive to disruptions of PARP1 enzyme function, often resulting in cell death. 55 Because of this synthetic lethality, researchers hypothesized that nanoparticle delivery of siRNA to knockdown PARP1 would prevent DNA repair and result in apoptosis. Delivery of siPARP1-NC100 knocked down 65% of PARP1 mRNA in BRCA1-deficient murine ovarian cell lines (BR5FVB1) in vitro, which correlated with increased apoptosis. In mice with BRCA1-deficient or BRCA1 wild-type ovarian tumors, siPARP1-NC100 treatment extended the survival of mice that had the BRCA1-deficient ovarian carcinoma allograft tumors by at least 50 days compared with mice that retained the BRCA1 gene, supporting their synthetic lethality hypothesis ( Fig. 3 ). As a result, PARP1 was confirmed to be a viable target for ovarian cancer treatment in the context of BRCA1 deficiency. 52
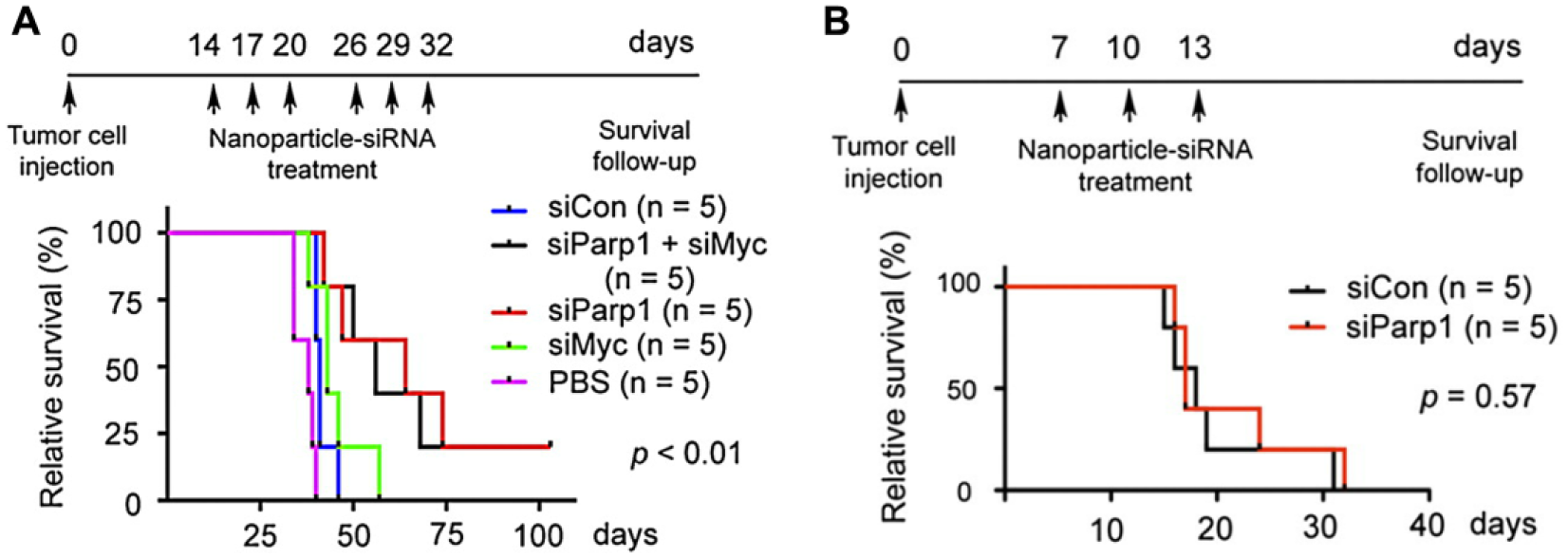
In vivo Kaplan-Meier survival curve after treatment with siParp1/NC100 in mice (
Other oncogene targets have been investigated and their role in ovarian cancer growth and metastasis confirmed with similar nanoparticle approaches. Notable strategies include silencing signal transducer and activator of transcription 3 and focal adhesion kinase with reconstituted high-density lipoprotein nanoparticles or down-regulating epidermal growth factor receptor with poly N-isopropylmethacrylamide nanogels.59,60 With increased survivability and low toxicity profiles, ID4, GAB2, and PARP1, among other genes, have the potential as personalized molecular therapeutic targets to complement the traditional use of surgery and chemotherapeutics.
Targeting Drug-Resistant Ovarian Cancer
Given that chemotherapy-resistant tumors stem from the selection of cancer cells with random secondary mutations, researchers are targeting genes behind the development of drug resistance in ovarian cancer cells. 61 By using nanoparticle-delivered siRNA to knockdown the expression of genes involved in resistance to chemotherapy, cancer cells could be resensitized to anticancer drugs, and future strategies using chemotherapy may be more effective.
To characterize genes involved in cisplatin resistance, Bao et al. 62 examined cisplatin-resistant ovarian cancer cells and found that genes encoding adenosine triphosphate–binding cassette (ABC) transporters in the subfamily F2 (ABCF2) were amplified. Using data from cell viability assays, the researchers hypothesized that the overexpression of ABCF2, a nuclear-related factor 2 (NRF2) target gene, was correlated with resistance to chemotherapy. 63 In vitro testing on A2780 cells revealed that cells became resistant to cisplatin treatment when overexpression of ABCF2 was induced. Lowering expression of ABCF2 on A2780 cells, on the other hand, led to the resensitization of such cells to cisplatin. Thus, the researchers concluded that ABCF2 expression levels may be correlated with cisplatin resistance and could be manipulated to resensitize cells to the traditional chemotherapeutic.62–64
Alternatively, p62, encoded by SQSTM1, is an autophagy adaptor believed to be related to the mechanism for platinum drug resistance. 65 p62 has been shown to be overexpressed in cisplatin-resistant ovarian cancer cells, and inhibition of p62 has resulted in reduced cisplatin resistance. 66 Higher levels of p62 activate NRF2, a downstream effector that conferred ovarian cancer cell resistance to platinum-based drugs. p62 has also been shown to promote cell proliferation and inhibit apoptosis, potentially leading to further growth of cancer cells. 67 In vitro studies using SKOV-3/DDP ovarian cancer cells with cisplatin resistance showed that the activation of the NF-kB pathway for transcription regulation was correlated with drug resistance. The study revealed that p62 was an upstream regulator for NF-kB signaling, suggesting a correlation between p62 up-regulation and drug resistance. 68
Another potential set of targets are multidrug resistance (MDR) genes, which lead to resistance against chemotherapy drugs such as cisplatin, paclitaxel, and doxorubicin when overexpressed. 69 Yang et al. 70 focused on the MDR gene product P-glycoprotein (P-gp), encoded by ABCB1. Hyaluronic acid (HA) nanoparticles with siRNA against ABCB1 were used to target cancer cells overexpressing cell surface protein CD44. 70 In OVCAR8TR paclitaxel-resistant ovarian cancer cells, fluorescence microscopy revealed that the HA-NP delivery suppressed P-gp levels for up to 120 h, suggesting HA-NPs could be used as a potential therapeutic to resensitize cells to paclitaxel treatment. In human MDR OVCAR8TR tumor xenograft models placed in mice, treatment was administered via intravenous injection, and the mice were monitored over 35 days. HA-NPs led to an increase of 150% in relative tumor volume compared with up to 300% growth for no treatment controls, displaying clear inhibition of tumor growth. 70
TWIST, encoding a developmental transcription factor that leads to chemotherapy resistance and cancer cell stemness, is also involved in ovarian cancer drug resistance. 71 Further, down-regulation of TWIST mRNA has been shown to increase the sensitivity of cancer cells to chemotherapeutics. 72 Roberts et al. used PAMAM dendrimers and mesoporous silica nanoparticles to carry the siRNA targeting TWIST (siTWIST) to cisplatin-resistant ovarian cancer cells (A2780R). 73 They hypothesized that the knockdown of TWIST would lead to resensitization to chemotherapeutics. The researchers used fluorescence microscopy to verify that the dendrimer-siTWIST complex was successfully taken up by A2780R cells. Two doses of the si419 and si494 treatments to target TWIST were administered at 50 nM and 100 nM in vitro, and subsequent Western blots revealed that the siRNA treatment successfully led to knockdown of the TWIST protein product after 1 week of treatment compared with nontargeting siRNA and untreated controls. 74 In vivo studies in mice injected intraperitoneally with OVCAR8 cells demonstrated that si419H with cisplatin led to decreased tumor weight, up to sixfold. The results confirmed TWIST to be a promising target for siRNA therapeutics and nanoparticle delivery. 73
Chen et al. aimed to knock down NOTCH3, which encoded a marker involved in ovarian cancer recurrence and chemotherapy resistance, with an aptamer-siRNA chimera delivery system using gold nanoparticles. 75 The gold nanoparticles were conjugated with iron(II,III) oxide, PEI, and aptamer-siRNA against NOTCH3 chimera modifications to target ovarian cancer cells via vascular endothelial growth factor (VEGF), a protein found to be overexpressed in ovarian cancer cells. 76 The aptamer was designed to be positively charged (+20 mV), which allowed for interaction with the negatively charged cell membrane and facilitated rapid clathrin-mediated endocytosis. 77 The aptamer successfully targeted VEGF signaling in cisplatin-resistant SKOV-3/DDP cells, and Western blot analysis confirmed that the nanoparticle-chimera delivery system was effective in knocking down NOTCH3 in SKOV-3 cells compared with a lipofectamine-mediated delivery system or the siRNA alone. Moreover, cell viability was decreased by a factor of 2 compared with the untreated control in SKOV-3/DDP cells. Thus, this aptamer-siRNA chimera delivery system presents a promising example of nanoparticle delivery of siRNA to reduce drug resistance in ovarian cancer. 75
Co-Delivery of siRNA and Anticancer Drugs
Although knockdown of genes involved in drug resistance in ovarian cancer may increase the efficacy of later rounds of chemotherapy, another promising course of treatment involves the co-delivery of siRNA with anticancer drugs for immediate action. Co-delivery of siRNA and anticancer drugs can improve the efficacy of chemotherapy by simultaneously inducing apoptosis and preventing protumor factors such as Bcl-2 or surviving.78–81 The major chemotherapeutic drugs reported in co-delivery with siRNA are cisplatin, doxorubicin, and paclitaxel. Cisplatin interacts with nucleotides and crosslinks DNA strands, thereby interfering with DNA replication and inducing apoptosis when repair mechanisms prove ineffective. 82 Doxorubicin intercalates between DNA strands to prevent DNA replication, up-regulate activation transcription factor 3 (ATF3), and to decrease the expression of phosphorylated protein kinase A and BCL2. 83 Paclitaxel destabilizes microtubules in cells, preventing cell replication. 78 However, the effects of these anticancer drugs may be limited by proteins that interfere with the drugs’ pathways. For example, BCL2 prevents the apoptotic effect of doxorubicin by preventing the disruption of the transmembrane mitochondrial potential,81,84 and overexpression of survivin has been shown to lead to high cancer cell viability. To combat this resistance, survivin siRNA has been employed to reduce cell viability by arresting the cell during mitosis. 85 Nanoparticles loaded with chemotherapy and siRNA may be directed toward cancer cells to knock down resistance against the drugs and induce apoptosis.
Cisplatin
He et al. 86 studied combination therapies involved with ovarian cancer drug resistance by using a nanoscale coordination polymer (NCP) system to deliver both cisplatin and siRNA. 86 They made use of pooled siRNA to target mRNA encoding survivin (BIRC5), apoptosis regulator BCL2 (BCL2), and P-gp (ABCB1)—products known to be related to the overexpression of MDR genes.79,80,87–90 The targeted siRNAs were attached to the surface of the self-assembled NCP consisting of metal ions and organic bridging ligands. This delivery system was effective in improving cellular uptake of cisplatin by 250% compared with free cisplatin over 24 h. After 24 h of NCP treatment on OVCAR3 and SKOV-3 cells, the cells had 50% higher rates of apoptosis than those treated with free cisplatin, illustrating the efficacy of the NCP delivery system. Subsequent in vivo studies in SKOV-3 subcutaneous xenografts in mice showed that the average tumor volume decreased from 97 mm3 to 38 mm3 after 28 days, and the therapy was more effective than control treatments (free siRNA, NCP-1, and Zn control with siRNA). However, the initial tumor volume was only ~0.1 cm3, which is not representative of the size at which ovarian tumors are found in humans, so additional testing to scale tumor regression is required before clinical application.86,91
Another potential gene target for resistant ovarian cancer is EZH2, a gene overexpressed in cisplatin-resistant cancer cells. 92 To deliver siRNA gene therapy to knockdown EZH2, Yu and coworkers 93 constructed a nanoparticle conjugated to peptides specific for luteinizing hormone-releasing hormone (LHRH) receptors, which are overexpressed in ovarian cancer cells. 93 A 400 nm iron(II,III) oxide core nanoparticle (NP-3) modified with PEI, PEG, LHRH, siEZH2, and cisplatin prodrug was compared with free cisplatin. NP-3 improved cisplatin uptake by 500% compared with free cisplatin in A2789/cisplatin (DDP)–resistant ovarian cancer cells 6 h after treatment. Further, the NP-3 delivery system significantly decreased EZH2 transcription and expression by 500% compared with the untreated control in A2780 and A2780/DDP cisplatin-resistant cells in vitro. In vivo studies in which mice were subcutaneously injected with A2780/DDP cells revealed that relative tumor volumes were approximately 300% lower after 15 days under nanoparticle treatment compared with the untreated control ( Fig. 4 ). The imaging modality of the iron(II,III) oxide core allowed researchers to develop a theranostic application and visualize nanoparticle accumulation at the tumor site via magnetic resonance imaging, thus allowing for the real-time evaluation of the therapeutic effect of NP-3. 94
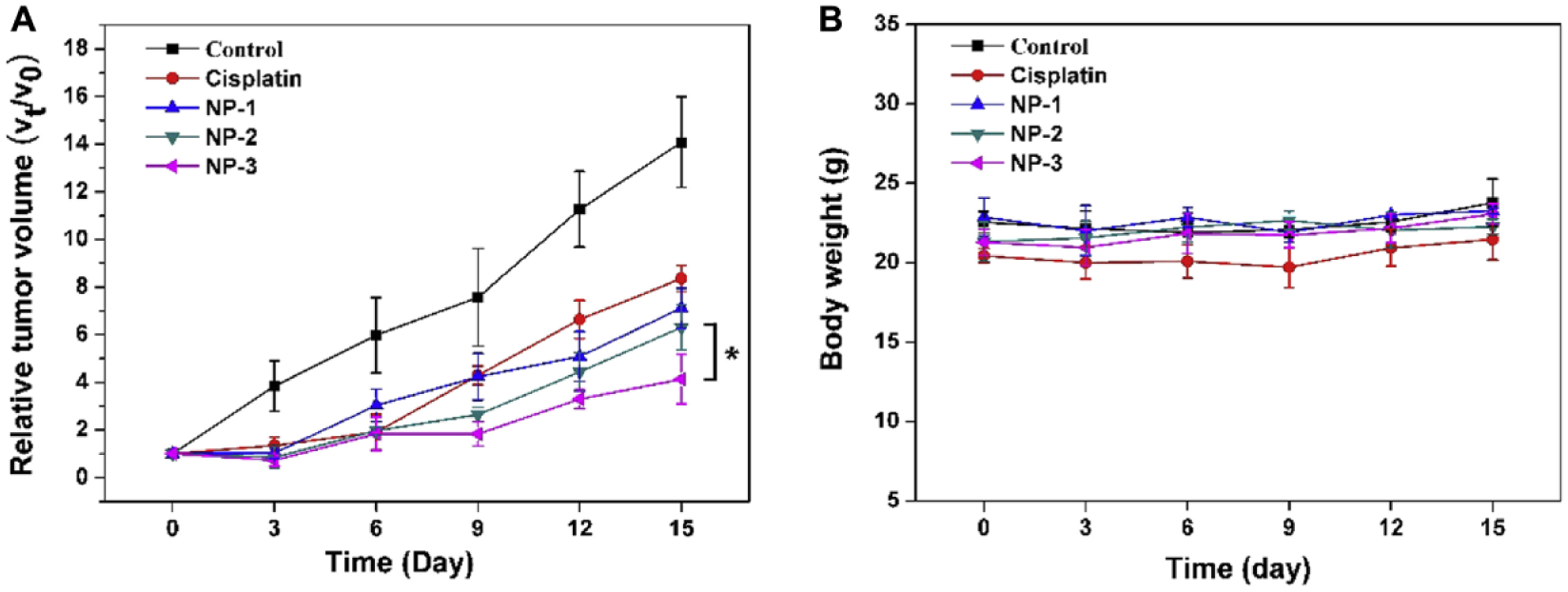
In vivo tumor studies in mice carrying A2780/cisplatin-resistant tumors, iron(II, III) oxide core nanoparticles modified with polyethylenimine, polyethylene glycol, luteinizing hormone-releasing hormone, siEZH2, and cisplatin prodrug show (
Doxorubicin
Zou et al. 81 designed a nanoparticle system to co-deliver doxorubicin with siRNA targeting BCL2, a gene that encodes an apoptosis-inhibiting protein involved in resistance to doxorubicin. The nanoparticle delivery system consisted of a folate-conjugated copolymer of PEG, PEI, and poly(ε-caprolactone; FA-PEG-PEI-PCL),80,81 self-assembled around doxorubicin, yielding a micelle that contained the anticancer drug. 81 The anionic siRNA was then adhered to the surface via electrostatic interaction by taking advantage of the nanoparticle’s cationic charge. The added folate group allowed the nanoparticle to target SKOV-3 cells with a high accuracy, as 85.7% cellular uptake with folate was observed compared with 43.9% without. 81 Moreover, cell uptake was reduced in a competition assay using excess folate, confirming the nanoparticles’ specificity to folate receptors. An in vitro terminal deoxynucleotidyl transferase dUTP nick end labeling (TUNEL) assay revealed that SKOV-3 cells treated with folate-targeted doxorubicin-loaded nanoparticles and BCL2 siRNA resulted in 77.5% apoptosis after 96 h of incubation, whereas only 50% of cells became apoptotic when treated with folate-targeted doxorubicin nanoparticles and scrambled siRNA. 81
Similarly, Chen et al. 95 synthesized a nanoparticle system for the co-delivery of doxorubicin and BCL2 siRNA consisting of copolymer PEG, poly(N-2,2′-dithiobis[ethylamine] aspartamide; PAsp[AED]), and pH-sensitive poly(2-[diisopropyl amino]ethyl methacrylate) (PDPA), termed PEG-PAsp(AED)-PDPA. The anionic siRNA was complexed to the surface of the positively charged nanoparticle derived from the reduction-sensitive layer of PAsp(AED). Because of the PDPA micelle core’s hydrophobicity at neutral pH, the nanoparticle was stable at a normal body pH of 7.4 but readily disassembled at a pH of 5.0 due to the protonation of tertiary amine groups, allowing for the release of siRNA and chemotherapy drug inside lysosomes. As observed through fluorescence imaging, nanoparticles released more doxorubicin in lysosome-mimicking environments of pH 5.0 and 10 mM glutathione compared with a more neutral 7.4 pH and no glutathione, thus indicating that the delivery system would preferentially release the drug intracellularly rather than in circulation. 95 In vivo fluorescence imaging in nude mice bearing subcutaneous xenografts of SKOV-3 cells illustrated that the nanoparticles were significantly more concentrated in the tumor site than any other tissue besides the liver after 24 h via the enhanced permeability and retention effect. At 1 µM doxorubicin concentration, MTT assays of SKOV-3 ovarian cancer cells revealed a 10-fold decrease in cancer cell viability in the doxorubicin/BCL2 siRNA nanoparticle compared with a doxorubicin/scrambled siRNA control, suggesting a greater antitumor effect with co-delivery of BCL2 siRNA. In nude mice bearing SKOV-3 xenografts, co-delivery of doxorubicin and siRNA via nanoparticles injected over 30 days resulted in the greatest survival: the treated mice had a 76% survival rate over 45 days compared with phosphate-buffered saline (PBS) control, in which no mice survived. Furthermore, co-delivery resulted in the least tumor growth, as mean tumor volume was only 110 ± 45 mm3 in mice receiving the doxorubicin and siRNA co-delivery treatment compared with 980 ± 60 mm3 for PBS control. Hence, anti-BCL2 siRNA delivered by nanoparticles has the potential to overcome drug resistance and sensitize ovarian cancer cells for doxorubicin treatment. 95
Another overexpressed protein in ovarian cancer is human epidermal growth factor receptor 2 (HER2). 96 Kotcherlakota et al. 97 developed a gold nanoparticle that delivered both doxorubicin and ERBB2 siRNA to SKOV-3 cells, designated as TDDS (Au-TR-DX-siERBB2). The ERBB2 gene, which leads to HER2 production, was selected because it leads to increased rates of cell proliferation and is overly expressed in SKOV-3 cells as well as other cancer cells. 98 In vivo studies in mice harboring SKOV-3 tumors demonstrated that the TDDS nanoparticle system limited tumor growth more effectively than controls including doxorubicin, siRNA, or doxorubicin and siRNA co-delivered without a nanoparticle carrier. Specifically, TDDS nanoparticles loaded with 2.5 mg/kg doxorubicin and 0.25 mg/kg ERBB2 siRNA were delivered intraperitoneally every other day over the course of 10 days, which resulted in a 600% smaller tumor volume compared with untreated controls and extended the mice’s life span by 50 days. Toxicological evaluations in C57BL/6 female mice confirmed that co-delivery of doxorubicin and ERBB2 siRNA resulted in low off-site toxicity because of the specific targeting to HER2. 97
Paclitaxel
Paclitaxel, another front-line chemotherapy agent, interferes with microtubule function during mitotic progression. 99 Hu et al. 100 developed a nanoparticle system termed PAP to co-deliver paclitaxel with survivin shRNA. These nanoparticles consisted of β-cyclodextrin and PEI surrounding an adamantine core conjugated with paclitaxel, and the survivin shRNA was adsorbed onto the surface. The rate of paclitaxel release was then studied at pH 7.4 and 5.0, representing normal body pH and tumor cell endosomal pH, respectively. The drug was observed to be continuously released even after 150 h, indicating that the nanoparticle system allowed for controlled and prolonged release of paclitaxel and protected it from degradation. In addition, paclitaxel was released from the nanoparticle 200% faster in pH 5.0 environments than pH 7.4 environments, indicating that nanoparticle delivery could enable preferential release within endosomes of cancer cells after internalization. Delivering 12 µg/mL of paclitaxel with shRNA targeting BIRC5 mRNA encoding survivin (shBIRC5) resulted in 82.93% apoptosis in SKOV cells after 24 h, compared with 22.15% apoptotic cells when paclitaxel was delivered alone and 30.35% apoptotic cells when the shRNA was delivered alone via the nanoparticle. In mice bearing SKOV-3 tumors, the PAP/shRNA paclitaxel system demonstrated the greatest antitumor effects and lowest rate of tumor growth, with a total cell viability of 2% with 2 µg/mL paclitaxel compared with 27% with just the shRNA or 30% with PAP alone at the same paclitaxel concentration after 30 days ( Fig. 5 ). 100
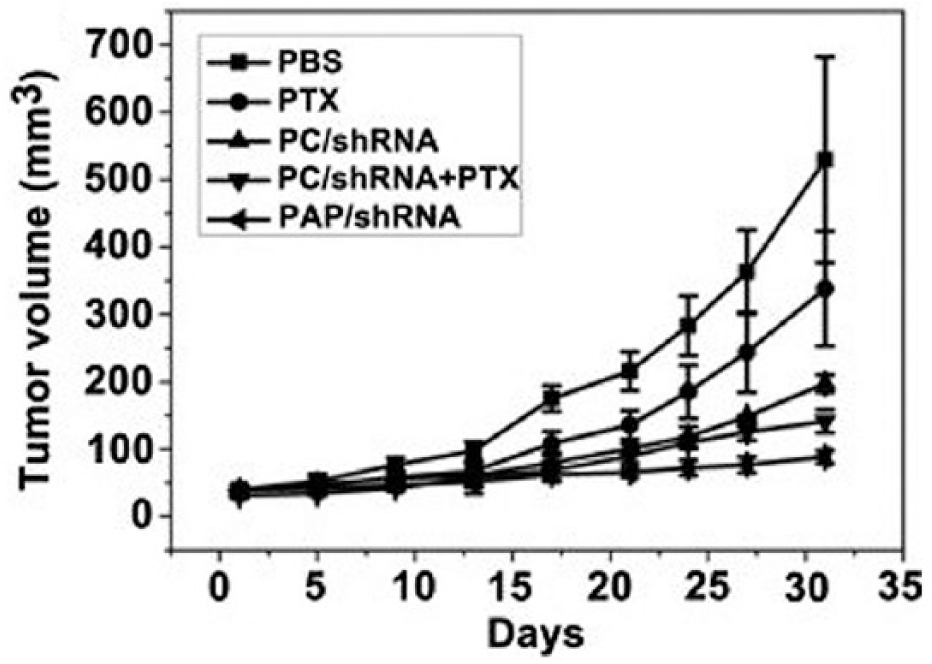
Tumor volume growth of a nanoparticle with a paclitaxel core conjugated with the survivin short-hairpin RNA (shRNA) compared with its controls. The nanoparticle with paclitaxel conjugated to the adamantine core and the shRNA adhered outside (PAP/shRNA) demonstrated the least amount of tumor growth. Reprinted with permission from Hu et al. 100 Biomaterials 33(27): 6580–6591. © 2012 Elsevier.
Survivin siRNA has also been examined in conjunction with paclitaxel therapeutics. Salzano et al. 78 synthesized a self-assembling polymeric micelle consisting of PEG 2000-phosphatidyl ethanolamine (PEG2000-PE). The micelle encapsulated both the survivin siRNA (siBIRC5) and paclitaxel, and the tumor-suppressing effects of the resulting nanoparticle were evaluated in vivo in mice hosting SKOV-3 tumors. The tumors were measured over a period of 30 days, and the results were consistent with Hu et al. as co-delivery of 1 mg/kg siBIRC5 with 10 mg/kg paclitaxel every 7 days for a total of five treatments resulted in maximum apoptotic effect, showing a relative tumor volume of 400% greater than original size after 30 days compared with a saline control with a 1200% size increase. Upon TUNEL assay, the co-delivery of survivin siRNA and paclitaxel resulted in the highest rate of tumor cell apoptosis compared with paclitaxel or survivin siRNA alone. Survivin expression was decreased significantly by the co-delivery nanoparticle, inhibiting survivin by 90% relative to PBS control, whereas BIRC5 levels were reduced by only 40% when siBIRC5 was delivered alone. By introducing siBIRC5 in combination with paclitaxel, cancer cells can became more sensitive to lower doses of chemotherapy, especially compared with previously ineffective dosages of paclitaxel alone. 78 Ultimately, co-delivery of siRNA and anticancer drug may increase the overall efficacy of treatment because of targeted delivery, reduced resistance to chemotherapy, and chemotherapeutic action against more-vulnerable cells.
Future Prospects
Ovarian cancer is a highly lethal disease responsible for thousands of deaths each year due to a lack of early detection methods and effective treatment options. The genetic nature of this cancer and well-established oncogene targets allow for the potential application of gene therapeutics such as RNAi for treatment. However, siRNAs are not stable in circulation, potentially toxic in vivo due to off-target effects, and unable to be taken up passively by cells. To that end, the use of nanoparticles for the delivery of siRNA to treat both drug-resistant and sensitive ovarian cancer is a rapidly developing field ( Table 1 ). Preclinical studies have shown promise in reducing tumor proliferation and increasing apoptotic effects of anticancer drugs; however, several steps must be taken before these therapeutics make an impact at the clinical level.
Summary of siRNA-Conjugated Nanoparticle Therapies in Treating Ovarian Cancer.
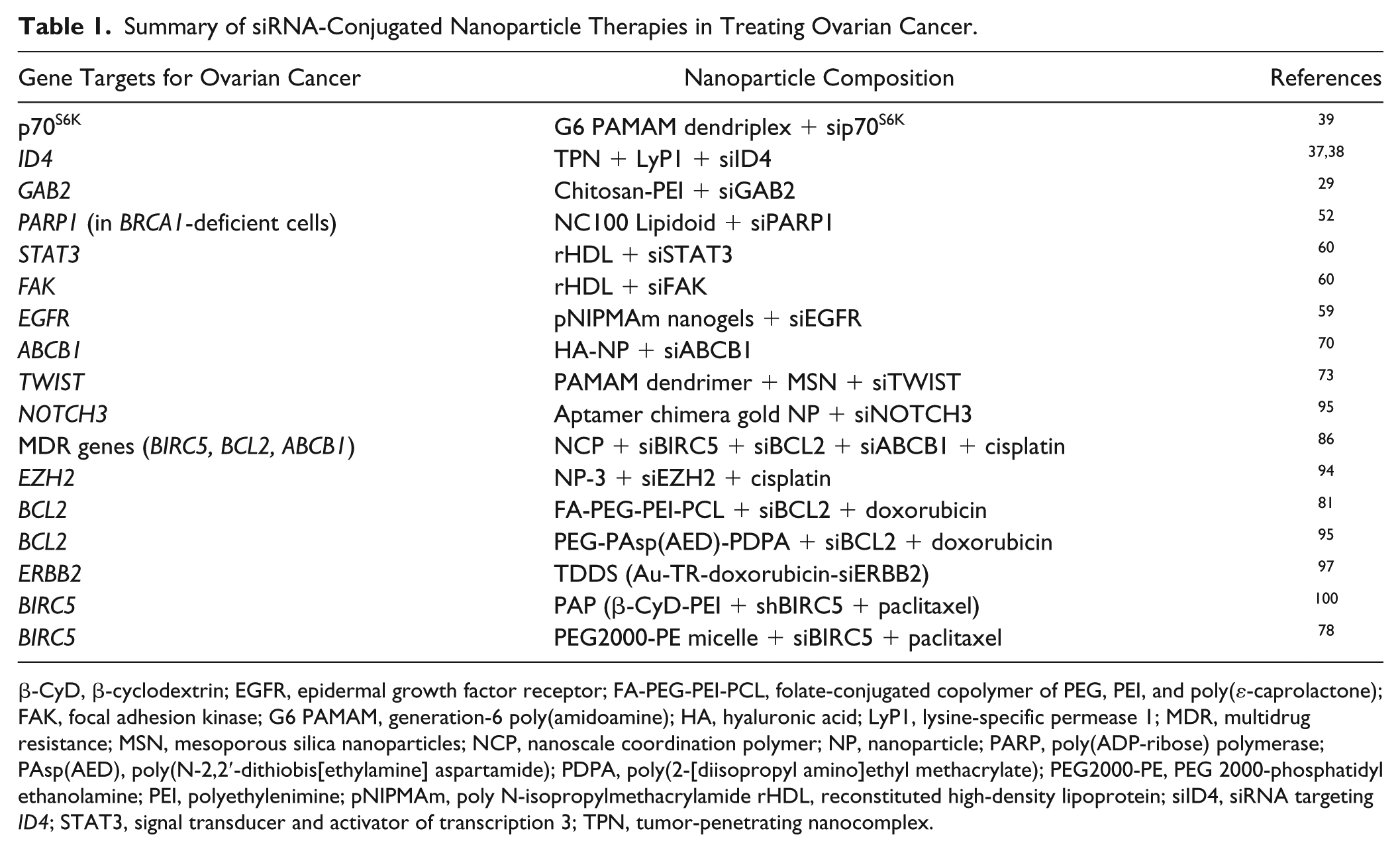
β-CyD, β-cyclodextrin; EGFR, epidermal growth factor receptor; FA-PEG-PEI-PCL, folate-conjugated copolymer of PEG, PEI, and poly(ε-caprolactone); FAK, focal adhesion kinase; G6 PAMAM, generation-6 poly(amidoamine); HA, hyaluronic acid; LyP1, lysine-specific permease 1; MDR, multidrug resistance; MSN, mesoporous silica nanoparticles; NCP, nanoscale coordination polymer; NP, nanoparticle; PARP, poly(ADP-ribose) polymerase; PAsp(AED), poly(N-2,2′-dithiobis[ethylamine] aspartamide); PDPA, poly(2-[diisopropyl amino]ethyl methacrylate); PEG2000-PE, PEG 2000-phosphatidyl ethanolamine; PEI, polyethylenimine; pNIPMAm, poly N-isopropylmethacrylamide rHDL, reconstituted high-density lipoprotein; siID4, siRNA targeting ID4; STAT3, signal transducer and activator of transcription 3; TPN, tumor-penetrating nanocomplex.
Future initiatives must continue to illuminate the molecular mechanisms and genes that underlie ovarian cancer. Through the characterization of better gene targets, various molecular pathways may be knocked down by combinatorial siRNA therapeutics to prevent cancer cell proliferation. In addition, the potential for reduction of drug resistance in ovarian cancer via nanoparticle-mediated siRNA therapy is promising in vitro; however, additional in vivo results must be obtained. Furthermore, multiple siRNAs could be employed at once in combination with chemotherapy agents to knock down multiple genes implicated in drug resistance to ensure maximum apoptotic effects. Because of the heterogeneous nature of ovarian cancer, future treatments should be mindful of molecular markers and combine multiple methods (chemotherapy, immunotherapy, gene therapy, etc.) for optimal treatment. Development of early detection strategies for ovarian cancer would have a remarkable impact on patient outcomes, as the 5-year survival rate for ovarian cancers detected in stage I-II is greater than 70%. 1 Potential nanotheranostic strategies for the early detection, characterization, and treatment of ovarian cancer could employ siRNA as a therapeutic modality for the knockdown of gene targets involved in early-stage cancer cell proliferation.
Progression from preclinical trials must occur to allow for evaluation of siRNA-based nanotherapeutics in vivo. First, in vivo safety of siRNA-based therapeutics and the nanoparticle delivery system must be ensured through consideration of off-target effects and potential toxicities. Pharmacological studies should be performed alongside current studies to ensure the mechanisms of distribution, clearance, and potential toxicity are well understood. Nanoparticle design must be carefully evaluated, as cationic nanoparticles, such as those used to adhere to anionic siRNA, have been shown to be potentially toxic in vivo. 101 Moreover, studies of potential long-term toxicity events should be completed to ensure clinical safety of siRNA-nanoparticle therapeutics. Organ-on-a-chip and organ-systems-on-a-chip technologies for the testing of potential therapeutics could be employed to better characterize off-target cytotoxicity and treatment efficacy and screen potential drug candidates in a high-throughput manner. Future applications of siRNA therapeutics in clinical trials must also consider patient preference, and administration strategies should be optimized to ensure patient compliance with a treatment schedule that requires repeat dosing. Challenges remain before nanoparticle-siRNA therapeutics are ready for clinical applications; however, the potential of nanoparticle-mediated delivery of siRNA-based therapies to treat ovarian cancer is emerging and shows promise.
Footnotes
Declaration of Conflicting Interests
The authors declared no potential conflicts of interest with respect to the research, authorship, and/or publication of this article.
Funding
The authors received no financial support for the research, authorship, and/or publication of this article.