
Abstract
Cystic fibrosis (CF) is a hereditary disease caused by mutations in the gene coding for the cystic fibrosis transmembrane conductance regulator (CFTR). A large number of nearly 2000 reported mutations, including the premature termination codon (PTC) mutations, urgently require new and personalized medicines. We have developed cell-based assays for readthrough modulators of CFTR PTC mutations (or nonsense mutation suppressors), based on the trafficking and surface expression of CFTR. Approximately 85,000 compounds have been screened for two PTC mutations (Y122X and W1282X). The hit rates at the threshold of 50% greater than vehicle response are 2% and 1.4% for CFTR Y122X and CFTR W1282X, respectively. The overlap of the two hit sets at this stringent hit threshold is relatively small. Only ~28% of the hits from the W1282X screen were also hits in the Y122X screen. The overlap increases to ~50% if compounds are included that in the second screen achieve only a less stringent hit criterion, that is, horseradish peroxidase (HRP) activity greater than three standard deviations above the mean of the vehicle. Our data suggest that personalization may not need to address individual genotypes, but that patients with different CFTR PTC mutations could benefit from the same medicines.
Introduction
Cystic fibrosis (CF) is an autosomal recessive disorder affecting 1 in 3500 newborns,1,2 which derives from mutations in the cystic fibrosis transmembrane conductance regulator (CFTR) gene.3–5 More than 10 million Americans are genetic carriers. The gene encodes a protein of the ATP binding cassette (ABC) protein superfamily, and the CFTR protein is unique, as it is the only ABC transporter known to function as an ion channel.6–9 CFTR is the primary mediator of chloride secretion in many epithelia. Mutated CFTR channels may result in malfunctioning lungs, pancreas, and other organs. In the lungs of CF patients, defective CFTR can lead to mucus-clogged airways, bacterial infection, extensive lung damage, and eventually respiratory failure. 10
Traditional CF therapies focus on symptomatic relief (i.e., mucociliary clearance, anti-inflammation, and anti-infection). Recent progress has resulted in two approved disease-modifying therapies by Vertex Pharmaceuticals. These CFTR modulators, lumacaftor and ivacaftor, improve protein folding and trafficking (correction)11,12 and increase chloride channel activity (potentiation),13–15 respectively.
There are about 2000 CFTR mutations reported, most of which cause CF by impairing protein translation, folding and stability, trafficking, and/or function.16,17 Many challenges remain in finding personalized therapies for CF patients with mutations in the CFTR gene that cannot be treated with the Vertex medicines. Premature termination codon (PTC) mutations, which prevent the translation of full-length functional CFTR protein, belong to this category. 18 Personalized medicine for the treatment of these patients is an urgent unmet need.
PTC mutations are found on at least one allele in approximately 10% of people with CF. 18 There are currently 69 PTC mutations in the CFTR2 mutation database (http://cftr2.org). The two most prevalent PTC mutations, G542X and W1282X, occur in about 5% and 2.4% of patients, respectively, of the total observed CF population; all other PTC mutations are much rarer (http://cftr2.org). The main challenge in addressing this group of rare mutations is finding a therapy that will bring clinical benefit to all or at least a large subset of all patients with PTC mutations (pan-PTC therapy).
We have developed high-throughput screening (HTS) assays for two representative CFTR PTC mutants (Y122X and W1282X)19–22 to evaluate (1) the feasibility of personalized therapy for CFTR PTC mutations and (2) the need for mutation-specific versus the potential of finding pan-PTC mutation therapies. Screening of about 85,000 small-molecule compounds in both assays yielded a significant number of common hits, suggesting that a focus on the discovery of pan-PTC therapies is a viable strategy.
Materials and Methods
Constructs and Cell Lines
The human CFTR cDNA (NM_000492) is the backbone for the constructs used in screening. The horseradish peroxidase (HRP) isozyme C coding sequence was fused into the CFTR open reading frame between T2694 and A2695, corresponding to an insertion between Ser898 and Arg899 in the fourth extracellular loop of the CFTR protein (CFTR-HRP), as described previously. 23 CFTR mutations (Y122X, TAA; G542X, TGA; and W1282X, TGA) were generated through site-directed mutagenesis. CFTR and CFTR-HRP were cloned into the vector pcDNA5/FRT (Thermo Fisher Scientific, Waltham, MA), under the control of the cytomegalovirus (CMV) promoter. Isogenic Fischer rat thyroid (FRT) cells were generated as described previously. 24
Cell Culture
FRT cells expressing CFTR and HRP fusion proteins (Y122X or W1282X) are cultured at 37 °C and 5% CO2 in Ham’s F-12, Coon’s Modification (Sigma-Aldrich, St. Louis, MO, #F6636) with 10% fetal bovine serum (FBS; Thermo Fisher Scientific, #26140-079), 1% penicillin-streptomycin (Thermo Fisher Scientific, #15140-122), and 100 µg/mL hygromycin B (Thermo Fisher Scientific, #10687010). These cells are maintained in 10-layer CellSTACK culture flasks (Corning, Corning, NY, #3320), and the media is replaced every 2–3 days. Cells are washed with Dulbecco’s phosphate-buffered saline (DPBS; Thermo Fisher Scientific, #14190); trypsinized for 10–15 min with Triple Express (Thermo Fisher Scientific, #12604021); pelleted at 1000 rpm for 5 min at room temperature; resuspended in 75% FRT growth media, 20% FBS, and 5% DMSO (Thermo Fisher Scientific, #BP231-100); and stored at −80 °C until use in HTS assays within 1–3 weeks.
HTS
Cells are removed from −80 °C freezers, thawed, and pelleted at 1000 rpm for 5 min at room temperature. The cells are resuspended in FRT growth media. For the Y122X screening, VX-809 (3 µM; Selleckchem, Houston, TX, #S1565) is added to the culture media. A multidrop reagent dispenser (Thermo Fisher Scientific, #22-387-053) is used to plate cells (25,000 cells/well, 50 µL/well) into 384-well assay plates (Corning, #3570BC). The plates are centrifuged at 500 rpm for 2 min at room temperature and incubated for 2 h at 37 °C and 5% CO2 before compound administration of 5 µL/well, to yield a final assay volume of 55 µL/well. The final compound concentration is 10 µM. The cells are incubated for 48 h at 37 °C and 5% CO2. Each assay plate includes as controls the vehicle (DMSO, 0.1%) and the combination of 3 µM VX-809 and 600 µM geneticin (G418; Sigma-Aldrich, #A-1720).
HRP Luminescence Assay
The HRP luminescence assay is prepared with a PlateMate Plus liquid handler (Thermo Fisher Scientific, #501-10001). The cells are washed three times with 1× DPBS, followed by incubation with SuperSignal West Femto HRP substrate (15 µL/well, Thermo Fisher Scientific, #34096) for 5 min at room temperature. The HRP-catalyzed luminescence is read with a Synergy Neo HTS microplate reader (Biotek, Winooski, VT) under the following conditions: room temperature; no shake/no delay; integration time, 0.1 s; read height, 8.00 mm.
FRT Cell Conductance Assay
FRT cells expressing CFTR mutants are seeded at a density of 3.8 × 105 cells/cm2 or 125,000 cells/filter on HTS Transwell 24-well filter inserts (Corning, #3378). Cells are submerged in Ham’s F-12, Coon’s Modification (Sigma-Aldrich, #F6636), and basolateral and apical media is changed every other day. One to two weeks after filter seeding, when cells have typically formed electrically tight epithelial cell monolayers with transepithelial electrical resistances of several kilo-ohms x square centimeter, they are assay ready. Cells are then treated on both sides with control (vehicle) or test compound and kept at 37 °C and 5% CO2.
Typically after 72 h, with a media change and fresh compound after 48 h, cell incubation medium is aspirated off and replaced with fresh HEPES-buffered (pH 7.4), compound- and bicarbonate-free F-12 culture medium. Cells are immediately mounted onto the assay platform and equilibrated to ~36 °C well temperature for 30 min. Transepithelial resistance is recorded using a 24-channel transepithelial current clamp amplifier (TECC-24, EP Design, Bertem, Belgium). Resistance values are collected at ~6 min intervals. Four to six measurements are used to determine baseline resistance, as well as the transepithelial resistances after each addition of CFTR agonists forskolin and VX-770 and antagonist CFTR-Inh172. Agonists and antagonist are prediluted to 10-fold concentrations in HEPES-buffered culture media and added to both basolateral and apical sides.
Transepithelial conductance (Gt) is calculated from resistance measurements corrected for series resistance. CFTR mediates chloride ion transport across the epithelial monolayer, and activation/inhibition of functional CFTR transport proteins results in a change in transepithelial conductance (ΔGt). Therefore, ΔGt can be used as a measure of functional CFTR surface expression or compound-mediated functional rescue of CFTR.
Western Analysis of Full-Length CFTR Protein
Cell lysates are separated by sodium dodecyl sulfate–polyacrylamide gel electrophoresis (SDS-PAGE) (3%–8% Tris-acetate gels) before being transferred to a polyvinylidene fluoride (PVDF) membrane. CFTR bands C and B are detected with antibody 596 (J. Riordan lab, University of North Carolina, Chapel Hill, NC). Na/K-ATPase, detected with a specific antibody (Thermo Fisher Scientific, #PA517251), is used as a loading control. Standard electrochemiluminescence (ECL)-based detection is performed using a Bio-Rad ChemiDoc XRS+ Gel Documentation System (Hercules, CA).
Results
Assay Design
CFTR is a multidomain membrane-spanning protein with complex regulation of its intracellular processing, trafficking, and channel activity. Many single amino acid changes in CFTR lead to disease-causing defects. Readthrough products of a PTC mutation-containing mRNA result from mispairing of the stop codon with an anticodon of a near-cognate aminoacyl-tRNA (usually complementary to two of the three bases). 25 Hence, readthrough protein products are likely not the wild type, but instead often will have other non-wild-type amino acids inserted in place of the PTC. In turn, this will lead to CFTR mutant proteins that may have trafficking and/or functional defects.
To develop a screening assay targeting PTC mutations in the context of full-length CFTR protein, the reporter protein HRP was fused into the fourth extracellular loop of CFTR (CFTR-HRP).23,26 We then introduced PTC mutations Y122X or W1282X into the fusion protein by site-directed mutagenesis ( Fig. 1 ). The Y122X PTC mutation (UAA, ochre) is located in the first transmembrane domain. W1282X (UGA, opal) is in nucleotide binding domain 2 (NBD2). HRP activity can be detected via a luminescence assay on live cells. The objective of this design is to detect PTC readthrough that yields trafficking-competent CFTR and filters out compounds that may provide readthrough but yield proteins with severe trafficking defects.
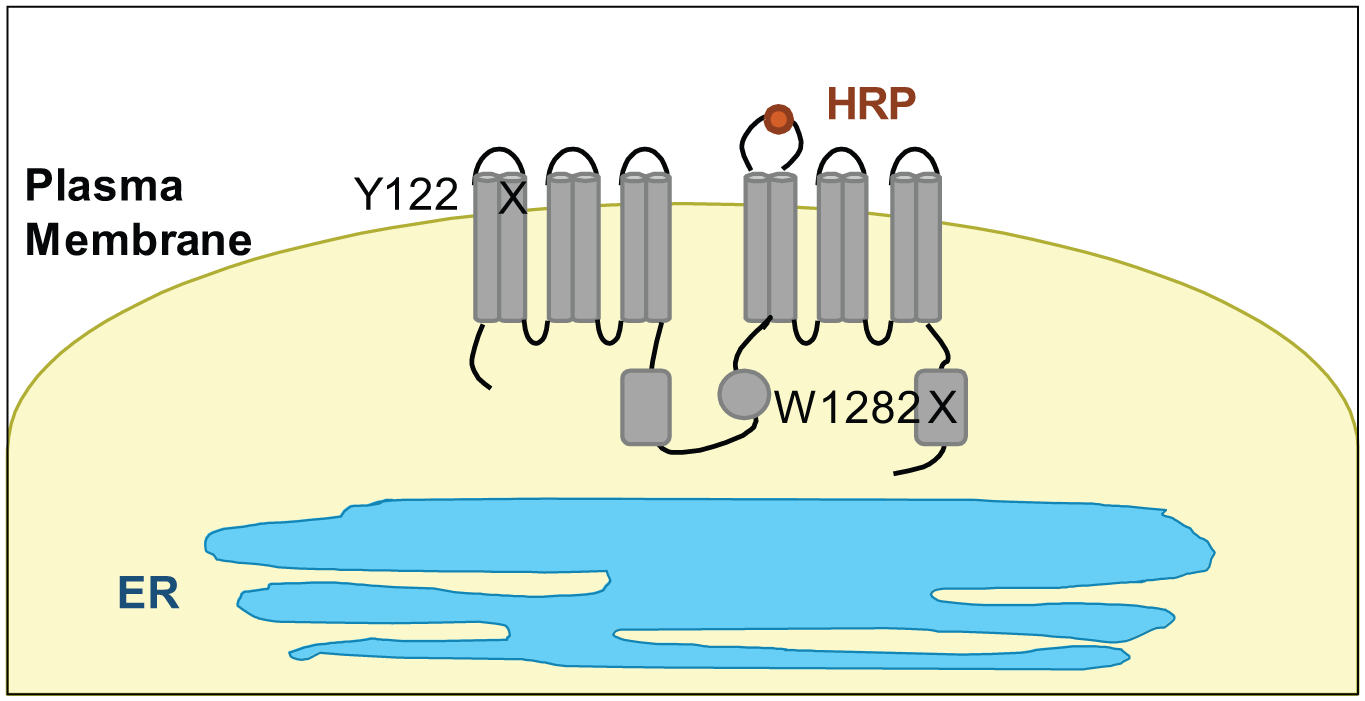
Reporter-based HTS assays using CFTR-HRP fusion proteins. Isogenic FRT cell lines were generated expressing CFTR-HRP fusion proteins with a PTC mutation (Y122X or W1282X). HRP is fused into an extracellular loop of the CFTR protein, yielding a robust reporter for CFTR channels at the plasma membrane. Cell surface HRP activity can be detected using a luminescence assay format.
Assay Development
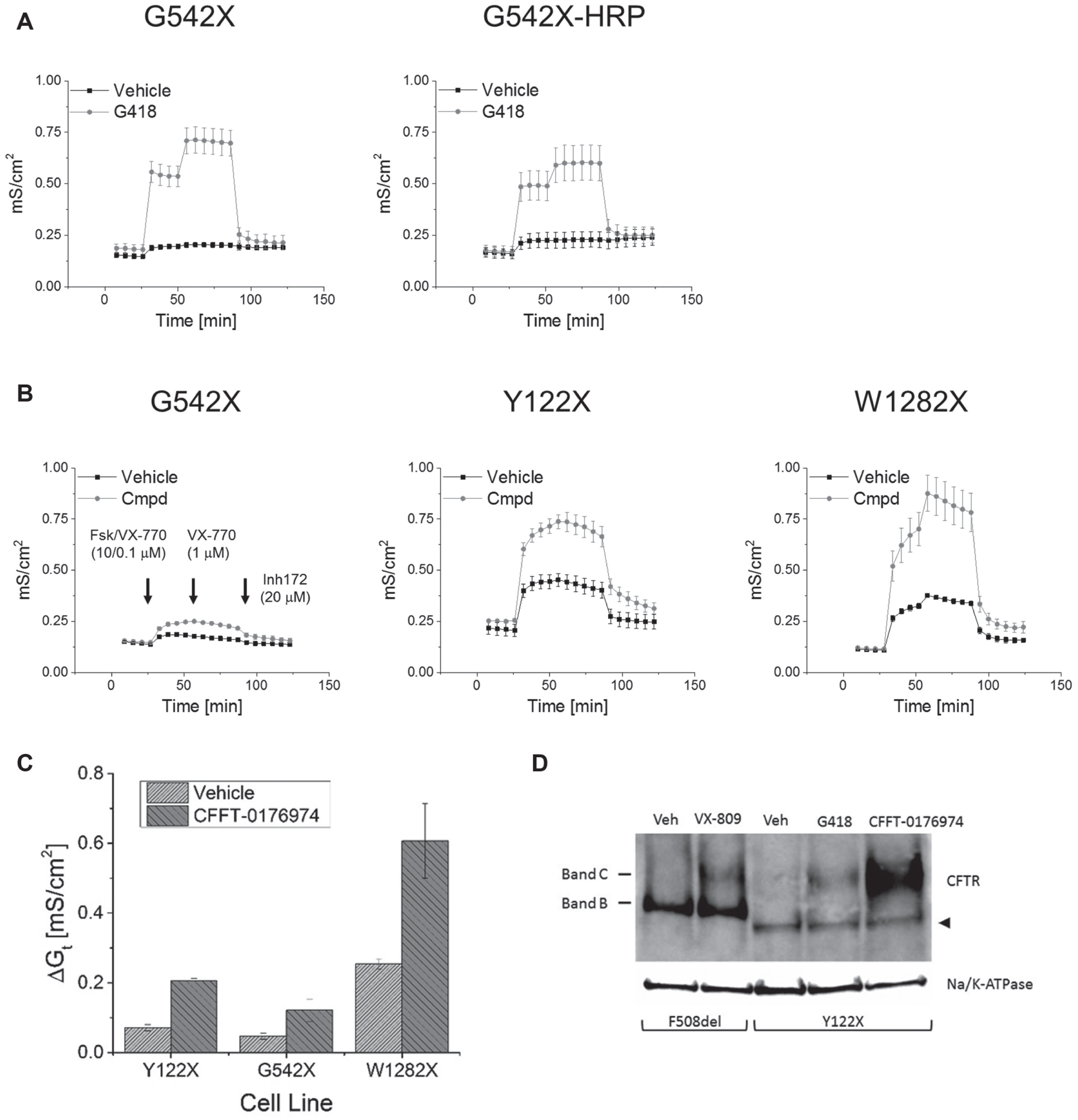
Isogenic stable cell lines expressing CFTR-HRP were developed in FRT epithelial cells, using the Flp-In System (Thermo Fisher Scientific). 27 Upon incubation with HRP substrate, luminescence levels measured for the CFTR-HRP Y122X cell line are similar to that of untransfected FRT cells, whereas the CFTR-HRP W1282X line has a measurable level of luminescence, about 5% of the activity of wild-type CFTR-HRP ( Fig. 2 ), resulting from some basal surface expression of the CFTR-HRP fusion protein truncated after amino acid 1281 ( Fig. 3A ).
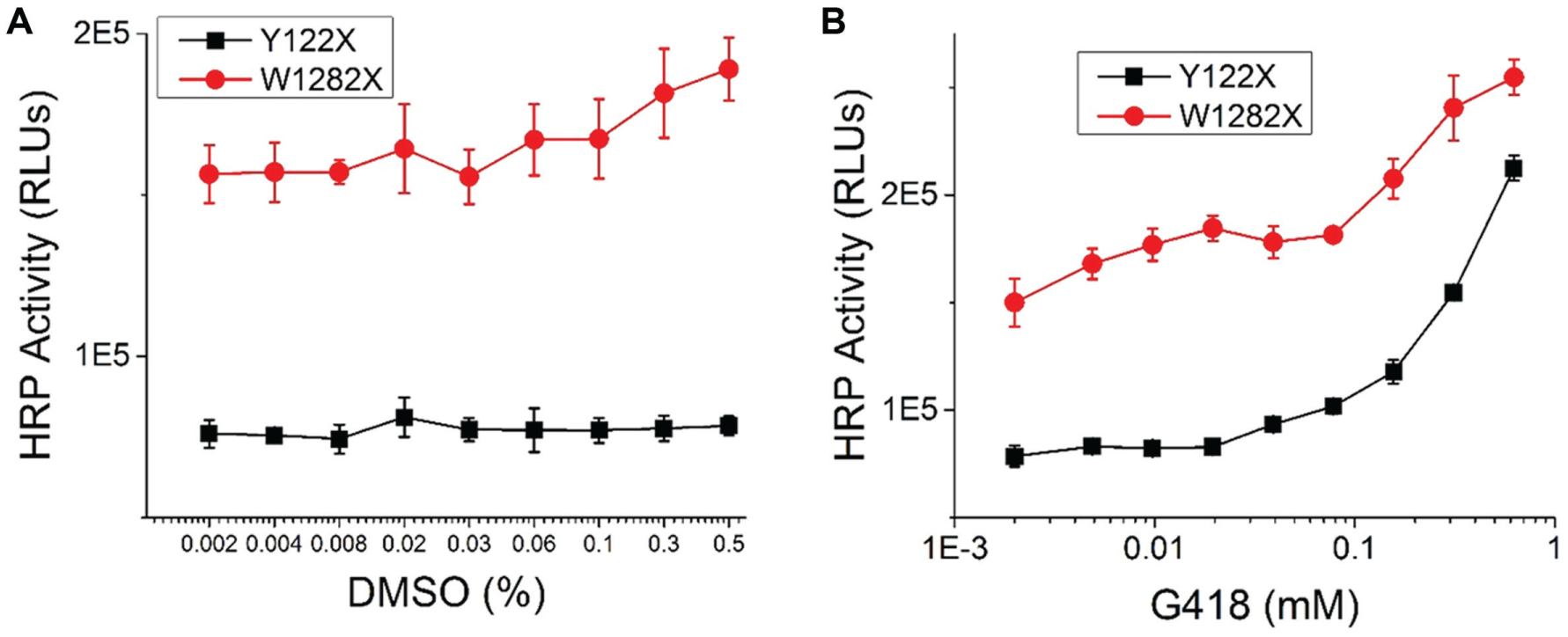
Screening assays have been developed in isogenic FRT cell lines expressing CFTR-HRP Y122X or W1282X. FRT cells were incubated with DMSO or G418 for 2 days at 37 °C and 5% CO2, prior to the detection of HRP activity. HRP activity is shown in relative luminescence units (RLUs). (
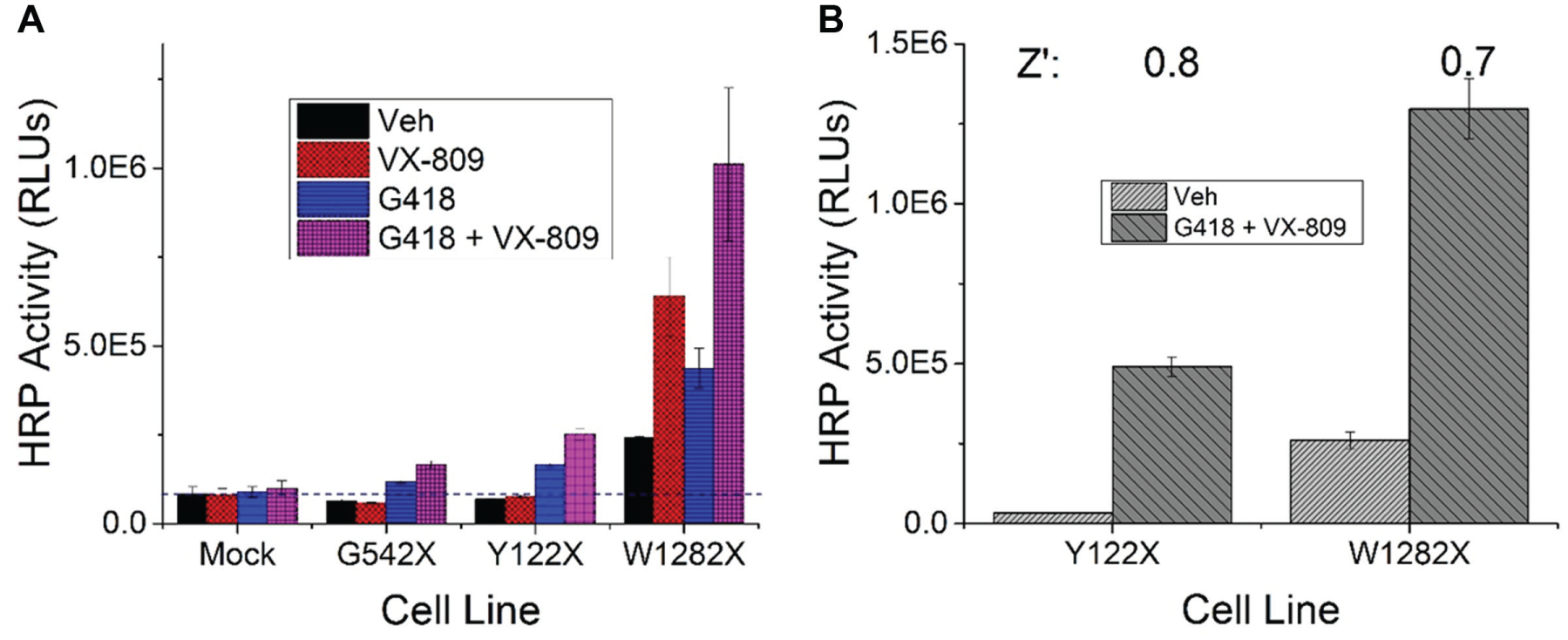
Dynamic window and analysis of assay plate uniformity. (
G418 (geneticin) has been reported as a readthrough modulator of PTC mutations, including PTC mutations in CFTR. 28 FRT cells expressing CFTR-HRP (Y122X or W1282X) were incubated with DMSO or G418 for 2 days at 37 °C, prior to the detection of HRP activity ( Fig. 2 ). DMSO is well tolerated at the HTS screening condition (0.1% total DMSO) in both cell lines ( Fig. 2A ). Dose–response analysis of G418 was performed by serial dilution ( Fig. 2B ). We observed a dose-dependent increase for the HRP-mediated luminescence with G418. Significant reporter activities were detected in both PTC assays at concentrations of 150 µM G418 and greater. At 600 µM, G418 generated favorable signal-to-background ratios, 1.7-fold and 2.7-fold for the W1282X and Y122X assay, respectively.
To enhance the performance of the HTS assay, we combined the CFTR folding and trafficking modulator VX-80911 (3 µM) with G418, utilizing this combination as the positive control. VX-809 does not increase PTC readthrough of either Y122X or G542X, but it can improve the intracellular trafficking of readthrough products of CFTR ( Fig. 3A ). In contrast, VX-809 increases surface luminescence in the CFTR W1282X assay in the absence of readthrough, indicating that other trafficking correctors may also show efficacy in this assay. Hence, we decided to supplement VX-809 (3 µM) in the growth media only for FRT cells expressing CFTR-HRP Y122X to amplify the readthrough signal, with the combination of G418 and VX-809 as positive control. As a result, the signal-to-background ratio significantly improved 15-fold ( Fig. 3B ). In the CFTR-HRP W1282X assay, compounds were screened in the absence of VX-809 so that all compounds that might increase trafficking of the truncated CFTR W1282X protein would be detected as hits. Assay plate uniformity was also tested in the 384-well format ( Fig. 3B ). Half of the cell samples in the whole assay plate were incubated with vehicle (DMSO, 0.1%) or the positive control (G418, 600 µM; VX-809, 3 µM). Signal is relatively uniform on the whole assay plate, and the edge effect is not significant. The Z factors (or Z′ values) 29 for the Y122X and W1282X assays are 0.8 and 0.7, respectively. From these results, we conclude that the reporter-based assays for the readthrough of CFTR PTC mutations are suitable for HTS.
HTS
The small patient populations for specific CFTR PTC mutations—many mutations have fewer than 100 patients worldwide—make it hard to develop genotype-specific therapies at both the research and development stages. One potential solution may be the development of personalized therapy for all or a larger group of CFTR PTC mutations, based on their common phenotype of prematurely terminated protein translation. We tested this possibility in HTS with about 85,000 compounds. Parallel screening with 10 µM compound was performed in Y122X and W1282X assays. The vast majority of our compound library is unbiased for therapeutic targets.
The performance of both HTS assays is robust ( Fig. 4 ). Z′ is analyzed with respect to the vehicle (DMSO, 0.1%) and reference (G418, 600 µM; VX-809, 3 µM) within the compound-treated assay plates. The average of Z′ in both screens is 0.8. Only three plates in the CFTR Y122X assay yielded Z′ values below 0.4, but this was only for one plate of a triplicate set, and after detailed inspection of the data, these plates were included in the accepted screening data. Both background and noise levels have been stable throughout the screening campaign.
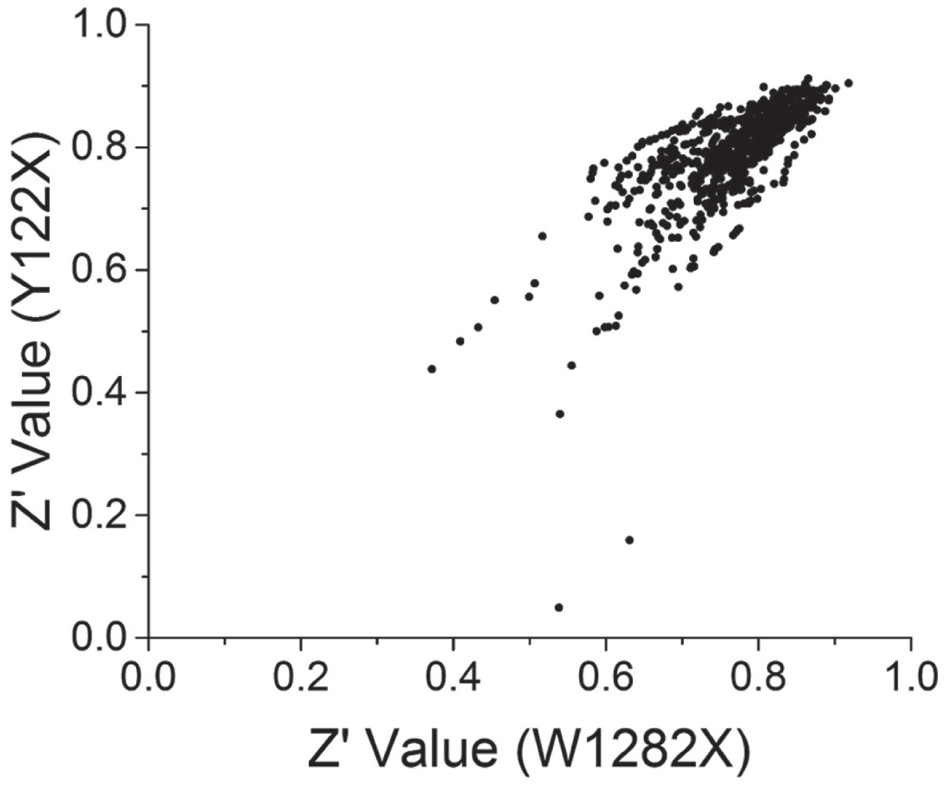
Z′ of all the accepted HTS assay plates from the two parallel PTC readthrough screens. Parallel screens of 85,000 compounds were carried out in two PTC readthrough assays (Y122X or W1282X) with FRT cells expressing CFTR-HRP fusion proteins containing the Y122X or W1282X nonsense mutations. VX-809 (3 µM) was added to the growth medium of FRT cells expressing CFTR-HRP Y122X to amplify the readthrough signal. The combination of VX-809 (3 µM) and G418 (600 µM) was used as a positive control. Z′ values are calculated for each assay plate between the vehicle and positive control. Z′ values in the two screens (W1282X, x axis; Y122X, y axis) are shown in a scatter plot. Each data point represents the Z′ values for the same compound source plate in the two assays (W1282X and Y122X).
Hit Profile
Luminescence was normalized to background, and hits have been selected with a cutoff of 1.5, that is, 50% over the average luminescence measured for vehicle-incubated cells on each assay plate ( Table 1 ). This analysis yielded 1767 hit compounds from the Y122X screen and 1197 hits from the W1282X screen, representing hit rates of 2% and 1.4%, respectively. In total, 2630 hits were selected for further dose–response analysis. The average of the triplicate luminescence reads from the screening assay for each hit compound are plotted for correlation analysis ( Fig. 5 ) between the two HTS assays representing CFTR Y122X (x axis) and W1282X (y axis). Only a small proportion of hits reveal similar levels of activities in both screens. At the stringent hit cutoff of 1.5, 13% of the total hits (334 out of 2630 compounds) met the cutoff in both assays. The hit overlap is significantly greater (~50%) if compounds are included that meet the stringent 1.5 signal-to-background cutoff in one screen and only 3 standard deviations above background (~1.2 signal-to-background ratio) in the second screening assay ( Table 1 ). The relatively small overlap at the more stringent hit threshold is likely due to varied efficacies of compounds in the two screens. Hence, to assess their potential as pan-PTC readthrough modulators, the HTS hits need to be further profiled against a broader range of CFTR PTC mutations, including amber codon PTCs (UAG).
Hit Selection Targeting Pan-PTC Readthrough Modulation.
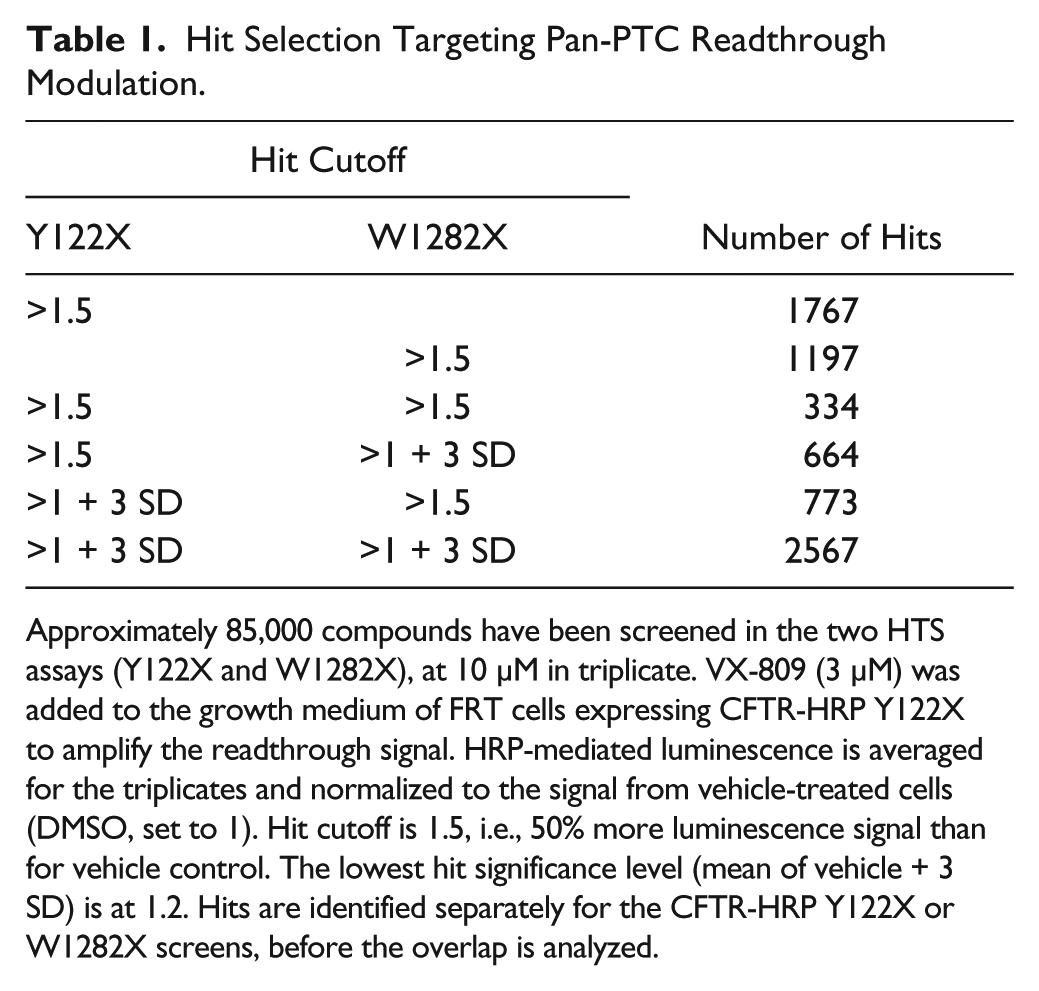
Approximately 85,000 compounds have been screened in the two HTS assays (Y122X and W1282X), at 10 µM in triplicate. VX-809 (3 µM) was added to the growth medium of FRT cells expressing CFTR-HRP Y122X to amplify the readthrough signal. HRP-mediated luminescence is averaged for the triplicates and normalized to the signal from vehicle-treated cells (DMSO, set to 1). Hit cutoff is 1.5, i.e., 50% more luminescence signal than for vehicle control. The lowest hit significance level (mean of vehicle + 3 SD) is at 1.2. Hits are identified separately for the CFTR-HRP Y122X or W1282X screens, before the overlap is analyzed.
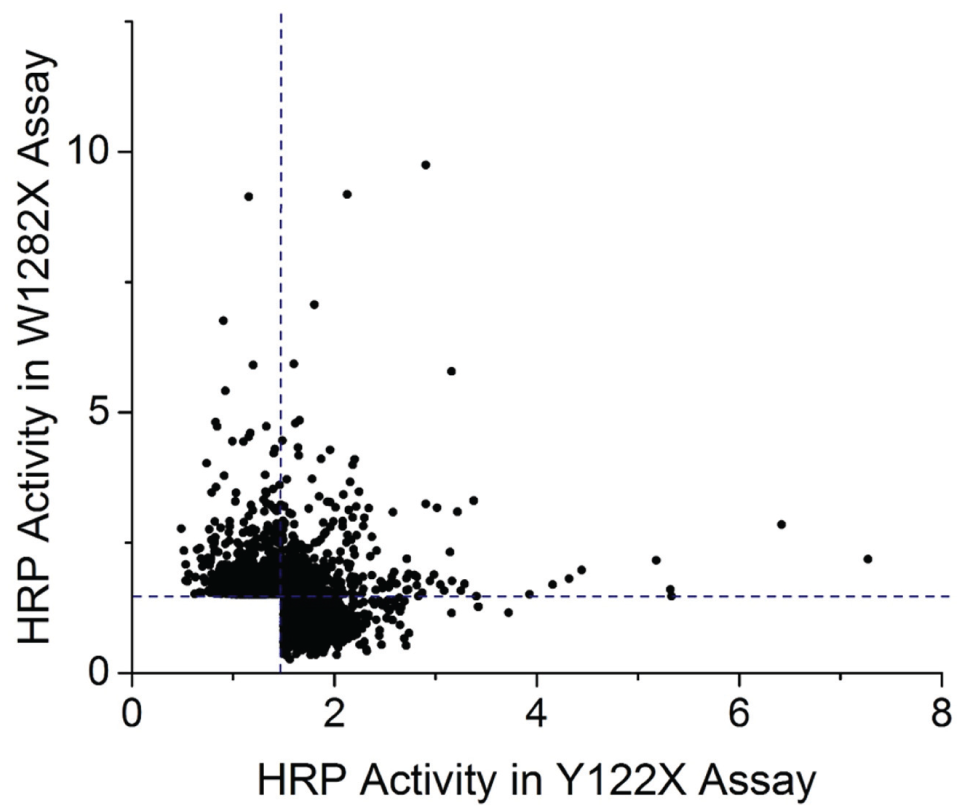
Normalized HRP activity for all hits from two parallel PTC readthrough screens. HRP-catalyzed luminescence for cell lines expressing CFTR-HRP Y122X or W1282X was normalized to vehicle signal (set to 1). The activity of a hit is the average of triplicate screening data points. A 1.5 cutoff for normalized activity (dashed lines) was used for hit calling. Each dot corresponds to HRP activities of one screening compound in two assays (Y122X, x axis; W1282X, y axis). VX-809 (3 µM) was added to the growth medium of FRT cells expressing CFTR-HRP Y122X to amplify the readthrough signal.
Hit confirmation was carried out in FRT cells expressing CFTR-HRP Y122X, W1282X, or G542X ( Table 2 ). VX-809 was supplemented in the growth medium of FRT cells expressing CFTR-HRP Y122X or G542X to amplify the readthrough signal. The hits with sufficient compound matter available, 2574 of 2630, were tested in dose–response titrations. Sixty-five percent of all primary hits (1661 of 2574) were confirmed in at least one of the HTS screening assays (Y122X or W1282X). Of the hits with activity in both Y122X and W1282X assays, the majority (72%, 642 of 892) also achieve significant activity in the G542X assay.
Profile of Confirmed HTS Hits.
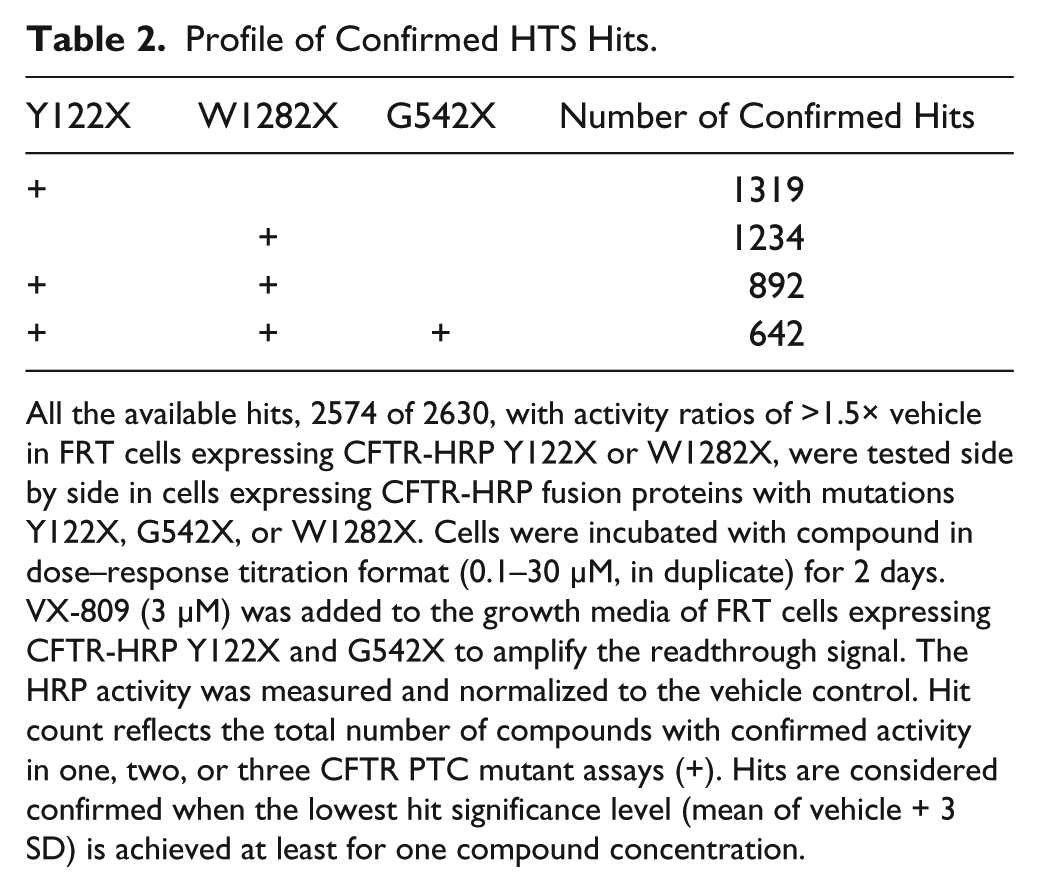
All the available hits, 2574 of 2630, with activity ratios of >1.5× vehicle in FRT cells expressing CFTR-HRP Y122X or W1282X, were tested side by side in cells expressing CFTR-HRP fusion proteins with mutations Y122X, G542X, or W1282X. Cells were incubated with compound in dose–response titration format (0.1–30 µM, in duplicate) for 2 days. VX-809 (3 µM) was added to the growth media of FRT cells expressing CFTR-HRP Y122X and G542X to amplify the readthrough signal. The HRP activity was measured and normalized to the vehicle control. Hit count reflects the total number of compounds with confirmed activity in one, two, or three CFTR PTC mutant assays (+). Hits are considered confirmed when the lowest hit significance level (mean of vehicle + 3 SD) is achieved at least for one compound concentration.
Some confirmed hits were further analyzed in FRT cell lines expressing CFTR PTC mutants without the fused HRP reporter protein. Such FRT cells expressing CFTR Y122X, G542X, or W1282X were treated with hit compounds. An example of these characterization studies is provided in Figure 6 . In Figure 6A , it is shown that CFTR constructs and CFTR-HRP fusion constructs behave similarly in functional assays, but the expression level of the CFTR-HRP fusion protein is likely somewhat reduced. Incubation with a hit compound increased CFTR-mediated chloride conductance ( Fig. 6B , C ), indicating functional expression of full-length CFTR. This expression of full-length CFTR is demonstrated by Western blot for the cell line expressing CFTR Y122X ( Fig. 6D ). These data demonstrate that HRP-based HTS can discover small molecules that can suppress PTC mutations in CFTR and yield functional CFTR chloride channels.
(
Discussion
Recent progress made in CFTR modulator therapy (lumacaftor and ivacaftor) is a major advancement from the traditional approaches targeting symptomatic relief. However, a large number of CFTR mutations result in altered pre-mRNA splicing or mRNA surveillance, incomplete protein translation, defective vesicular trafficking, and/or decreased channel function. All these defects can cause CF. Many of the CF patients with such mutations will not experience clinically meaningful benefit when treated only with the currently approved drugs that improve CFTR folding and trafficking (lumacaftor) or channel activity (ivacaftor). There is an urgent need for drug discovery to address the currently not treatable mutations.
The vast majority of reported CFTR modulator screens focus on trafficking correction of F508del CFTR and channel activity of G551D CFTR.12,23,30–33 New HTS assays are needed that target the translational defects caused by CFTR PTC mutations. To that end, we have developed HTS assays based on the trafficking and surface expression of CFTR for PTC mutation readthrough modulators. The luminescence-based assays utilize the reporter enzyme HRP, which is fused into an extracellular loop of the CFTR protein, yielding a robust readout for CFTR channels at the cell surface. With a different HTS assay, Vertex published a hit rate for its original F508del CFTR corrector of 0.11%, 12 and an HTS campaign for F508del CFTR correctors with a HRP cell surface readout (similar to our assay) resulted in a hit rate of 0.15%, 23 which are lower hit rates than we report for our screens (1.4% and 2%). The different hit rates may reflect differences in cell lines, execution of the assays, the compound library, and the nature of the mutations (F508del vs. PTC).
The most efficacious HTS hits restore surface expression of CFTR PTC mutants more effectively than the reference readthrough modulator G418. The top five W1282X hits increase surface CFTR to more than 20% of wild-type CFTR in the CFTR-HRP assay. It is also encouraging to observe that hit compounds can yield full-length functional CFTR protein. Future chemical matter structure–activity relationship (SAR) exploration will shed light on the therapeutic potential of the existing hits.
The CFTR PTC mutations in this study have been selected based on multiple criteria, including mutation frequency, stop codons, and protein structure. Both UAA (ochre, Y122X) and UGA (opal, W1282X) are represented in the screening. We also developed an assay based on CFTR G542X (opal, UGA) 34 that is used in hit profiling. The selection of these PTC mutants also covers different domains of the CFTR protein. Y122 is located in the transmembrane region, and G542 is in NBD1, while W1282 is located in the NBD2 of CFTR. Y122X is the second most abundant UAA PTC, with an allele frequency of 0.06% (http://cftr2.org). It has been suggested the CFTR Y122X is one of the most easily suppressed nonsense mutations, whereas it is much harder to obtain functional CFTR from G542X readthrough. 19 Thus, we chose Y122X as a potentially more sensitive assay, assuming that we will find hits that will translate to G542X, but that they may be more easily discovered in a Y122X assay. Furthermore, G542X and W1282X share the same opal UGA stop codon, and it was attractive to start the search for a pan-PTC readthrough compound with at least two different stop codons. Meanwhile, recent work has suggested that UAA may be the most difficult stop codon to read through, at least with the drugs ataluren and G418. 35 The efficiencies for readthrough of specific stop codons may in the end depend not just on the PTC itself, but also on the context and the PTC readthrough modulator. An assay for the rarer UAG amber codon (in 0.58% of all CFTR PTC mutant alleles; http://cftr2.org) needs to be developed for further evaluation of pan-PTC hits.
Opal PTC mutations are 10-fold more frequent than either ochre or amber mutations (http://cftr2.org). In the recently reported clinical trial of ataluren as a CFTR PTC mutation readthrough drug, the majority of patients (209 of 232) have an opal codon (W1282X, G542X, R1162X, and R553X). 36 Further profiling of the HTS hits in these most frequent CFTR PTC mutations will shed light onto the potential clinical developmental path for CFTR PTC mutation therapies.
There is a significant number of hits with allelic preference for either Y122X or W1282X. This is partly due to the C-terminal truncated CFTR of W1282X being able to traffic to the plasma membrane in the absence of any PTC readthrough, and this trafficking can be enhanced by VX-809 or other compounds with trafficking corrector activity. Further characterization of W1282X-specific hits for trafficking enhancing or readthrough activity will be important.
Defects caused by CFTR PTCs are affected by multiple factors, including codon usage, the PTC position within the CFTR gene, and mRNA levels. The relative impact of each of these factors on each CFTR PTC mutation is likely not the same and not easily deconvoluted, and more hit profiling on different PTC mutations may be needed. One limitation of the described screening approach is the lack of introns in the screening constructs, and thus the absence of mRNA splicing. Exon junction complexes (EJCs) appear to be an important component for at least one type of nonsense-mediated mRNA decay (NMD), 37 and this biology is not present in our HTS assay.
The defect of PTC mutations of CFTR is at the level of protein translation. PTC readthrough therapy has been hypothesized to require modulation of the essential cellular function of ribosomes. Concerns have been raised that suppression of nonsense mutations may be toxic due to interference with essential functions within the cell. The current cell-based HTS format does not take into account cell viability, so the hits need to be counterscreened for cytotoxicity. Hits that confer readthrough of more than one PTC mutation will be further profiled in secondary assays.
PTC mutations in other genes cause many other genetic rare diseases, for example, Duchenne’s muscular dystrophy, beta-thalassemia, and Hurler syndrome. While the objective of our screening is the restoration of functional CFTR protein from CFTR PTC mutant genes, we do expect that molecules with pan-PTC readthrough activity will also be able to rescue other gene products. However, the ability to achieve clinically beneficial readthrough of other gene products will need to be assessed case by case, as every PTC mutation will have different requirements and tolerance for specific amino acid substitutions that may lead to rescue of functional protein. The reference PTC readthrough modulator G418 lacks gene specificity and can produce different amino acid substitutions for a given PTC. Novel readthrough modulators may be less promiscuous.
The data presented demonstrate that it is feasible to identify novel compounds that modulate PTC readthrough for three different CFTR nonsense mutations, supporting the possibility of developing a pan-PTC therapy. As we have shown, the efficacy ranking of HTS hits for the two readthrough screens (Y122X and W1282X) varies significantly, influenced by the specific mutation. A large number of hits appear to have a preference for a specific nonsense mutation. This is a challenge in the hit selection in pursuit of potential pan-PTC drug leads. The use of additional PTC mutations, such as G542X or amber codon PTCs, in the secondary assays will be important for the identification and prioritization of compounds with broad pan-PTC efficacies.
Footnotes
Acknowledgements
We thank our colleagues Laura Fitzpatrick and Chloe Schneider for compound management, Thomas Ferrucci for database support, and Dr. Hillary St. John for critical review of the manuscript. We thank Drs. Jeong S. Hong and Eric J. Sorscher (Emory University School of Medicine) for generating the FRT cell lines. We also thank Drs. Guido Veit and Gergely L. Lukacs (McGill University) for providing a CFTR-HRP DNA template and sequence information.
Declaration of Conflicting Interests
The authors declared no potential conflicts of interest with respect to the research, authorship, and/or publication of this article.
Funding
The authors are employees of Cystic Fibrosis Foundation Therapeutics, Inc., and received no external or third party financial support for the research, authorship, and/or publication of this article.