
Abstract
One of the main reasons for the lack of drug efficacy in late-stage clinical trials is the lack of specific and selective target engagement. To increase the likelihood of success of new therapeutics, one approach is to conduct proximal target engagement testing during the early phases of preclinical drug discovery. To identify and optimize selective IRAK4 inhibitors, a kinase that has been implicated in multiple inflammatory and autoimmune diseases, we established an electrochemiluminescence (ECL)-based cellular endogenous IRAK1 activation assay as the most proximal functional evaluation of IRAK4 engagement to support structure–activity relationship (SAR) studies. Since IRAK1 activation is dependent on both the IRAK4 scaffolding function in Myddosome formation and IRAK4 kinase activity for signal transduction, this assay potentially captures inhibitors with different mechanisms of action. Data from this IRAK1 assay with compounds representing different structural classes showed statistically significant correlations when compared with results from both IRAK4 biochemical kinase activity and functional peripheral blood mononuclear cell (PBMC)-derived tumor necrosis factor α (TNFα) secretion assays, validating the biological relevancy of the IRAK1 target engagement as a biomarker of the IRAK4 activity. Plate uniformity and potency reproducibility evaluations demonstrated that this assay is amenable to high throughput. Using Bland–Altman assay agreement analysis, we demonstrated that incorporating such proximal pharmacological assessment of cellular target engagement to an in vitro screening funnel for SAR studies can prevent compound optimization toward off-target activity.
Introduction
Interleukin-1 receptor-associated kinase 4 (IRAK4) is a serine/threonine protein kinase that plays an essential role in transducing signals from pathogen recognition toll-like receptors (TLRs) and the interleukin-1 (IL1) receptor superfamily to bridge innate and adaptive immune responses against microbial infection. 1 Dysregulation of the IRAK4 signaling pathway triggers aberrant expression of inflammatory cytokines/chemokines and hyperactivation of B cells, leading to multiple chronic inflammatory and autoimmune diseases such as multiple sclerosis (MS), systemic lupus erythematosus (SLE), rheumatoid arthritis (RA), and inflammatory bowel disease.2–5 IRAK4 signaling has also been implicated in the initiation, development, and therapeutic responses of various cancers.6–9 Abrogation of IRAK4 activity using either selective IRAK4 inhibitors or kinase-inactive mutants has demonstrated attenuation of disease activity in mouse models of RA, SLE, and experimental autoimmune encephalomyelitis (EAE).4,10–12 In contrast to Irak4–/– mice that suffer severe defects in response to broad bacterial and viral challenges, 1 human IRAK4 deficiency predisposes patients to a narrow range of pyogenic bacterial infection without severe disease manifestation to viral, parasitic, or fungal infection. Although life-threatening, such infections occur only in infancy and early childhood, which can be prevented with prophylactic antibacterial treatments. 13 As patients grow into adolescence and adulthood, IRAK4 becomes dispensable for host defense against most pathogens.13,14 These observations suggested that IRAK4 could be an attractive target for treating inflammatory, autoimmune diseases or cancer without increasing the risk of microbial infections.
To identify and optimize IRAK4 inhibitors, we have implemented a biochemical IRAK4 IMAP fluorescence polarization (FP) screening assay to assess compound binding and inhibition of IRAK4 kinase activity, followed by a phenotypic human peripheral blood mononuclear cell (PBMC) tumor necrosis factor α (TNFα) secretion assay to demonstrate functional perturbation of the IRAK4 pathway. 15 Although biochemical and biophysical assays using purified recombinant protein to evaluate target engagement provided a robust, high-throughput, first-pass screening, the results did not always translate into cellular efficacy due to the lack of a physiological cellular context for the target. 16 In addition, relying on distal phenotypic cellular readouts while omitting cellular proximal target engagement during SAR studies may derail the optimization of the molecule toward the off-target activity, as multiple signaling pathways could contribute to the regulation of a functional phenotype.
One approach to improve the probability of success is to measure direct drug–target interaction in living cells. Multiple methods have been developed, such as proximity detection using either labeled ligands, 17 resonance energy transfer like fluorescence resonance energy transfer–fluorescence lifetime imaging (FRET-FLIM) and bioluminescence resonance energy transfer (BRET),18,19 or protein stability-based enzyme fragment complementation (EFC). Although these approaches become pivotal in target identification and target validation, they require modifications of the ligand and/or target. Assays that depend on protein modification require generation of stable cell lines that may alter the physiological cellular context due to overexpression of the tagged target. Recent adaptation of thermal shift assay to cells (CETSA) allows label-free investigation of drug binding to native target in the physiological cellular environment. 20 Coupled with immunodetection, robust CETSA can be performed in high throughput, as demonstrated through library screening to identify B-Raf and poly (ADP-ribose) polymerase 1 (PARP1) binders. 21 However, because CETSA measures changes in thermostability of the protein upon drug interaction, such changes do not always translate into functional modulation of the target in a cellular environment. 21 It is also possible that small-molecule binding that results in a functional modulation of the target may not induce detectable changes in thermostability. Therefore, evaluation of functional modulation on the most proximal event following target activation is of paramount importance during lead optimization.
To complement the IRAK4 recombinant kinase assay and the functional PBMC-derived TNFα secretion assay, we considered a proximal cellular approach to demonstrate IRAK4-mediated drug–target engagement under physiological conditions. In the TLR/IL1R-initiated signaling cascade, multiple myeloid differentiation primary response factors (MyD88) bind to the receptor and assemble into a scaffold to promote the recruitment of other adaptor proteins and kinases, including IRAK-1 and IRAK-4, forming the Myddosome complex.22,23 Within this complex, dimerized IRAK4 undergoes activation through autophosphorylation,24,25 leading to subsequent IRAK1 phosphorylation and activation. 26 Hyperphosphorylated IRAK1 attracts and activates the E3 ubiquitin ligase tumor necrosis factor–associated factor 6 (TRAF6), which further cascades the signal to nuclear factor κB (NF-κB) and mitogen-activated protein kinase (MAPK) pathways following K63-mediated polyubiquitination.27,28 Activated IRAK1 subsequently undergoes K48-linked polyubiquitination that directs it to proteasome-mediated degradation.28–30 Although in mouse cells IRAK4 catalytic activity is critical for maximal production of inflammatory cytokines,31–33 pharmacological inhibition with IRAK4 inhibitors or rescue of IRAK4-deficient cells with kinase-inactive IRAK 4 mutants illustrated that an IRAK4-mediated scaffold function was critical for Myddosome assembly and NF-κB or MAPK pathway activation, while human IRAK4 kinase activity was dispensable for cytokine production.35–36 Hence, an assay that measures IRAK 4 phosphorylation may not fully capture different mechanisms of IRAK4 inhibitors. However, the recruitment of IRAK1 and its subsequent activation depends on both the scaffold functionality and kinase activity of IRAK4.30,37,38 Evaluating IRAK1 activation, which is the most proximal event mediated by IRAK4, would potentially capture multiple mechanisms of inhibitors, including the disruption of IRAK4 catalytic activity and the Myddosome assembly.
Immunodetection has been used for quantitative measurement of protein markers in various complex biological matrices such as serum, tissues, or cellular lysates. However, conventional methods like enzyme-linked immunosorbent assay (ELISA) utilizing absorbance readouts require multiple time-consuming steps, a major disadvantage when adapting to automation and throughput testing, which are the key considerations when developing an assay for drug screening and development. The electrochemiluminescence (ECL)-based mesoscale assay (MSD) utilizes a streptavidin-coated microplate with built-in carbon electrodes at the base in close proximity to the analytes captured by analyte-specific biotin-labeled antibodies. Using a ruthenium-labeled detection antibody, luminescence signals are generated via multiple rounds of oxidative-reductive electrochemical reactions of ruthenium with tripropylamine provided in the buffer. High sensitivity and a wide dynamic range from the luminescence signal make the assay amenable to miniaturization. Here we describe the development of a high-throughput MSD-based human cellular endogenous IRAK1 activation assay. We validated the assay through an orthogonal method and demonstrated the biological relevance, as well as the potential value for inclusion in the SAR screening funnel for selective IRAK4 inhibitor optimization.
Materials and Methods
Cell Culture and Stimulation
Human mantle cell lymphoma JEKO-1 cells (ATCC, Manassas, VA) and diffuse large B-cell lymphoma line OCI-LY3 (DSMZ, Germany) were maintained in RPMI-1640 (Gibco, Gaithersburg, MD) with 20% fetal bovine serum (FBS; Fisher Scientific, Pittsburgh, PA). Human myeloma cell line RPMI-8226 (ATCC) was maintained in RPMI-1640 supplemented with 10% FBS. To stimulate the cells with TLR7 agonist R848 (Tocris, Minneapolis, MN), cells were cultured in 384-well plates in RPMI-1640 supplemented with 1× NEAA (nonessential amino acids; Gibco), 1 mM sodium pyruvate, 10 mM HEPES, and 0.5% charcoal/dextran-treated FBS (Fisher Scientific) at a density of 8 × 105/mL for 16 h. Cells were treated with IRAK4 inhibitors for 1 h before stimulation with R848. Human lung fibroblast MRC5 cells (ATCC) were maintained in MEM (minimum essential medium; Gibco) with 10% FBS. To stimulate MRC5 cells with IL1-β, cells were plated in 384-well plates at a density of 50,000/well in MEM (Gibco) supplemented with 1× NEAA, 1 mM sodium pyruvate, 10 mM HEPES, and 0.5% charcoal/dextran-treated FBS overnight. Cells were treated with IRAK4 inhibitors for 1 h before stimulation with recombinant human IL1-β (R&D, Minneapolis, MN).
Immunoprecipitation
JEKO-1 cells were lysed in lysis buffer containing 150 mM NaCl, 20 mM Tris-HCl, pH 7.5, 1 mM EGTA, 1 mM EDTA, 1% Triton X-100, EDTA-free protease inhibitor cocktail (Roche, St. Louis, MO), phosphatase inhibitor II (Sigma-Aldrich, St. Louis, MO), phosphatase inhibitor III (Sigma-Aldrich), and 1 mM NaF (Sigma-Aldrich). Cells were lysed on ice for 30 min, followed by centrifugation at 4 °C, 14,000g for 10 min. Supernatants were collected and stored at –80 °C. Total protein concentration was quantified using Pierce’s BCA reagent (Thermo Fisher, Waltham, MA). IRAK1 in the cell lysate was pulled down with anti-IRAK1 antibody using the Dynabead Protein G immunoprecipitation kit protocol (Invitrogen, Carlsbad, CA). Briefly, 50 µL of Dynabeads Protein G was resuspended in 200 µL of antibody binding and washing solution containing 3 µg of anti-IRAK1 antibody. Antibody binding to the beads was facilitated by incubation for 10 min at room temperature on a rotator. Excess antibody was removed after placing the tube on the magnet and washing with antibody binding and washing buffer. Cell lysate with 500 µg of total protein was added to the beads/antibody complex and incubated overnight at 4 °C on a rotator. After washing with washing buffer three times, the Dynabeads/IRAK1/anti-IRAK1 complex was resuspended in 15 µL of elution buffer. The complex was separated from the beads after addition of 15 µL of premixed 2× NuPAGE LDS Sample Buffer (Invitrogen) and NuPAGE Sample Reducing Agent (Invitrogen), followed by heating for 10 min at 70 °C. Twenty-five microliters of eluants was loaded to sodium dodecyl sulfate–polyacrylamide gel electrophoresis (SDS-PAGE) gel for electrophoresis and Western blotting probing with corresponding antibodies.
Western Blot Analysis
Cell lysates were heat denatured at 70 °C for 10 min with 1× NuPAGE LDS sample buffer. Twenty micrograms of total proteins was loaded per lane of a 4%–12% NuPAGE Bis-TRIS mini gel (Invitrogen). Proteins on the gel were then electrophoretically transferred to 0.45 µm nitrocellulose membrane in 1× NuPAGE transfer buffer (Invitrogen) with 20% methanol for 1 h. The membrane was rinsed in 1× phosphate-buffered saline (PBS) and blocked with Odyssey blocking buffer (LI-COR Biosciences, Lincoln, NE) for 1 h at room temperature, followed by incubation with primary antibody diluted in Odyssey blocking buffer with 0.2% Tween-20 overnight at 4 °C on a shaker. The antibodies used were anti-human IRAK1 (1:1000; SC7883, Santa Cruz Biotechnology, Dallas, TX), anti-human IRAK4 (1:1000; ADI-KAP-ST206-E, Enzo Life Sciences, Farmingdale, NY), and anti-human phospho-IRAK4 (1:350; 11927, Cell Signaling Technology, Danvers, MA). Antibody reactive bands were detected after incubation with IRDye 800CW goat anti-rabbit IgG secondary antibody diluted in Odyssey blocking buffer with 0.2% Tween-20 at 1:15,000 dilution for 1 h at room temperature and scanned on an Odyssey CLx infrared imaging system (LI-COR Biosciences). The membrane was washed for 5 min in 1× PBS-T (PBS supplemented with 0.1% [v/v] Tween-20) three times after each antibody incubation. For reprobing, the blot was stripped with NewBlot IR stripping buffer (LI-COR Biosciences) for 20 min at room temperature. For capillary-based simple Western assays, samples were run on Peggy Sue system using the anti-rabbit detection module (ProteinSimple, San Jose, CA) according to the manufacturer’s protocol. The antibodies used were anti-IRAK1 SC7883 (1:50) and anti-β-actin (1:50; 4967, Cell Signaling Technology). Protein expression was quantified as the area under the curve of chemiluminescence signal using Compass software (ProteinSimple).
ECL Mesoscale Assay
Antibodies were labeled with biotin using the ChromaLink Biotin Antibody Labeling Kit (TriLink BioTechnologies, San Diego, CA) according to the manufacturer’s protocol. Briefly, bovine serum albumin (BSA)-free antibodies were buffer exchanged into 1× modification buffer through a Zeba 7K MWCO column (Thermo Fisher Scientific), adjusted to 1 mg/mL concentration following protein quantification using BCA reagent. Antibodies were incubated with sulfo-chromaLink biotin for 1 h at room temperature. Excess labeling reagents were removed by passing the reaction mixture through a Zeba 40K MWCO column (Thermo Fisher). Labeling antibodies with ruthenium (II) tris-bipyridine, N-hydroxysuccinimide followed the MSD Gold Sulfo-TAG-NHS-Ester labeling protocol provided by the vendor (Meso Scale Discovery, Rockville, MD). BSA-free antibodies were buffer exchanged into 1× PBS (pH 7.9) through a Zeba 7K MWCO column. Antibody concentrations were adjusted to 2 mg/mL following protein quantification with BCA reagent. Sulfo-TAG was dissolved in distilled H2O. Based on the total amount of antibody being labeled, an appropriate amount of the Sulfo-TAG was added to the antibody solution, followed by an incubation at 23 °C for 2 h protected from light. Unlabeled Sulfo-TAG was removed by passing the solution through a 40K MWCO Zeba column. Labeled antibodies were stored at 4 °C in 1× PBS (pH 7.4 with 0.01% sodium azide).
To test the inhibition of IRAK1 activation by IRAK4 inhibitors, compounds were serially diluted in DMSO and predispensed into each well of a 384-well tissue culture plate using Echo Acoustic Liquid Handlers (Labcyte, Indianapolis, IN). Cells were seeded in corresponding culture media at 35 µL/well, and the plate was incubated for 1 h in a humidified 37 °C incubator with 5% CO2. Cells were stimulated by the addition of 4 µL of 10× stimulants and incubated under the same conditions for the required amount of time. To lyse the cells, 35 µL of media was removed, followed by the addition of 25 µL of lysis buffer. After incubation on a shaker for 30 min at 4 °C, the plate was centrifuged at 1000g for 5 min. Twenty microliters of cell lysate was transferred to a 384-well MSD streptavidin-coated plate (Meso Scale Discovery), which was preblocked with 3% BSA for 1 h at room temperature. Biotin-labeled capture antibody and ruthenium-labeled detection antibody were diluted in PBS, 0.2% Tween-20, and 2% BSA at 3× final concentration. After adding 10 µL of antibody mixture to each well, binding was facilitated by incubation at 4 °C on a plate shaker overnight and protected from light. The plate was washed three times in buffer containing 50 mM Tris-HCl, pH 7.5, 150 mM NaCl, and 0.1% Tween-20 and read on a MESO SECTOR S600 plate reader (Meso Scale Discovery) to record the ECL signal following addition of 35 µL of 2× MSD Read Buffer T (Meso Scale Discovery).
IRAK4 Fluorescence Polarization Kinase Assay
IRAK4 kinase activity was measured as described by Lim et al. 15 Briefly, in 20 µL of reaction mixture, 0.5 nM GST-tagged human recombinant IRAK4 (SignalChem, Richmond, BC, Canada) was incubated with 100 nM 5-FAM-labeled peptide substrate (Molecular Devices, San Jose, CA) in a buffer containing 10 mM Tris-HCl, pH 7.2, 1 mM DTT, 1 mM MgCl2, and 0.01% Tween-20. The kinase reaction was initiated by the addition of 100 µM ATP, and after 30 min of incubation at 25 °C, the reaction was stopped with 60 µL of IMAP Progressive Binding Reagent (Molecular Devices). Phosphorylated peptide binding to metal Mn(III) immobilized on nanoparticles was conducted by incubation for 1 h, and the change of FP of the peptide was quantified on Analyst HT (Molecular Devices).
Human PBMC TNFα Release in Response to IL1β Stimulation
Frozen human PBMCs were recovered overnight in RPMI culture medium supplemented with 0.5% FBS, 2 mM Glutamax, 1× MEM NEAA, and 1 mM sodium pyruvate. Twenty thousand PBMCs in 36 µL of culture medium were seeded per well of a 384-well plate and preincubated with test compounds for half an hour in 5% CO2 incubator at 37 °C. Cells were then stimulated with 4 µL of IL1β at a final concentration of 2 ng/mL for 5 h in the incubator, followed by the addition of 50 µL of medium and mixing. After centrifugation at 1000g for 5 min, 15 µL of medium was transferred to a human TNFα 384-well plate that was precoated with TNFα capture antibody (Meso Scale Discovery) and blocked with 3% BSA. TNFα was detected followed the manufacturer’s instructions. Briefly, after overnight at 4 °C, ruthenium (II)-labeled detection antibody was added and incubated for 2 h at room temperature, followed by three washes with 1× PBS–0.05% Tween-20. Data were acquired using a MESO SECTOR S600 plate reader.
Kinase Profiling
Kinase profiling was performed by Z′-LYTE Screening using Thermo Fisher’s SelectScreen Kinase Profiling Services. Two hundred fifty-six kinases (
Statistical Analysis
All plots and statistical analysis were performed in Prism (GraphPad Software, San Diego, CA). Test compound potencies (IC50) were calculated from four-parameter logistic regression curve fitting of normalized concentration response data. For normalization, 100% inhibition was defined by signals from no stimulation control, while 0% inhibition was defined by signals from agonist stimulation in the absence of compound treatment. Both controls received 0.1% DMSO as in the compound treatment. The Pearson correlation coefficient was calculated from active compounds in both assays analyzed. For kinase profiling, the efficient IC50 of each kinase was derived from variable slope logistic regression curve fitting with a defined top of 100% inhibition and bottom of 0% inhibition.
For assay performance analysis, Z′ was calculated as described by Zhang et al. 39
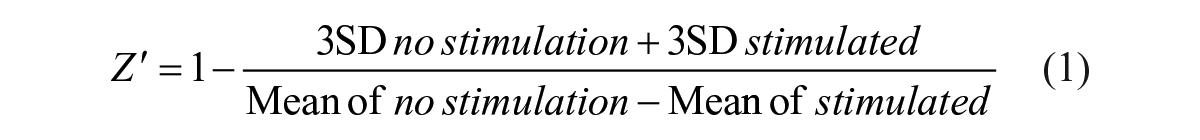
To evaluate reproducibility of the assay, 24 compounds were subject to two independent concentration response runs. The minimum significant ratio (MSR) from this test–retest experiment was calculated as per the Assay Guidance Manual, 40 where the difference of paired log potency (IC50) from each compound was first calculated to derive the standard deviation (SD) for MSR calculation:

The limit of agreement (LsA), defined by Bland–Altman, 41 was computed to determine statistical prediction of the potency ratio limits and calculated as described by Eastwood et al. 42
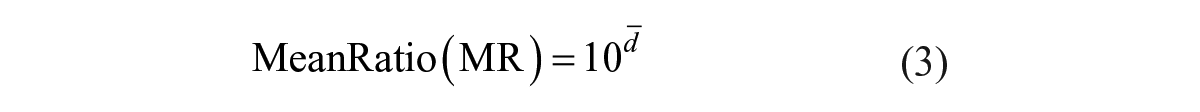
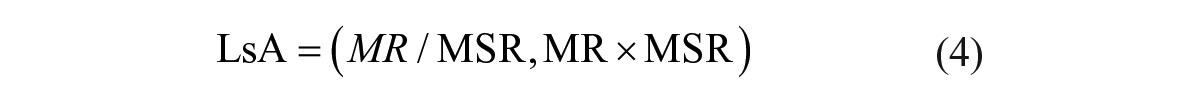
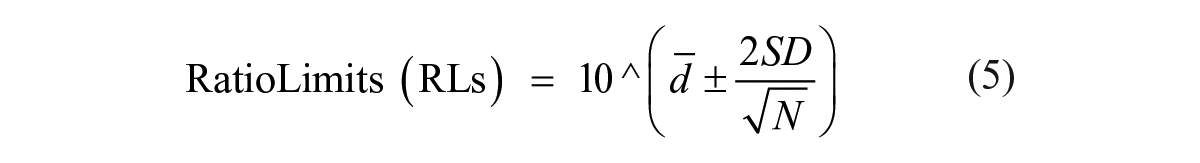
Bland–Altman analysis was used to quantify the agreement of data sets by plotting each tested compound’s mean log potency from the two assays against their difference. Acceptable limits of agreement were defined by 2SDagree that took into consideration the combined variation from both assays. To obtain SDagree, each assay’s precision level was first determined through repeated testing of reference compounds and calculated as SDpooled as described by Sun et al.: 43
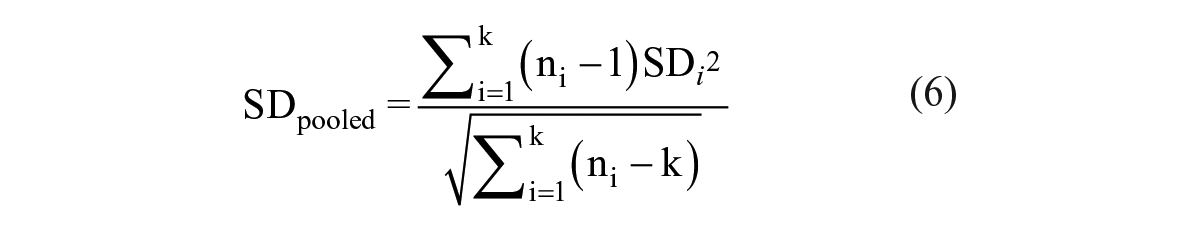
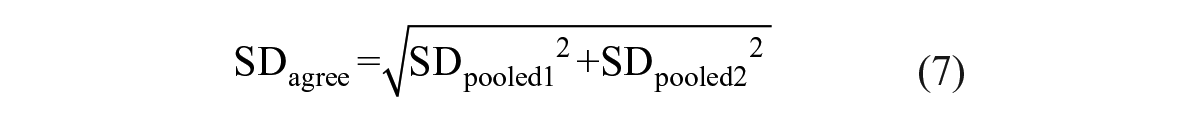
where k is the number of reference compounds tested, n is the number of repeats for each compound, and SD is the standard deviation of log potency.
Results
Development and Optimization of a High-Throughput IRAK1 Activation Assay
Signal transduction through IRAK4 is stimulated upon ligand activation of the TLR/IL1 receptor superfamily. To identify human cells that are responsive to IRAK4 pathway activation, we treated several cell lines that express either TLR7 or IL1R with selective agonists, including IL1β, R848, and lipopolysaccharide (LPS) to stimulate IL1R, TLR7, and TLR4, respectively. The experimental hallmark of IRAK1 activation is the disappearance of an unphosphorylated IRAK1 band on immunoblot due to activation through hyperphosphorylation, ubiquitination, and subsequent proteasomal degradation.
31
Using Western blotting, we evaluated unphosphorylated IRAK1 levels in cell lysates following receptor activation, along with induction of phospho-IRAK4. Although we did not observe R848-dependent phospho-IRAK4 upregulation over time in mantle cell lymphoma JEKO-1 cells, progressive disappearance of unphosphorylated IRAK1 was detected as early as 20 min and sustained for up to 3 h poststimulation (
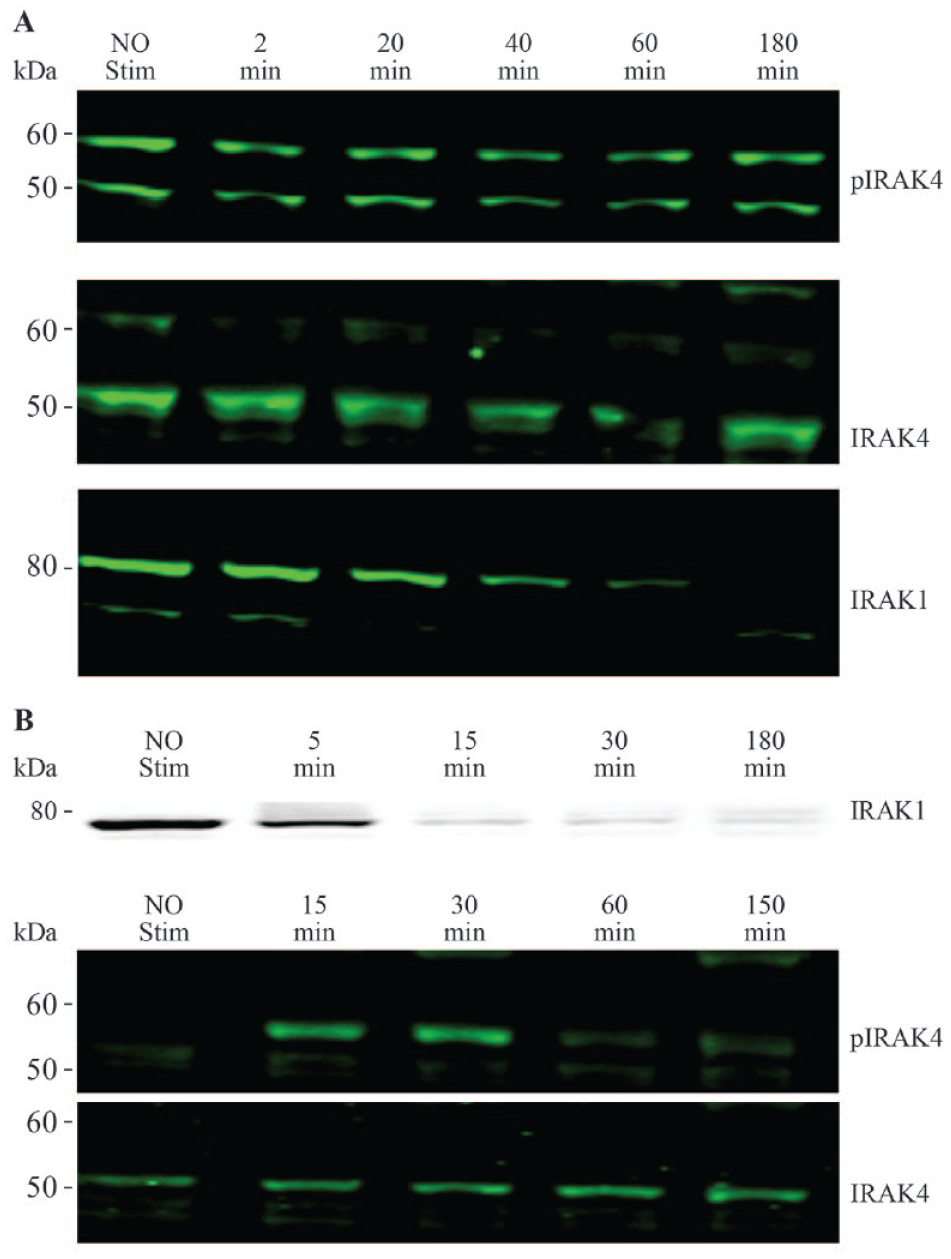
Analysis of IRAK1 activation and IRAK4 phosphorylation to identify cell lines responsive to IRAK4 pathway stimulation. (
To identify antibodies that recognize native IRAK1 in cell lysate for developing the MSD-based immunoassay, we screened 18 commercially available antibodies by immunoprecipitation of JEKO-1 cell lysate (
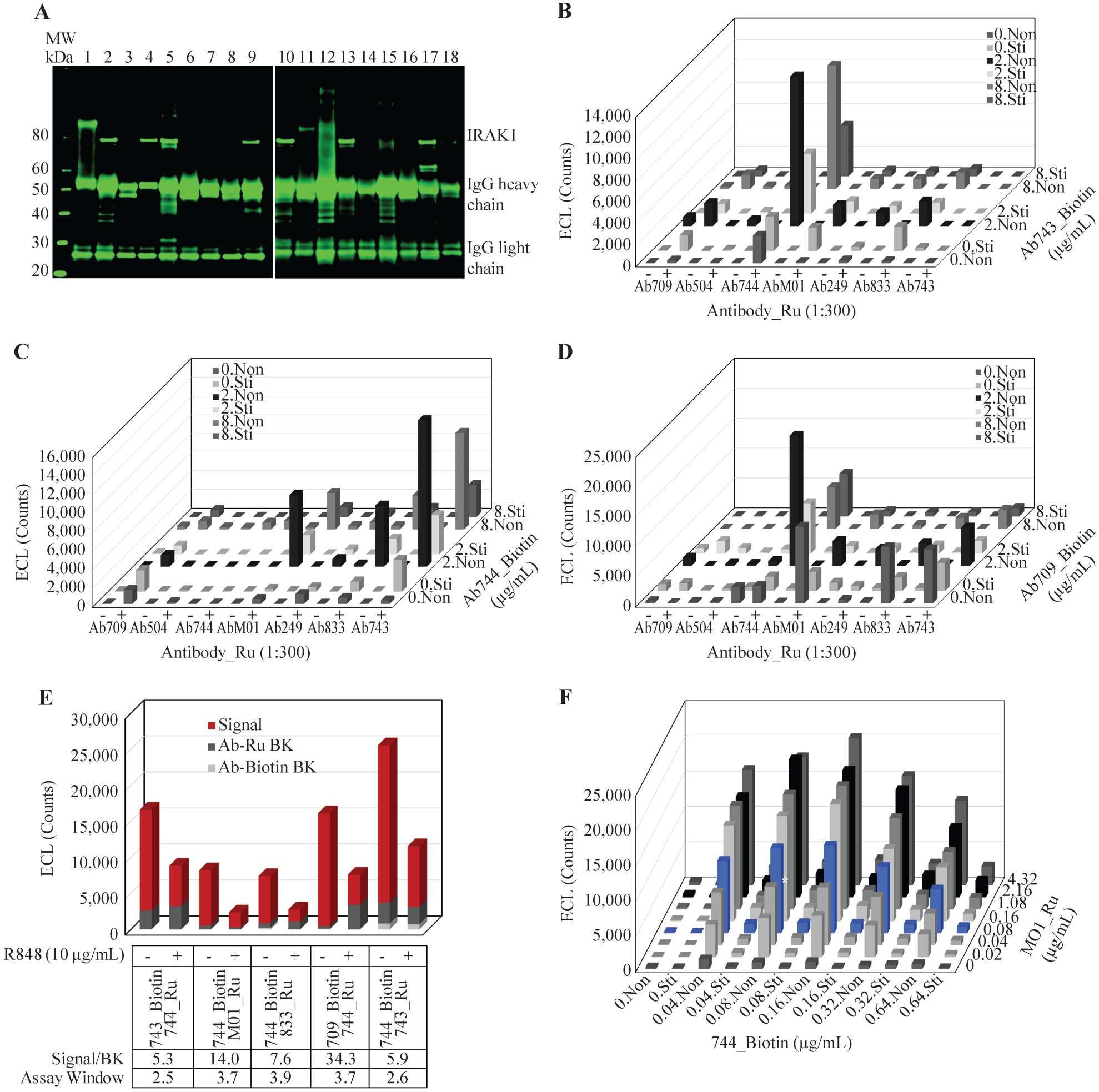
Antibody selection and optimization for the high-throughput IRAK1 activation assay. (
Discordance between ECL and Western Blot Quantification to Assess IRAK4 Inhibition of IRAK1 Activation in JEKO-1 Cells Stimulated with TLR7 Agonist R848
With the optimized assay conditions established for quantifying IRAK1 activation stimulated by R848 in JEKO cells, we next titrated R848 in order to determine the maximal stimulation concentration.
Figure 3A
shows that 50% stimulation (EC50) was achieved with 0.32 µg/mL R848, while 10 µg/mL induced a maximal activation of IRAK1. To evaluate whether inhibition of IRAK4 activity can prevent IRAK1 activation, cells were pretreated with IRAK4 inhibitor A (
Fig. 3B
) at 10 µM for 1 h before stimulation with R848 for 2 h. IRAK4 inhibitor was chosen for its potency on inhibiting IRAK4 kinase activity as evaluated by an in vitro IMAP FP kinase assay using recombinant IRAK4 and its substrate peptide (IC50 = 4.5 ± 1.6 nM). This inhibitor also showed high selectivity over other kinases in a counterscreen panel including 265 kinases (96% >100-fold selectivity) (
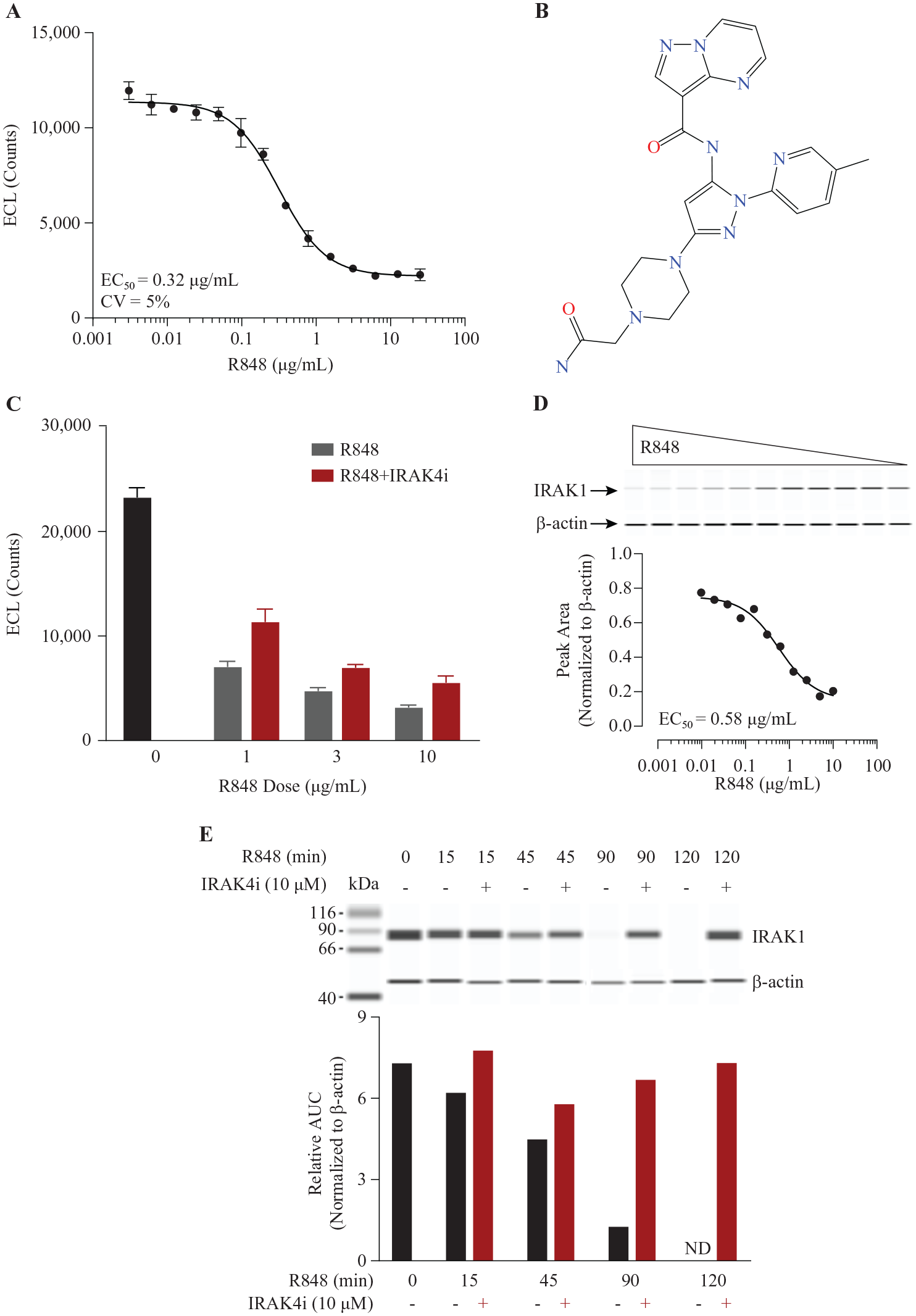
Effect of IRAK4 inhibitor on IRAK1 activation in JEKO-1 cells stimulated with TLR7 agonist R848. (
Effect of IRAK4 Inhibition on IRAK1 Activation Mediated by IL1β in MRC5 Cells
To evaluate how IRAK4 inhibition rescues IRAK1 in MRC5 cells, we pretreated MRC5 with IRAK4 inhibitor at 10 µM for 1 h, followed by IL1β (100 ng/mL) stimulation; R848 was omitted as MRC5 cells were unresponsive to this treatment. A time course of stimulation was performed and unphosphorylated IRAK1 was quantified with simple Western detection run as shown in Figure 4A ; more than 80% of IRAK1 was lost at 10 min of stimulation, which was sustained up to 3 h ( Fig. 1B ). IRAK4 inhibitor effectively blocked IRAK1 activation, with a recovery of unphosphorylated IRAK1 to 70% of the prestimulation level at 10 min ( Fig. 4A ). Although titration of IL1β resulted in similar EC50 values in stimulating IRAK1 activation throughout the time course study ( Fig. 4B ), a decreased level of IRAK1 rescue by IRAK4 inhibitor at maximal stimulation was observed with longer IL1β treatment ( Fig. 4A ). We then performed a stimulation time course study by reducing the IL1β to 30 ng/mL at the EC90 level and reevaluated IRAK4 inhibition on IRAK1 using ECL detection. In contrast to JEKO-1 cells stimulated with R848, where there was a disconnect between simple Western detection and ECL; we observed a full rescue of unphosphorylated IRAK1 following IRAK4 inhibition after 10 min of stimulation with IL1β ( Fig. 4C ). Compound titrations of IRAK4 inhibitor were carried out concomitantly with both simple Western detection and ECL. To calculate IC50 for the inhibitor from the concentration–response curve quantified by simple Western detection, we normalized IRAK1 signal to total protein loading and obtained an average IC50 of 300 ± 92 nM ( Fig. 4D ), which was similar to the IC50 obtained by ECL detection (averaged 439 ± 9.9 nM) ( Fig. 4E ). The concordance between the two detection methods on MRC5 cells validated the ECL approach for a high-throughput assay.
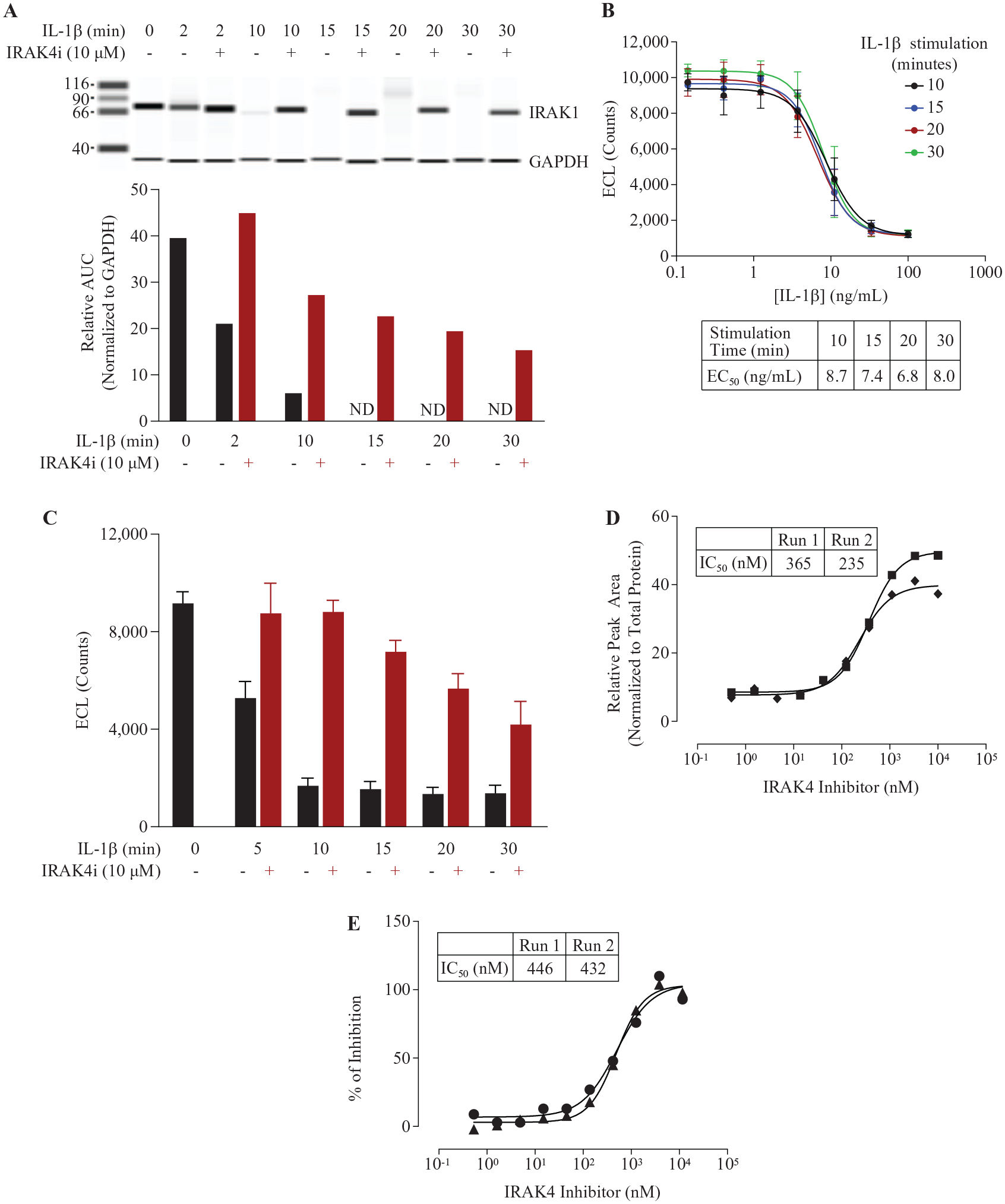
Effect of IRAK4 inhibitor on IRAK1 activation in MRC5 cells stimulated with IL1β. (
Determination of Performance of IRAK1 Activation ECL Assay in High-Throughput Format
To assess the robustness of the ECL assay in high-throughput format, MRC5 cells were seeded into every well of a 384-well plate except the edge wells. Alternate columns of the cells were stimulated with either IL1β at 30 ng/mL corresponding to EC90 or buffer control for 10 min, and the total IRAK1 was detected in cell lysates by the ECL method. With manual operation of the assay, the run resulted in a mean maximum signal of 7974 RLU from no IL1β stimulation with a coefficient of variation (CV) of 10%, and a minimum signal of 1795 RLU from IL1β stimulation with a CV of 14% ( Fig. 5A ). Excluding one obvious outlier from the high signal, the Z′ calculated from eq 1 was 0.47. The maximal column-to-column or row-to-row drifts from high signals were 15.3% and 19%, respectively ( Fig. 5A ). This plate uniformity and signal variability assessment demonstrated that the assay was acceptable for high-throughput screening with a CV <20% for both high and low signals, Z′ ≥0.4, and no significant material drift (<20%) according to the Assay Guidance Manual. 40 To test the reproducibility of IC50, 24 compounds went through a 10-point half-log concentration response in two independent runs. A potency plot showed agreement between run 1 and run 2 with a correlation coefficient of log potency (Pearson r) of 0.94 (p < 0.0001) ( Fig. 5B ). MSR and LsA were also calculated according to the Assay Guidance Manual using eqs 2–5: 46 MSR = 2.5 and LsA = 0.45–2.83 ( Fig. 5C ). The reproducibility test illustrated that the assay passed both test criteria of MSR <3.0 and LsA between 0.33 and 3.0, per the Assay Guidance Manual. 40
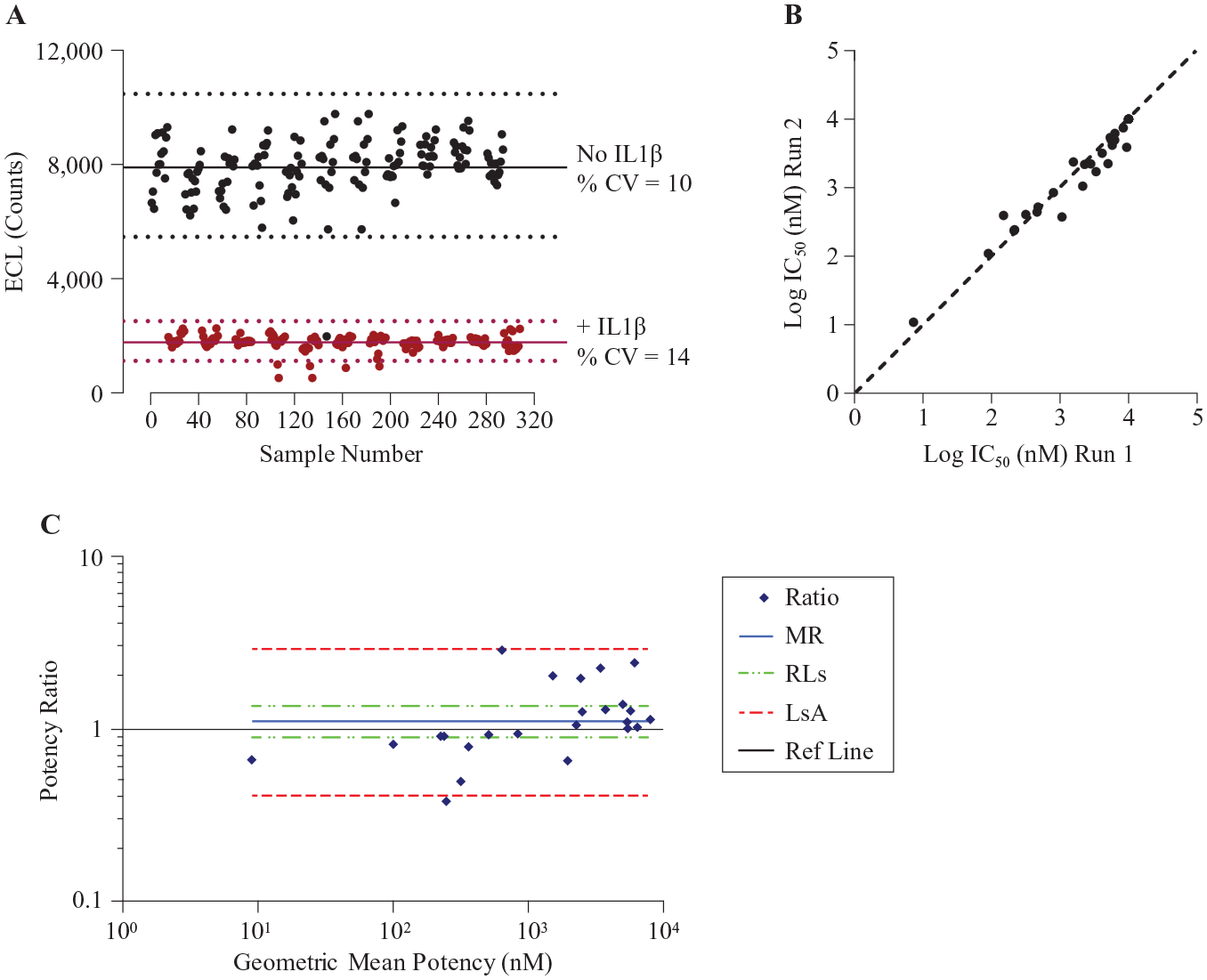
Quality assessments of the high -hroughput mesoscale IRAK1 activation assay in MRC5 cells. (
IRAK1 Activation Assay Evaluation for Biological Relevancy
Although we have demonstrated that the performance of the cell-based IRAK1 activation assay meets the criteria of a high-throughput screening assay, the biological relevance of the measured signal to IRAK4 and associated signaling pathway also needed to be validated in order to deploy the assay in SAR studies for the discovery of novel IRAK4 inhibitors. Twenty-seven compounds were evaluated in 10-point half-log concentration titrations starting at 10 µM, to determine their potency for inhibiting IRAK4 kinase activity in a biochemical IMAP FP assay, cellular IRAK1 activation in MRC5 stimulated by IL1β, and TNFα secretion from IL1β-treated human PBMCs. The selected test set included developing IRAK4 inhibitors from several structural series whose potencies span a range of 3 logs, and two negative control compounds, as demonstrated by their activities in the biochemical IRAK4 kinase IMAP FP assay ( Fig. 6A ). A scatterplot of potencies between the IRAK4 kinase assay and IRAK1 activation showed a positive correlation with the Pearson correlation coefficient (r = 0.64, p = 0.0072) and a bias toward higher potency in the IRAK4 kinase assay ( Fig. 6A ); this is comparable to the association observed between the IRAK4 kinase assay and IL1β-induced human PBMC TNFα secretion (r = 0.68, p = 0.0004) ( Fig. 6C ). There is a stronger positive correlation between the potencies from IRAK1 activation and PBMC TNFα secretion assays (r = 0.81, p = 0.0002) ( Fig. 6E ), both of which measured cellular signaling events following IRAK4 activation, with IRAK1 activation being a proximal functional biomarker while TNFα secretion is a phenotypical outcome of IRAK4 engagement. In addition, both negative control compounds failed to rescue the loss of unphosphorylated IRAK1 stimulated by IL1β in MRC5 cells ( Fig. 6A ), further demonstrating that the measured signal from the IRAK1 activation assay depends on IRAK4 target engagement.
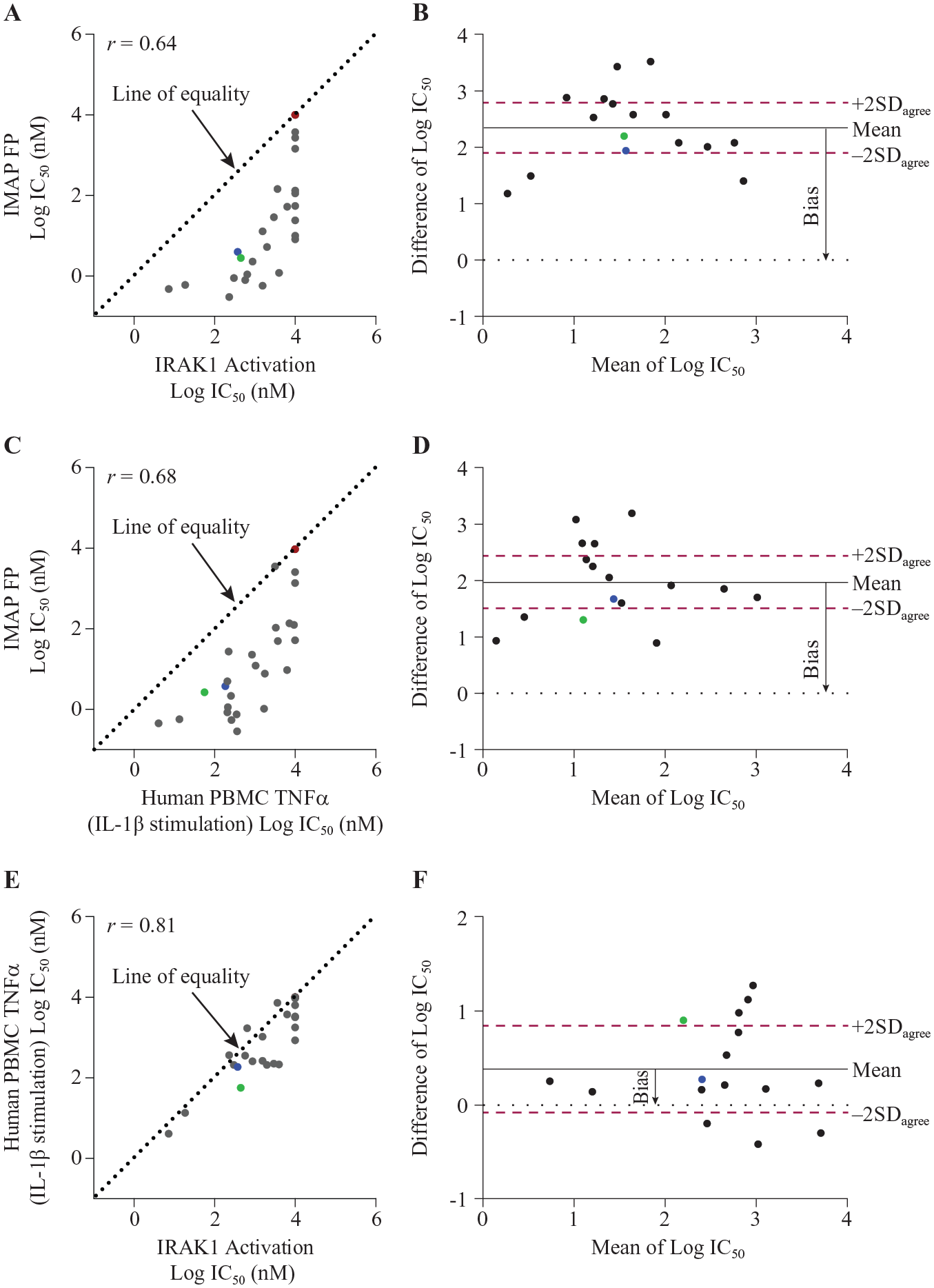
Biological validation of IRAK1 activation assay using agreement analysis. Twenty-seven compounds were run through the IRAK4 in vitro kinase activity IMAP FP assay, IRAK1 activation in MRC5 cells was stimulated by IL1β, and human PBMC TNFα secretion was stimulated by IL1β. Each compound’s IC50 was calculated from a 10-point concentration titration and four-parameter logistic regression curve fitting. (
To define the degree of agreements between the assays, we performed Bland–Altman analysis with the 16 compounds that showed activity in all three assays and calculated acceptable limits of agreement defined by 2SDagree that considers combined variations of the two plotted assays.
43
We first determined each assay’s precision level using SDpooled (eq 6) from two reference compounds that were run multiple times in either the IRAK4 kinase IMAP FP or human PBMC TNFα secretion assay, and the IC50 of 24 compounds from the test–retest runs of the IRAK1 activation assay described above (
Discussion
A review of clinical trials found that a lack of efficacy accounted for more than 50% of phase II and III failures; 44 this is partly attributed to the absence of drug target engagement or target-specific pharmacological intervention in humans.45,46 For example, several unsuccessful phase III clinical trials of iniparib, a compound initially developed as a non-NAD+ competitive PARP prodrug that showed tumoricidal activity in cancer cell lines, was later demonstrated not to be a bona fide PARP inhibitor, but rather it nonselectively modifies cellular cysteine-containing proteins,47,48 highlighting the importance of confirming drug target engagement and mechanism of action in cells during early phase preclinical drug development. To support IRAK4 inhibitor discovery, we developed an ECL-based endogenous IRAK1 activation assay as a proximal functional target engagement assessment for IRAK4 inhibitors.
During the assay development, we observed a disconnect between ECL and simple Western results on evaluating IRAK4 inhibitor in JEKO-1 cells. This phenomenon could be attributed to the difference between the two assay conditions. In ECL, a mild nondenaturing lysis buffer containing nonionic detergent Triton X-100 likely preserved IRAK1 in three native conformations: free IRAK1, recruited to the Myddosome, or activated through phosphorylation and ubiquitination. In simple Western immunoblot analysis, addition of ionic detergent and sample denaturing disrupted all protein–protein interactions. Comparable EC50 values of the agonist R848 stimulation obtained from both detection methods (0.58 µg/mL vs 0.32 µg/mL) indicated that the two antibodies used in ECL were able to capture both free IRAK1 and IRAK1 in the Myddosome complex. However, binding of the IRAK4 kinase inhibitor likely affected interactions among the proteins in the Myddosome, rendering the antibodies incapable of recognizing IRAK1 in the complex. Hence, a partial recovery of IRAK1 signal following complete inhibition of IRAK4 kinase activity was observed in ECL, while a complete recovery was detected by Western blot. Indeed, inactivation of IRAK4 kinase activity has been reported to change the binding affinities among the proteins and increase the stability of the Myddosome structure in LPS-stimulated TLR4 signaling.
34
The assembly of different adaptor proteins into Myddosomes during various TLR or ILR activations
49
could also affect antibodies’ ability to recognize IRAK1 in the complex and would explain the different results of IRAK4 inhibitor on IRAK1 detection by ECL in MRC5 cells. Although IRAK1 levels could be fully recovered by IRAK4 inhibitor under short-term IL1R stimulation in MRC5 cells, receptor endocytic regulation following persistent stimulation and lysosome degradation
50
could cause the observed progressive loss of full recovery on IRAK1 (
Plate uniformity and potency reproducibility studies showed that the assay is acceptable for testing IRAK4 inhibitors in high throughput. Sixteen IRAK4 inhibitors from multiple structural series and with potencies covering a wide range were selected to perform assay agreement analysis. We observed a large systematic potency shift toward the recombinant IRAK4 biochemical kinase assay performed at an ATP Km of 100 µM. Such a bias could be caused by compound cellular permeability or a higher cellular ATP concentration (1–10 mM) that decreases the apparent potency of ATP competitive inhibitors when compared with the biochemical assay result that measured IRAK4 catalytic activity in the presence of 100 µM ATP. With the consideration of variability from both assays, about half of the tested compounds were found to fall outside the limits of agreement between the biochemical assay and either the cellular IRAK1 activation or PBMC TNFα secretion assay. However, Pearson correlation plots showed that in evaluating IRAK4 inhibitors, the results from the IRAK1 assay to either the biochemical or PBMC TNFα cellular phenotypic assay are positively correlated, validating the biological relevancy that IRAK1 activation depends on IRAK4 engagement.
The decision on incorporating the assay into SAR studies depends on whether additional information can be provided for compound assessment. Theoretically, when the two assays measure signals that are purely generated from the same biological target, the results should be in agreement so that one assay can replace the other without losing information. 41 However, between the two cellular assays, only 56% of the compounds tested were within the limits of agreement. This could be due to various causes, including selectivity and less optimized physiochemical properties during the early stage of lead discovery. For example, two compounds (A and B) were selected for a broad kinase panel profiling. Compound A turned out to be a highly selective IRAK4 inhibitor, with IC50 >100-fold selective against 98% of the kinases in the 265-kinase panel, while compound B was >100-fold selective against 66% of the kinases tested. In assay agreement analysis, compound A was within the limits of agreement among all three assays. Interestingly, compound B was within the limits of agreement between biochemical and IRAK1 activation assays, yet fell out of the limit of agreement for the biochemical to PBMC TNFα phenotypic assay or IRAK1 activation to the phenotypic assay analyses, a result reflecting that the phenotypic measurement was influenced by off-target activities. Although the IRAK1 activation assay is less prone to false-positive inhibitors than the downstream phenotypic assay, we should be aware that compounds stimulating dephosphorylation or preventing ubiquitination and proteasome degradation will appear as false positives. Our results suggested that proximal pharmacological assessment of cellular target engagement will be part of the in vitro screening funnel for SAR studies during early preclinical drug development, along with biochemical/biophysical assays and cellular phenotypic assays, especially at the early stage of lead optimization when compound cellular permeability and off-targets are largely unknown.
Supplemental Material
sj-pdf-1-jbx-10.1177_24725552211021074 – Supplemental material for Establishing and Validating Cellular Functional Target Engagement Assay for Selective IRAK4 Inhibitor Discovery
Supplemental material, sj-pdf-1-jbx-10.1177_24725552211021074 for Establishing and Validating Cellular Functional Target Engagement Assay for Selective IRAK4 Inhibitor Discovery by Yiping Chen, Dongyu Sun, Ruojing Yang, Jongwon Lim, Christopher Sondey, Jeremy Presland, Larissa Rakhilina, George Addona, Ilona Kariv and Hongmin Chen in SLAS Discovery
Footnotes
Acknowledgements
We thank Bruce Beutel and Melanie Kleinschek for their support of the project and Merck’s compound management group for compound preparation.
Supplemental material is available online with this article.
Declaration of Conflicting Interests
The authors declared the following potential conflicts of interest with respect to the research, authorship, and/or publication of this article: All authors are current employees of Merck & Co., Inc. (Kenilworth, NJ) and may own stock or stock options in Merck & Co.
Funding
The authors disclosed receipt of the following financial support for the research, authorship, and/or publication of this article: This work was supported by Merck & Co., Inc. (Kenilworth, NJ, USA).
References
Supplementary Material
Please find the following supplemental material available below.
For Open Access articles published under a Creative Commons License, all supplemental material carries the same license as the article it is associated with.
For non-Open Access articles published, all supplemental material carries a non-exclusive license, and permission requests for re-use of supplemental material or any part of supplemental material shall be sent directly to the copyright owner as specified in the copyright notice associated with the article.