
Abstract
Community-acquired pneumonia (CAP) is a common infectious disease linked to high rates of morbidity and mortality. Fast and accurate identification of the pathogens responsible for CAP will aid in diagnosis. We established a capillary electrophoresis-based multiplex PCR (CEMP) panel to enable the detection of viral and bacterial pathogens associated with CAP. The assay simultaneously detects and identifies the 13 common unculturable CAP viral and bacterial pathogens within 4 h. We evaluated the performance of a commercially available panel with 314 samples collected from CAP patients. We compared the results to those obtained with the liquid chip-based Luminex xTAG Respiratory Viral Panel (RVP) Fast Kit (for viruses) and the agarose gel-based Seegene PneumoBacter ACE Detection Kit (for atypical bacteria). All positive samples were further verified by the Sanger sequencing method. The sensitivity, specificity, positive predictive value, and negative predictive value of CEMP were 97.31%, 100%, 100%, and 99.85%, respectively. CEMP provides a rapid and accurate method for the high-throughput detection of pathogens in patients with CAP.
Introduction
Community-acquired pneumonia (CAP), one of the most common infectious diseases, carries a significant healthcare burden worldwide.1,2 Susceptible populations include children, the elderly, and patients with underlying conditions. People taking immunosuppressive therapy are at higher risk for CAP than immunocompetent patients due to the inflammatory response and defective immune functionality.3–5 CAP remains a major cause of morbidity and mortality due to complications including sepsis, multiple organ failure, and acute respiratory distress syndrome. CAP-related complications are particularly high in developing countries where medical resources are limited. 6 Diagnosis is challenging when based solely on clinical observations since the common symptoms of CAP, especially at an early stage, are similar to those of ordinary respiratory tract infections. Without reliable tests, it is difficult to determine the best care management. Physicians have to select the treatment empirically based on the nature of pneumonia, and antibiotic treatments are typically prescribed in worst-case scenarios. Injudicious treatment by clinicians, as a result of lack of objective diagnosis at first hand and significant delay of standard procedures, has contributed to the emergence of CAP with severe symptoms that is directly associated with a higher mortality rate. 7 Thus, it is of great benefit for patients and providers to have a rapid and precise diagnosis to guide treatment decisions. 8
Standard clinical microbiology laboratory methods culture bacteria from patients’ respiratory tracts and identify the pathogens by the morphological characteristics of the colonies. 1 This is time-consuming, labor-intensive, and unreliable. 9 Viral pathogens and some bacteria require molecular approaches for identification and characterization.10–12 Multiplex PCR methods, such as Seegene Seeplex and Luminex xTAG, have been used to detect CAP-associated respiratory pathogens.11,13–18 Both culturing and existing multiplexed PCR methods are time-consuming and have limited ability to identify CAP pathogens. Additionally, CAP patients may be infected with multiple pathogens concurrently. 19 However, most diagnostic panels integrate an insufficient number of pathogens to guide the clinician. Pathogens responsible for CAP vary with geography, and this is an important consideration when diagnosing a patient. Current commercial assays do not account for these geographical variations. Therefore, multiple tests are often needed to cover pathogens common to the patient’s locale. Some PCR assays cannot differentiate pathogens that share common genomic features. For example, human rhinovirus (HRV) presents a high genomic sequence similarity to human enterovirus (HEV) and some PCR methods cannot distinguish between the two of them.20,21 Another drawback of existing assays is that they cannot identify pathogens in samples with degraded nucleic acid. This can increase the false-negative results. All these limitations dramatically increase the diagnostic time and healthcare cost. While emerging microfluidic technologies, such as electrothermal flow and electrochemical biosensor technologies, have the potential to deliver point-of-care diagnostics for CAP, these methods suffer from matrix effects in patient samples.22–26 Some applications integrated with microfluidic techniques, such as FilmArray PCR, have been commercialized. However, a well-established, generic, high-throughput, microfluidic-based product remains unavailable to clinical CAP testing.
Here, we report a clinical study of a capillary electrophoresis-based multiplex PCR (CEMP) assay in patients with CAP. We used a capillary electrophoresis-based genetic analysis system and a commercially available PCR panel. The panel covers the 13 common unculturable CAP viruses and atypical bacterial pathogens in East Asia. Included in the panel are influenza A virus (flu A, subtypes 2009H1N1 and H3), influenza B virus (flu B), respiratory syncytial virus (RSV), human parainfluenza virus (HPIV), human metapneumovirus (HMPV), human adenovirus (HAdV), HRV, human bocavirus (HBoV), human coronavirus (HCoV), Mycoplasma pneumoniae (Mp), and Chlamydia (Ch) (Chlamydia pneumoniae [Cp] and Chlamydia trachomatis).27–31 We did not include Streptococcus pneumoniae (Sp) and Haemophilus influenzae (Hi) in the panel because these two pathogens reside asymptomatically in healthy carriers and would generate false-positive results. The extraction kit of the panel can process the nucleic acids of both viral and bacterial pathogens in one procedure. We achieved multiplex pathogen detection with the electrophoretic separation of specific PCR products ( Fig. 1 ).32,33 CEMP can process 96 samples simultaneously and identify the pathogen in as little as 4 h, compared with days in standard clinical procedures. The presence of a specific pathogen is objectively determined by comparison to the size standard (containing different fragments of sizes) migration times. In addition to the specific PCR products of the pathogens, nucleic acid degradation of the sample can be detected in the electropherogram. This establishes the quality of the sample and determines the validity of the test. This diagnostic test provides a comprehensive approach to detect multiple pathogens and sample degradation simultaneously.
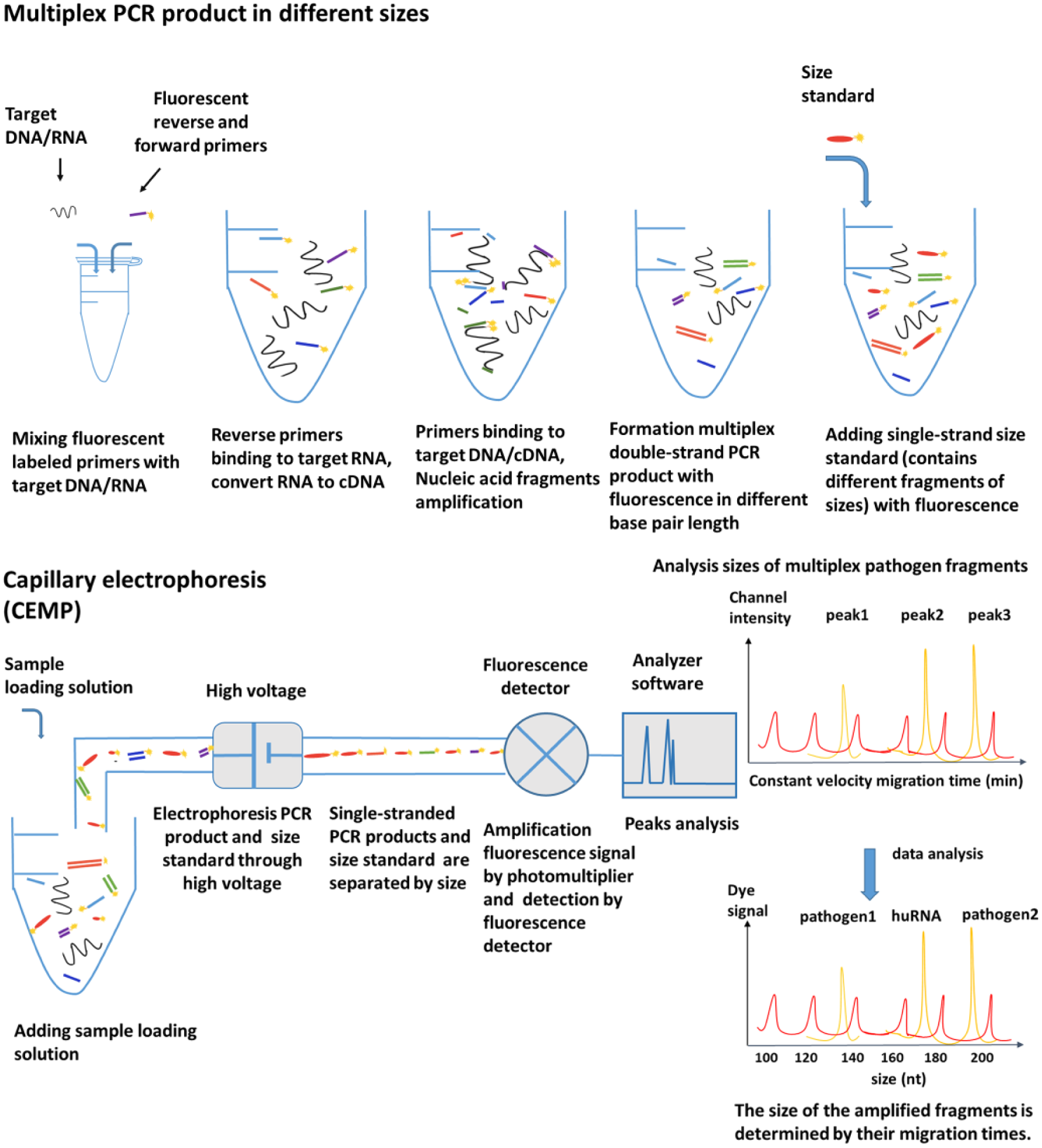
Multiplex pathogen detection scheme using CEMP. The nucleic acids of samples were mixed with fluorescently labeled primers. There were 15 different pairs of primers (13 target pathogens, human DNA, and human RNA) in the CEMP assay. Each pair of primers (a forward primer and a reverse primer) amplified one target fragment. Different amplification products have different lengths. A fluorescently labeled size standard was added to the product. We analyzed these samples using a capillary electrophoresis analyzer. Smaller fragments move quickly, and larger fragments move slowly. By comparison to the migration time of the size standard, the various lengths of PCR product fragments were determined and specific pathogens were detected.
Materials and Methods
Ethics Statement
Patient respiratory specimens were collected with approval from the Ethics Committee of Beijing Tongren Hospital Affiliated with Capital Medical University (ECBTH). Informed consent forms were obtained from all study subjects with CAP. Consent was approved by the ECBTH and documented with a written record of the participant’s name, medical record number, and date of enrollment.
Sample collection
Respiratory samples were collected from Beijing Tongren Hospital affiliated with Capital Medical University and Beijing Luhe Hospital affiliated with Capital Medical University between January 2014 and December 2016. Samples were collected based on the clinical judgment of the treating physician in accordance with the Infectious Diseases Society of America/American Thoracic Society consensus guidelines on the management of CAP in adults. 34 All patients were over 18 years old and diagnosed with CAP within 48 h of admission.
For each patient, a sample was collected within 48 h of admission to the hospital. During the study, a total of 314 respiratory samples were collected. Depending on the patient’s status, induced sputum, bronchoalveolar lavage, or pharyngeal swab was obtained by sputum induction, bronchoscopy, or FLOQSwabs (Copan, Italy), respectively. Table 1 details the collection methods used in the study. All samples were sent for clinical laboratory pathogen detection at the Tongren clinical microbiology laboratory. Those samples not assayed immediately were stored at −80 °C until testing. Qualitative detection was done with the CEMP method.
Patient Samples Detected in the Study.
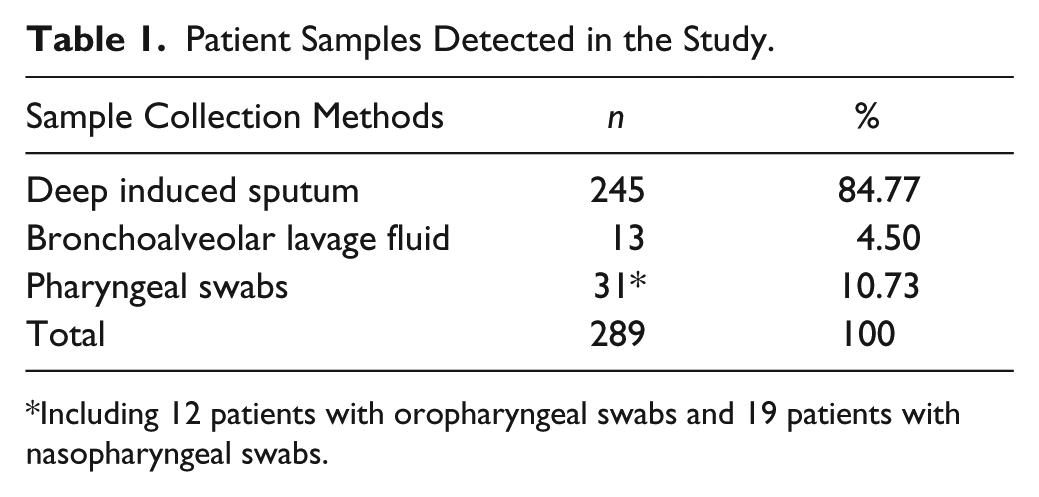
Including 12 patients with oropharyngeal swabs and 19 patients with nasopharyngeal swabs.
Nucleic Acid Extraction
Total DNA/RNA was extracted with QIAamp MinElute Virus Spin Kit (QIAGEN, Hilden, Germany) according to the manufacturer’s protocol. With this kit, all the nucleic acids of both viral and bacterial pathogens were extracted simultaneously, eliminating multiple lysis processes for different pathogens. For each specimen, the genomic material was extracted from a 200 µL sample suspension and the extracted nucleic acids were eluted in 60 µL of distilled water.
CEMP Assay for Clinical Specimens
We used the CEMP-compatible assay Respiratory Pathogen Multiplex Detection Kit (Ningbo HEALTH Gene Technologies Ltd., Ningbo, China) for our multiplex PCRs. The assay includes PCR enzyme, 0.25 μM of each of the 15 pairs of primers for each targeted pathogen, dNTPs, MgCl2, and buffer. Each pair of primers (a forward and a reverse primer) amplifies one target fragment. The 15 pairs of primers detect 13 pathogens as well as human DNA and human RNA. The volume of the PCR was 20 µL containing 14 µL of PCR mix buffer, 1 µL of PCR enzyme mix (containing uracil-DNA glycosylase enzyme, reverse transcriptase, and DNA polymerase), and 5 µL of extracted nucleic acids. Multiplex PCR was completed on a T100 thermal cycler (Bio-Rad, Hercules, CA) with this procedure: step 1, 25 °C for 5 min, 50 °C for 15 min, 95 °C for 2 min; step 2, 94 °C for 30 s, 60 °C for 30 s, 72 °C for 30 s. Step 2 was repeated for 35 cycles. The samples were then incubated at 72 °C for 10 min and held at 4 °C.
After PCR amplification, each sample was prepared for capillary electrophoresis by mixing 1 µL of the amplified product with 30 µL of GenomeLab sample loading solution, 0.3 µL of GenomeLab DNA size standard 400, and one drop of mineral oil (all from Beckman Coulter, Fullerton, CA). PCR products were separated by size on the GeXP capillary electrophoresis system (Sciex, Concord, ON, Canada) in less than 60 min. The signals of the 15 labeled PCR products were measured by fluorescence.31,35 Given by the kit instructions, the positions of pathogen amplicons are as follows: flu A 105 nt (2009H1N1 163.3 nt, H3N2 244.9 nt), HAdV 110.2/113.9 nt (represents different subtypes), HBoV 121.6 nt, HRV 129.6 nt, HPIV 181.6 nt, Ch 190.5 nt, HMPV 202.8 nt, flu B 212.7 nt, Mp 217 nt, HCoV 265.1 nt, and RSV 280.3 nt. The unit “nt” is the abbreviation of “nucleotide.” The number of nucleotides of a fragment is used as a measure of the length of a fragment. In this experiment, the lengths of amplified fragments are calculated by the migration time instead of direct measuring, which may result in the nonintegral length in nucleotides. The difference between actual fragment size and the reference fragment size has to be within 1.5 nt. 35 The CEMP assay included positive and negative controls and was tested to acquire control values prior to the tests with patient samples. The test was positive if the peak value of the targeted pathogen in the amplification product was higher than the peak value of the positive control. The results were considered negative if the peaks were smaller than a certain value. In the cases that yielded intermediate peak values, the samples were considered indeterminate and would be retested.
Patient samples invariably contain some human nucleic acid. We used the presence of human DNA and RNA in the sample as proof that the sample had not degraded. If we found no evidence of human genomic material in the sample, it was considered degraded and not used for pathogen detection.
Comparison to Other Multiplex PCR Assays
As a comparison to the CEMP method, all samples were processed by two other multiplex PCR assays. For viruses, we used the xTAG Respiratory Viral Panel (RVP) Fast Kit (Luminex Molecular Diagnostics Corporation, Toronto, ON, Canada) to detect and quantify the amplicons produced by targeted pathogens.36,37 The xTAG RVP Fast Kit is based on the Liquid Chip technology integrated in the Luminex MAGPIX system (Austin, TX). The xTAG RVP Fast Kit includes the following 18 virus targets: flu A (subtypes H1, H3, and 2009H1N1), flu B, RSV A and B, HPIV 1–4, HMPV, HAdV, HRV/HEV, HBoV, and HCoV 229E, NL63, OC43, HKU1. 17 Fluorescence intensity results on the MAGPIX were categorized as positive (median fluorescence intensity ≥300), negative (median fluorescence intensity <150), or “no call.”15,38
For atypical bacteria, we used the Seeplex PneumoBacter ACE Detection Kit (Seegene Inc. Seoul, Korea) to generate amplicons that were separated by agarose gel electrophoresis following the instructions provided by the manufacturer. Agarose gel (2%) was prepared by mixing 30 mL of 1× Tris-acetate-EDTA (TAE) buffer (Beijing Solarbio Science & Technology Corporation, Beijing, China) and 0.6 g of agarose powder (TransGen Biotech Corporation, Beijing, China) and microwaving for 3 min. The agarose solution was allowed to cool down to about 50 °C. Three microliters of GelStain (10,000×) (TransGen Biotech Corporation) was added into the agarose solution. The agarose solution was poured in a casting mold (6 × 6 cm) with an appropriate well comb in place. It was allowed to sit at room temperature for 20–30 min, until it completely solidified. For PCR amplification electrophoresis, the voltage was set at 110 V for 45 min via 2% agarose gel.34,39,40 The PneumoBacter ACE Detection Kit targets six bacteria: Mp, Legionella pneumophila (Lp), Sp, Hi, Bordetella pertussis (Bp), and Cp.
The sizes of the amplicon characteristics of Mp, Lp, Sp, Hi, Bp, and Cp were 583, 472, 350, 257, 200, and 146 bp, respectively.41–43 The agarose gel was read on a Tanon 1600 Gel Image System (Tanon Inc., Shanghai, China). Signals were considered positive if they were greater than the signal of the internal size marker. Each amplification reaction contained plasmid DNA as an internal control; if the results of internal controls were negative, the PCR was considered to have failed.
A selection table is presented in Table 2 to illustrate the corresponding targeted pathogens for each kit. The xTAG RVP Fast Kit covers the most common viruses, but it misses some pathogens commonly linked to CAP in the Asian area. The PneumoBacter ACE Detection Kit is specially used as a comparison for the atypical pathogens included in the CEMP panel. The characteristics and detectable pathogens of the three kits are summarized in Table 2 .
Comparison of Three Commercial Kits for Detecting Respiratory Pathogens.
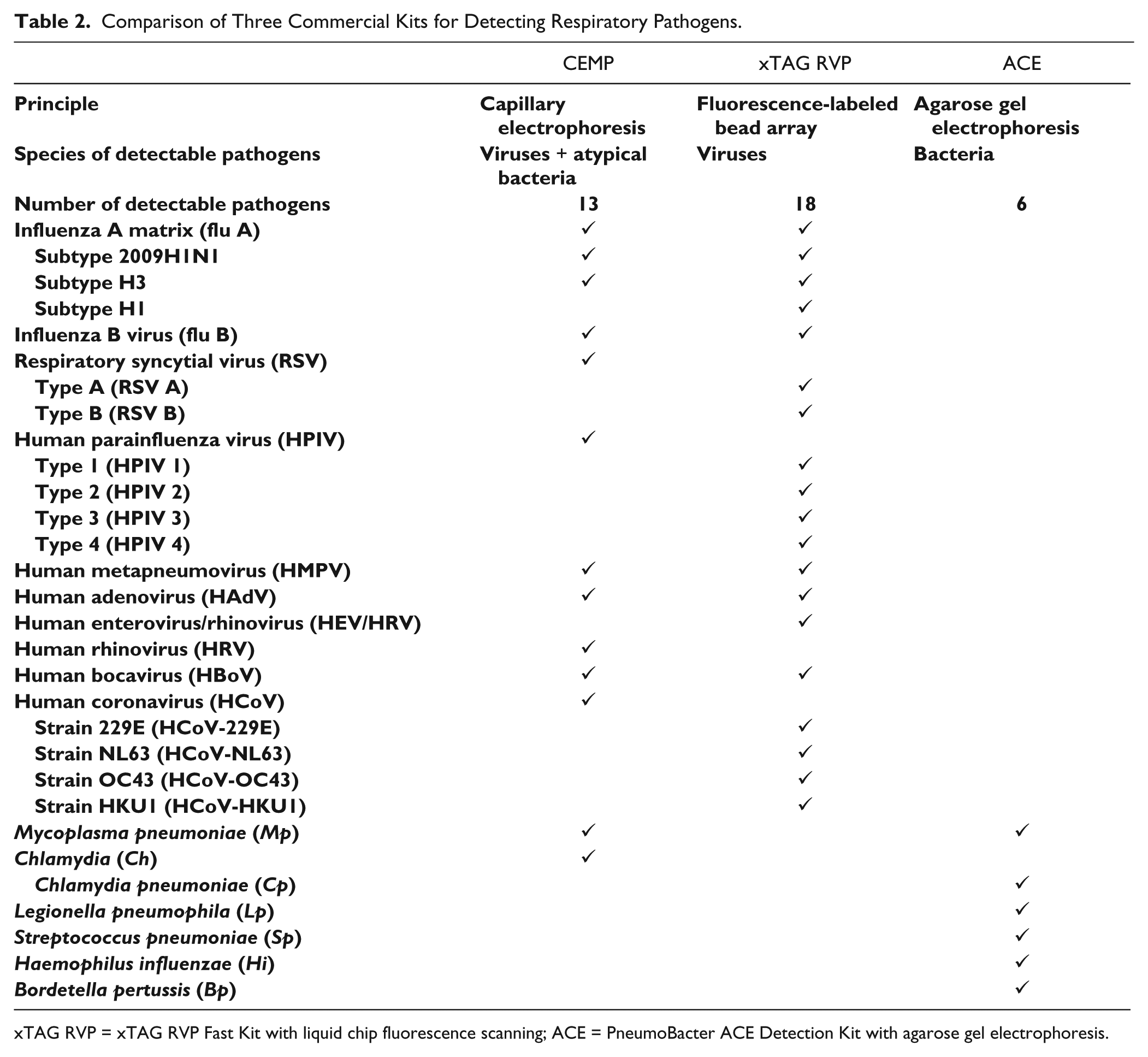
xTAG RVP = xTAG RVP Fast Kit with liquid chip fluorescence scanning; ACE = PneumoBacter ACE Detection Kit with agarose gel electrophoresis.
Verification by Amplicon Sequencing
We verified all positive samples by sequencing the amplicon products with the GenomeLab Dye Terminator Cycle Sequencing Quick Start Kit (Beckman Coulter) on a GeXP system. The sequences were compared with those in GenBank using BLAST 44 ( Fig. 2 ). Sequencing results served as the gold standard with which to compare our experimental results. 45
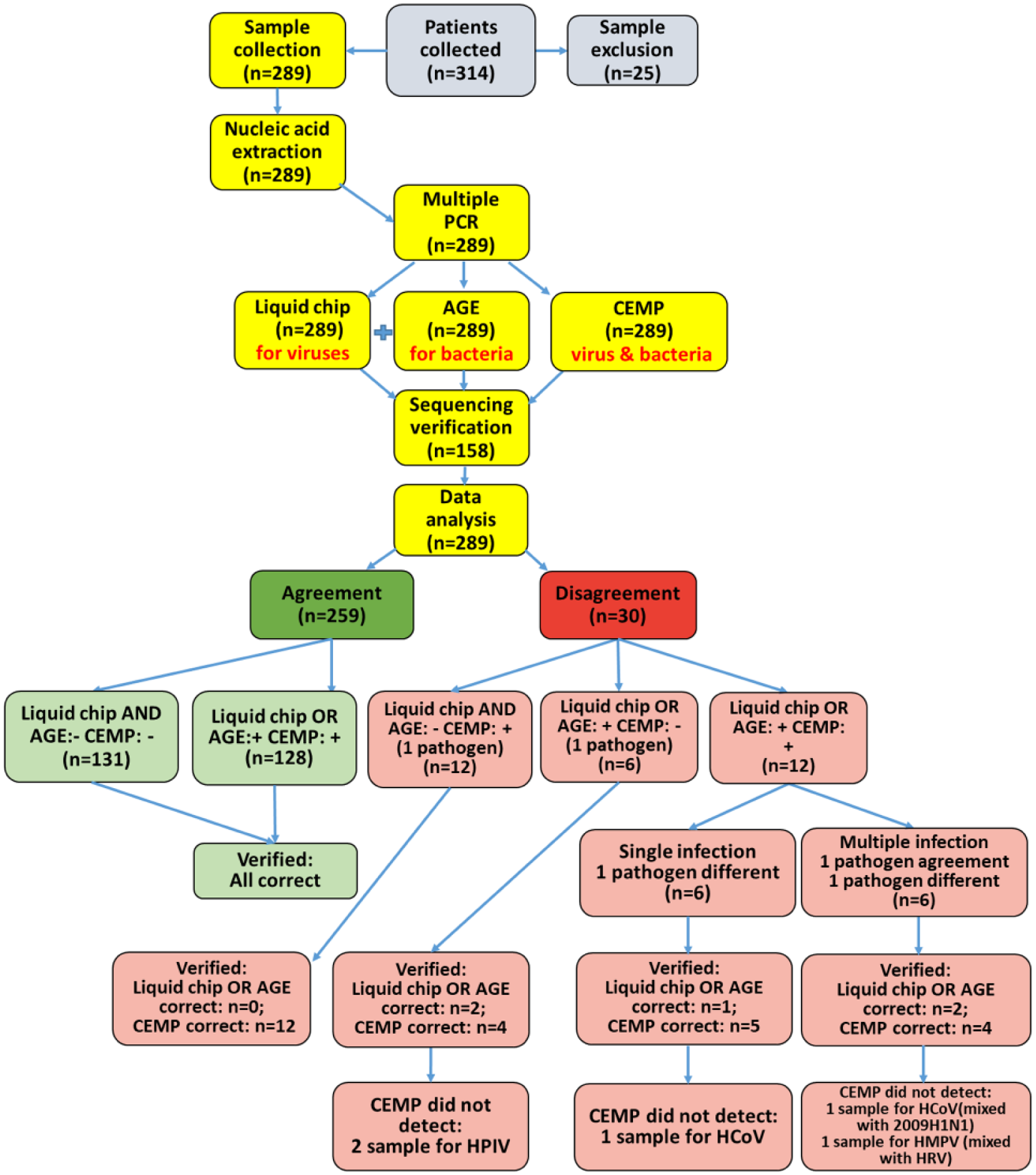
Clinical study flowchart. Of 314 samples collected from 314 CAP patients, 259 specimens identified by CEMP were in complete agreement with those identified by liquid chip and agarose gel electrophoresis analysis, including 131 negative samples and 128 positive samples. Twelve samples were in partial agreement, including six single infections and six multiple infections.
Data Analysis
We assessed the performance of our method by examining the sensitivity, specificity, positive predictive value (PPV), and negative predictive value (NPV). In the experiment, we used the gene sequencing result as the reference standard (REF). All the CEMP results were verified by the gene sequencing. The kappa values were calculated to determine the agreement of CEMP and sequencing. Some samples experienced nucleic acid degradation due to temperature variation and sample contamination, which were detected by CEMP. Therefore, these samples were omitted from data analysis due to the lack of valid gene sequence information. All statistical analyses, including kappa values, positive and negative predictive values, agreement/consistency rate, and true-positive and true-negative rates, were calculated by the Statistical Package for Social Sciences (SPSS) software (version 22.0; IBM Corp., Armonk, NY).
Results
Study Population and Sample Characteristics
A total of 314 patients were recruited as study participants between January 2014 and December 2016. A single sample was collected from each participant for a total of 314 samples. Twenty-five samples were excluded due to excessive sample degradation as indicated by the loss of human DNA or RNA from the CEMP results. The samples and their respective sources (sputum induction, bronchoscopy, and pharyngeal swab) are listed in Table 1 .
Of the 289 samples used in this study ( Table 3 ), 54.67% (158/289) had viruses or atypical bacteria known to cause CAP. Of the 158 pathogen-positive cases, 82.91% (131/158) indicated the presence of a single pathogen and 17.09% (27/158) had more than one pathogen. One sample was infected with three pathogens (flu A/2009H1N1, H3, and HRV). All viruses included in the CEMP assay were detected except HBoV. Flu A and HRV were the most common viruses found in single infections and co-infections.
Respiratory Pathogens Detected in Enrolled Samples.

Including one sample of 2009H1N1, H3, and HRV triple infection.
Capillary Electrophoresis Clinical Validation
We tested 289 patient samples with our CEMP assay for 13 respiratory pathogens commonly associated with CAP. Examples are shown in Figure 3 . Additionally, we tested the same samples by two other techniques. Finally, we sequenced the amplicons generated by each sample to verify the results of all methods.
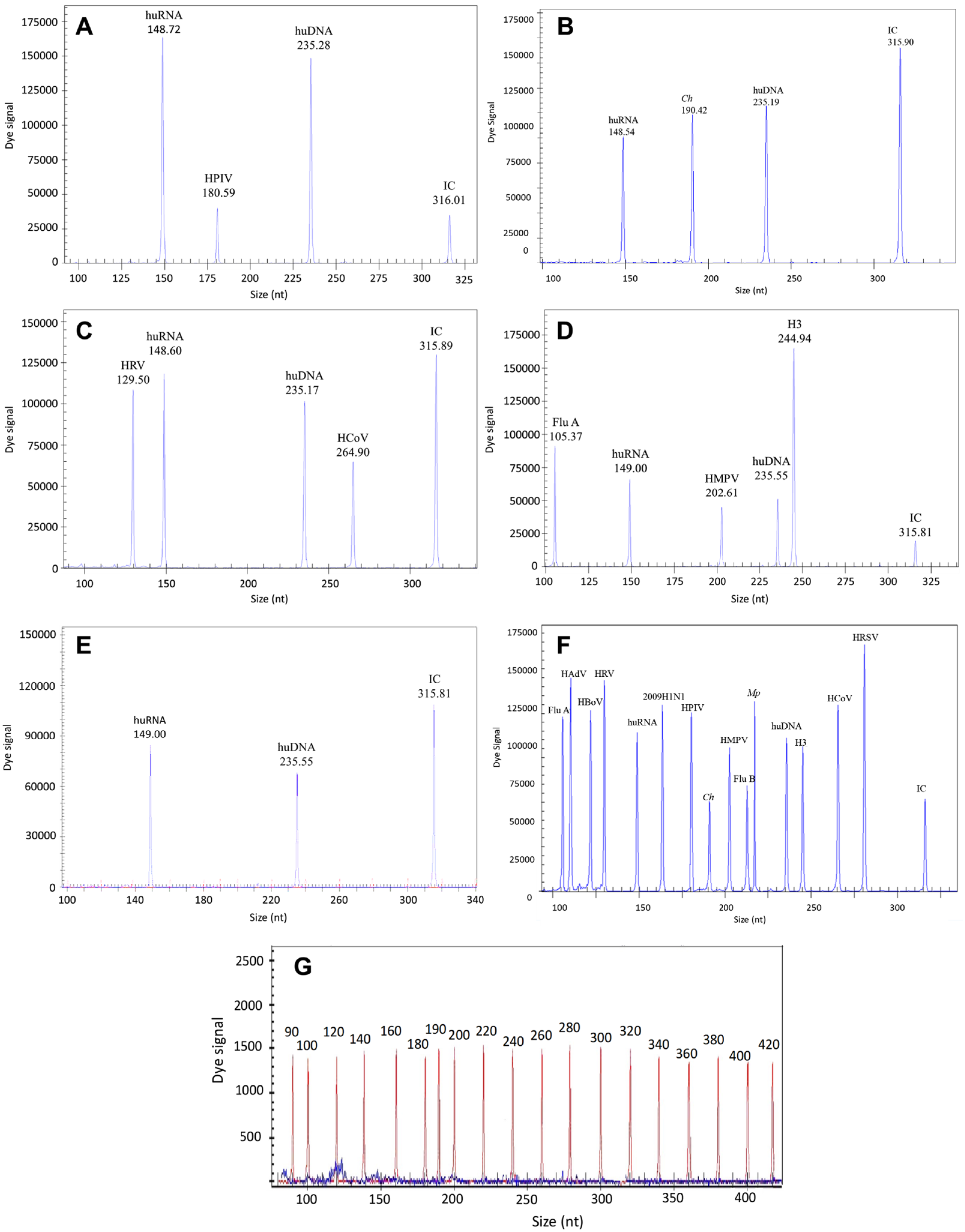
Quantitative detection of bacterial and viral pathogens in the respiratory samples from CAP patients by CEMP. These electropherograms show pathogens detected in the samples from CAP patients. The Y axis indicates the fluorescent signal, and the X axis indicates the size of the PCR products, including huRNA (human RNA), huDNA (human DNA), and IC (internal control). (
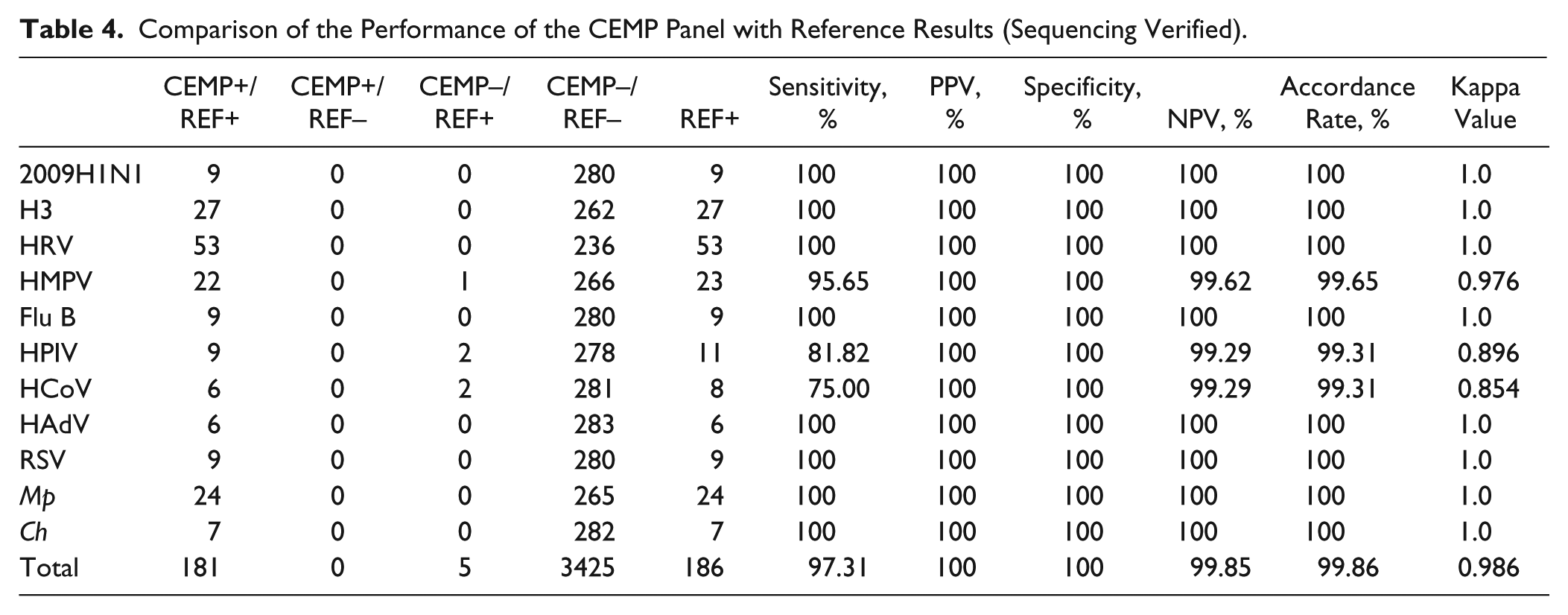
Of the 289 samples tested with CEMP, 89.62% (259/289) generated the same results when tested with the liquid chip and the agarose gel methods. Figure 2 shows that the 131 samples that CEMP found to be negative were confirmed to be negative with sequencing results, indicating that no false positives were detected by CEMP. A total of 128 samples tested positive for all three experimental assays. The overall kappa value of CEMP compared with the reference was 0.986. The overall sensitivity, specificity, PPV, and NPV of the CEMP were 97.31%, 100%, 100%, and 99.85%, respectively ( Table 4 ). Even the lowest kappa values (for HPIV and HCoV samples) were greater than 0.85.
Comparison of the Performance of the CEMP Panel with Reference Results (Sequencing Verified).
In 30 out of 289 results, the three nonsequencing methods were inconsistent. There were 18 instances of detection mismatches in single infection samples between CEMP and the alternative liquid chip or agarose gel electrophoresis assay. In 12 of those 18 tests, CEMP detected a pathogen while neither alternative method did. Sequencing confirmed the positive CEMP results. In the remaining six of those tests, either the liquid chip or the agarose gel method identified a pathogen while CEMP did not. Subsequent sequencing showed that the CEMP method was correct in four tests while it was incorrect in two instances, both for HPIV.
Of the remaining 12 of 30 inconsistent results, sequencing confirmed that the CEMP method was correct in 9 cases while the alternative methods were correct in 3 ( Fig. 2 ).
Of the 289 samples, CEMP correctly detected all 20 flu A H3N2-positive samples. Only 15 samples were H3N2 positive by liquid chip. For HAdV, CEMP correctly detected two positive samples, while liquid chip falsely identified three samples as positive. Moreover, both false positives and false negatives were found in the detection of eight samples for HRV, HMPV, and flu B by liquid chip. In addition, seven Mp samples and two Ch samples were falsely identified as negative by gel electrophoresis but were correctly identified by CEMP. Nevertheless, CEMP also had false negatives for five samples, including two for HPIV, two for HCoV, and one for HMPV. Among those five samples, one was an HCoV and 2009H1N1 co-infection. Another one was an HMPV and HRV co-infection. In spite of the false-negative results, CEMP correctly detected the 2009H1N1 and HRV in two samples ( Fig. 2 ).
Discussion
We report a clinical study of CEMP, a rapid molecular assay for respiratory pathogen identification in patients with CAP. This assay is based on multiplex PCR amplification and the capillary electrophoretic separation of PCR amplicons by length. Multiplex PCR uses PCR to amplify several different DNA sequences simultaneously. It reduces both the reagent usage and the time required for analysis. The capillary electrophoretic separation of fluorescently labeled amplicons has previously been clinically applied to the detection of pathogens such as human papillomavirus and flu A H1N1.46,47 By comparing the results with a standard size marker of targeted pathogens, pathogens in the samples can be identified as expected.33,45,48,49 To our knowledge, this is the first time that CEMP has been used to detect the pathogens of CAP adult patients.
None of the 289 tested samples produced false positives from nonspecific amplification. The experiment results show that the overall sensitivity, specificity, PPV, and NPV of the CEMP were above 97%. The kappa value comparing CEMP with the reference was above 0.9. This was sufficiently sensitive and specific for the detection of CAP pathogens. The two lowest kappa values from HPIV and HCoV samples were both greater than 0.85.
Compared to liquid chip method, the CEMP panel provides more reliable results when detecting the H3N2 subtype of flu A. This is most likely due to the primer specificity. A certain degree of diversity and mutation was observed in flu A virus,50,51 especially for H3N2, which usually mutates into many strains (NIAID Influenza Genome Sequencing Project Data Releasing Status, influenza virus resource, NCBI). The primers in the panel of xTAG RVP (liquid chip technology) could not amplify all forms of the mutated strains of the subtype.52,53 The CEMP panel gives more specific results in the detection of the genus Enterovirus. The Enterovirus genus includes nine enterovirus species and three HRV species. All 53 HRV-positive samples were detected by CEMP, while the liquid chip could merely identify the pathogens as genus Enterovirus, unable to differentiate between HRV and HEV.21,54
In previous clinical research, nasopharyngeal aspirates were taken from the children with pneumonia or bronchopneumonia. In that case, the sensitivity of a CEMP method run on a GeXP system was reported to be similar to that of the liquid chip assay. However, no verification process was included in that report. 32 Moreover, flu A/H3, one of the most common subtypes of flu A, and Mp, the most common atypical bacteria of pneumonia, were not included in the assay.55,56 In contrast, not only did we study the performance of CEMP in the detection of Mp and flu A, as well as its subtypes, but we also sequenced the samples to verify the results. In the works by Chen et al. and Tang et al., a new liquid chip panel, the Luminex NxTAG RVP assay, was used to detect viruses and atypical bacteria. These papers demonstrated that the sensitivity and specificity of the liquid chip with the new panel were 80%–100% and 98.9%–100%, respectively.57,58 In their studies, the samples were collected with nasopharyngeal swabs from patients with respiratory tract infection, which may result in a lower overall specificity than that of lower respiratory samples. In our study, deep induced sputum and the bronchoalveolar lavage fluid were the primary selective specimens. Pharyngeal swabs were suitable only for virus detection, while lower respiratory samples were suitable for the detection of atypical bacteria and intracellular pathogens other than virus.2,41,59 In addition, the use of lower respiratory samples can effectively avoid the influence of colonized pathogens.60–62 The Seeplex PneumoBacter ACE Detection Kit was a useful screening tool for the rapid detection of respiratory bacterial pathogens, but the sensitivity and specificity for Mp detection were reportedly 52.4% and 92.6%, respectively, both less than those with CEMP.43,63 Yang et al. also reported that a GeXP PCR assay had comparable sensitivity and specificity to those of our CEMP assay. 47
Under the circumstances of handling, storage, and testing, the nucleic acids of some pathogens can degrade before testing, generating false-negative results. In this study, we found that the nucleic acids in 8% of the samples had degraded. Unlike the liquid chip and agarose gel electrophoresis methods, CEMP allowed us to determine and exclude the samples in which nucleic acids are degraded. In addition, the uracil-DNA glycosylase enzyme added in the Respiratory Pathogen Multiplex Detection Kit panel employed in CEMP prevents the carryover contamination of PCR amplification product (nucleic acid containing d-UTP) of the previous experiment, which commonly occurs during multiplex PCR. We can detect co-infections with multiple pathogens with one test, considerably increasing the clinical efficiency. It takes the operator only 4 h to finish one testing process.
While the overall concordance of CEMP in this study is high, the concordance for HPIV and HCoV is not as impressive. Considering the small number of HPIV and HCoV samples in this study, we need to investigate further the performance of CEMP on these pathogens. Another limitation of this study is that Sp and Hi, two common pathogens causing CAP, were not included in the present CEMP panel.29,64 We chose not to include Sp and Hi because they are frequently found in disease-free individuals. PCR-based detection for Sp and Hi with samples from nasopharynx and oropharynx may not indicate disease. 65 Besides, unlike the 13 unculturable pathogens included in the CEMP panel, Sp and Hi can be identified by culturing, making the demands of detection on these two pathogens less urgent. We expect to include the analysis of these pathogens in future assay panels. Other point-of-care biosensor technologies may also play important roles for pathogen identification and are being developed for Sp to expand the CEMP assay for unexplained severe pneumonia in the future. 25
This CEMP assay is highly suited to CAP diagnosis, especially in patients at risk for multiple pathogen infections. With short time-consuming and extraordinary concordance, the CEMP assay provides a rapid, accurate, and high-throughput pathogen-detecting method for patients with CAP.
Footnotes
Acknowledgements
The authors thank all the study participants: Dr. Lixin Tao for data analysis; Zhi Xu for helpful support in drafting this manuscript; Dr. Joe Olechno, Prof. Pak Kin Wong, and Stephen for manuscript suggestions and modifications.
Declaration of Conflicting Interests
The authors declared no potential conflicts of interest with respect to the research, authorship, and/or publication of this article.
Funding
The authors disclosed receipt of the following financial support for the research, authorship, and/or publication of this article: This work was supported by the Ministry of Science and Technology of the People’s Republic of China for the national key technology research and development program “The Control and Prevention of Major Infectious Diseases” (2014ZX10004005).