
Abstract
Classical high-throughput screening (HTS) technologies for the analysis of ionic currents across biological membranes can be performed using fluorescence-based, radioactive, and mass spectrometry (MS)-based uptake assays. These assays provide rapid results for pharmacological HTS, but the underlying, indirect analytical character of these assays can be linked to high false-positive hit rates. Thus, orthogonal and secondary assays using more biological target-based technologies are indispensable for further compound validation and optimization. Direct assay technologies for transporter proteins are electrophysiology-based, but are also complex, time-consuming, and not well applicable for automated profiling purposes. In contrast to conventional patch clamp systems, solid supported membrane (SSM)-based electrophysiology is a sensitive, membrane-based method for transporter analysis, and current technical developments target the demand for automated, accelerated, and sensitive assays for transporter-directed compound screening. In this study, the suitability of the SSM-based technique for pharmacological compound identification and optimization was evaluated performing cell-free SSM-based measurements with the electrogenic amino acid transporter B0AT1 (SLC6A19). Electrophysiological characterization of leucine-induced currents demonstrated that the observed signals were specific to B0AT1. Moreover, B0AT1-dependent responses were successfully inhibited using an established in-house tool compound. Evaluation of current stability and data reproducibility verified the robustness and reliability of the applied assay. Active compounds from primary screens of large compound libraries were validated, and false-positive hits were identified. These results clearly demonstrate the suitability of the SSM-based technique as a direct electrophysiological method for rapid and automated identification of small molecules that can inhibit B0AT1 activity.
Introduction
At least 5% of the human genome encodes membrane-embedded transport proteins, such as solute carriers (SLCs), ion channels, ion pumps (ATPases), and ABC transporters. 1 Among them, SLC proteins represent the largest group with more than 450 members distributed across 65 subfamilies (http://slc.bioparadigms.org). Given the tissue- and cell-specific expression patterns of SLCs and their remarkable repertoire of substrates, this family of membrane proteins plays key roles in various metabolic pathways. Despite findings implicating SLCs in various human pathophysiologies, including respiratory, cardiovascular, gastrointestinal, and neurological dysfunction,2,3 the SLC family is still underrepresented in research and drug development.4,5
Due to their biological characteristics, transporters are challenging targets for drug development. Several approaches have been developed for the primary screening and profiling of compounds that can affect ion transporter activity: (1) fluorescence-based assays using voltage-sensitive dyes, fluorescent substrate analogs, or ion-specific fluorescent probes; (2) radioactive ligand binding and flux assays; (3) electrophysiological technologies like patch or voltage clamp; (4) atomic absorption spectrometry; and (5) mass spectrometry (MS)-based assays. 2 Most are suitable for high-capacity screening formats (libraries containing >106 compounds using 384- or 1536-well microplates), but these assays can have limited assay sensitivity or specificity that results in a significant error rate.6,7 In addition, radioactive assays require extensive safety measures. Even patch clamp applications, which are still the gold standard for direct real-time recording of ion channel activity and recently available as automated medium- to high-throughput workstations, 8 are often not suitable for the screening of electrogenic transporters. Many transporters are characterized by very slow substrate transport rates (approximately 10–103 molecules per second), up to 105 times less than ion channels. 2 Consequently, highly sensitive assay techniques that have very low background signals are needed for reliable and robust screening assay performance. In addition, some SLC proteins, such as the vesicular zinc transporter ZnT8 in pancreatic β cells, 9 are expressed in internal membrane compartments and are therefore not accessible by standard electrophysiological techniques.
The Surfe 2 r technology (Nanion Technologies GmbH, Munich, Germany) addresses the requirements for an automated and sensitive electrophysiological approach for direct analysis of electrogenic transporter activity, and the technological setup was described in detail previously. 10 The assay principle is based on the ability to detect transient electrical currents of activated transporters. For this purpose, membrane vesicles are immobilized on solid supported membranes (SSMs).10–13 The SSM is applied atop a gold-coated polyethylene surface (sensor) by covalently linking an alkanethiol monolayer to the gold surface and covering it with a phospholipid monolayer. Membrane fragments or proteoliposomes containing the target of interest adsorb to these hybrid layers. Generation of a substrate gradient by rapid exchange of nonactivating (without substrate) for activating (with substrate) buffers induces synchronized activation of numerous transporters, causing movement of charged substrates or ions into the vesicles. Additional ionic movement is prevented by the nonconducting SSM. Through capacitive coupling, the charge accumulation inside the vesicles induces a detectable electric current between the gold electrode and the reference electrode that is submerged in the buffer. The electric current is recorded and amplified. The magnitude of the electric current correlates with charge accumulation inside the vesicles. The final exchange of activating for nonactivating buffers inverts the concentration gradient, resulting in opposing currents and restoration of the initial conditions.
The advantages of the SSM-based assay technology are (1) direct, label-free detection of transporter-induced electrical currents, which are less prone to artifacts; (2) a several-millimeter large sensor diameter that allows adsorption of large amounts of membrane material, and correspondingly high numbers of transporter proteins on the sensor; (3) synchronized transporter activation that induces electrical currents between pico- to nanoamperes, allowing real-time recording of currents by electrical amplifiers, which is particularly beneficial for proteins with low conductance;10,14 (4) a cell-free method that permits batch production and long-term storage of isolated membrane fragments such that continuous cell culture is not required; and (5) the ability to examine transporters embedded in intracellular membranes or reconstituted into liposomes. 15
In recent years, SSM-based electrophysiology was successfully used for biophysical characterization of various transporters, including the human peptide transporter PepT1, 12 the mammalian Na+/K+ATPase, 16 the prokaryotic sodium–calcium exchanger NCX_Mj, 14 and the Ca2+ ATPase SERCA. 17 However, early generations of SSM-based electrophysiology devices offered only a limited degree of automation and sample output, and thus were not applicable for large-scale profiling purposes.18–20 The current generation of SSM-based devices, the Surfe 2 r 96SE, an automated electrophysiology-based high-throughput instrument, allows simultaneous measurement of up to 96 sensors as well as buffer exchange and optional automated sensor preparation with the pipetting unit, significantly increasing assay performance—a prerequisite for medium- to high-throughput approaches.
The objective of this study was to evaluate the SSM-based workstation in the context of a secondary assay to validate actives identified in a primary high-throughput screening (HTS) campaign. The human amino acid transporter B0AT1 (SLC6A19) was chosen as a model target because this transporter is comparatively well characterized, and a limited number of pharmacological modulators suitable for cross-validation are known. B0AT1 is a secondary active, sodium-dependent transporter for neutral amino acids. 21 Expression of this transporter is restricted to the small intestine and kidney and mediates epithelial uptake of neutral amino acids from the intestinal lumen and renal proximal tubules. 22 B0AT1 transport cycles are electrogenic with cotransport of one Na+ per neutral amino acid. 23 Membrane expression and catalytic function of B0AT1 depend on ancillary proteins such as the transmembrane protein collectrin (also known as TMEM27) in the kidney24,25 and angiotensin-converting enzyme 2 (ACE2) in the intestine. 26 Recently, the structure of the ACE2-B0AT1 complex was elucidated, thus allowing a more detailed view of the molecular basis of this interaction. 27
An inactivating dysfunction of the transporter results in Hartnup disease.22,28 The phenotype of this disorder is largely benign, manifesting as massive urinary excretion of neutral amino acids (aminoaciduria) and, in rare cases, with photodermatitis. Surprisingly, mouse models with genetic disruption of SLC6A19 demonstrated improved glycemic control and resistance to high-fat-diet-induced obesity. 29 These findings suggest that reduced systemic levels of amino acids arising from decreased intestinal uptake and increased renal loss associated with SLC6A19 mutation could trigger release of the metabolic regulator FGF21, which induces ketone and fat tissue metabolism. Furthermore, increased GLP1 levels were observed in these mice that stimulate insulin-independent glucose uptake. 29 These findings sparked interest in B0AT1 as a potential new target for diabetic therapies. Recently, inhibition of B0AT1 was also discussed as a potential new approach for the treatment of aminoacidopathies like phenylketonuria. 30 Blocking of neutral amino acid transport in phenylketonuria mouse models by B0AT1 knockout or knockdown resulted in increased excretion rates of phenylalanine in the urine and amelioration of associated neuropathologies. 30
For this study, human and mouse B0AT1 were heterologously expressed using a mammalian expression system (Chinese hamster ovary [CHO] cells). An SSM-based assay was developed and optimized such that real-time current recordings reflect the transport characteristics of B0AT1-induced currents to ensure signal specificity. The assay should guarantee high sensitivity and specificity, as well as produce reproducible and robust results. Finally, the protocol should permit a high degree of automation and routine workflow, thus allowing considerable throughput and implementation of the method into the screening campaign for B0AT1 hit validation and optimization.
Material and Methods
Cell Line Generation and Cultivation
The Flp-In T-REx system (Thermo Fisher Scientific, Waltham, MA) was utilized for stable expression of B0AT1 (SLC6A19) and collectrin (TMEM27) from human or mouse, respectively, according to the manufacturer’s instructions. The cell line was generated in two steps. In brief, cDNA encoding B0AT1 was cloned into the expression vector pcDNA5-FRT-TO-Dest and subsequently transfected into CHO Flp-In host cells using Lipofectamine 2000 (Thermo Fisher Scientific). After successful selection of B0AT1-transfected cells based on resistance to hygromycin B, the cells were cotransfected with pcDNA3.1 collectrin and finally selected by exposure to hygromycin B and geneticin. CHO Flp-In host cells (hereafter referred to as parental cells) and cells expressing B0AT1 collectrin were cultured in Ham’s F-12 media supplemented with 10% (v/v) fetal bovine serum and kept under standard conditions in a humidified incubator at 37 °C and 5% CO2. Culture media for parental cells was supplemented with zeocin, while media for transfected cell lines contained hygromycin B and geneticin. Cultures were passaged every 3–4 days using Detachin (Genlantis, San Diego, CA) for cell detachment. If not otherwise mentioned, antibiotics, culture media, and agents were purchased from Thermo Fisher Scientific.
Isolation of B0AT1 Containing Membrane Fragments
Confluent CHO cells overexpressing human SLC6A19 were harvested from 175 cm2 culture flasks and 50–100 Mio cells were resuspended in 1 mL of cold lysis buffer (10 mM Tris, pH 7.2, 5 mM EDTA, 1× complete protease inhibitor cocktail). Cells were disrupted using Minilys tissue homogenizer (Precellys, Bertin Instruments, Montigny-le-Bretonneaux, France). The homogenate was precleared by centrifugation at 300g for 10 min at 4 °C, and membranes were then isolated by centrifugation at 48,000g for 90 min at 4 °C. To increase the membrane fraction purity, homogenization and reisolation of membranes were repeated twice. The membrane pellet was resuspended in lysis buffer. The total protein concentration was adjusted to 20 mg/mL, and the fractions were stored at −80 °C.
Thiol Coating of 96-Well Sensor Plates
Electrophysiological measurements were performed in 96-well sensor plates (Nanion Technologies). Each sensor well was first filled with 100 µL of 0.5 mM thiol solution (1-octadecanethiol diluted in 2-propanol) and incubated for 2 h or overnight at room temperature (RT). The plate was rinsed twice with 2-propanol, followed by two washing steps with Milli-Q water and drying for 15 min at RT.
Preparation of SSMs
Each thiol-coated sensor was covered with 2 µL of lipid solution (7.5 mg/mL 1,2-diphytanolyl-sn-glycero-3-phosphocholine dissolved in n-decane), followed by immediate addition of 100 µL of loading buffer (30 mM HEPES, pH 7.4, 140 mM NaCl, 4 mM MgCl2, 0.5 mM CaCl2). To increase homogenization, thawed membrane fractions were diluted to 4 mg/mL protein with assay buffer (140 mM NaCl, 4 mM MgCl2, 0.5 mM CaCl2, 30 mM HEPES, pH 7.4, adjusted with N-methyl-d-glucamine [NMG]) and sonicated 10 times using an ultrasonic processor (Hielscher UP50H equipped with MS1 tip, Dr. Hielscher Ultrasonic GmbH, Teltow, Germany) at an amplitude of 20% and a 0.5 s cycle time ratio. It is recommended that the protein concentration for each membrane preparation be optimized individually. Membrane suspensions (5 µL, including 20 µg of total protein) were dispensed directly into the buffer in each well and the sensor plate was centrifuged at 845g for 30 min at 20 °C. Before starting the measurement, the plates were stored at RT for 2 h.
Electrophysiological Analysis and Assay Parameters
All measurements were performed using a Surfe 2 r 96SE instrument (Nanion Technologies). The 96-well sensor plate was placed into the measurement chamber and analysis was executed according to a predefined assay protocol. Before starting the electrophysiological analysis, the electrical properties of the SSM sensor were controlled by determination of conductance and capacitance of the prepared sensors. 14 The SSM-based device includes default protocols to determine these values. Only sensors with conductance below 3 nS and capacitance below 20 nF were used for final data analysis.
The assay protocol defines buffer addition sequence and volumes, application speed and time, wash steps, and operation of the robotic pipetting unit. The current response was recorded during the complete buffer addition sequence. Each run started with the application of assay buffer to remove the loading buffer and to establish a clear baseline before transporter stimulations (low control). Next, leucine-containing activation buffer (140 mM NaCl, 4 mM MgCl2, 0.5 mM CaCl2, 1 mM leucine, 30 mM HEPES, pH 7.4, with NMG) was added to induce transporter-dependent electric currents (high control). In the last step, the activation buffer was replaced by assay buffer and a new cycle of transporter activation could be achieved by repeated addition of activation buffer. In the daily lab routine, activation cycles were consecutively repeated two to three times and the measurements from the repeated cycles were subsequently averaged. If compounds were analyzed either as a single dose or dose–response analysis, substances (up to 30 µM in assay buffer) were added after several repetitions of high control measurements and incubated for 30 min. Buffers were subsequently replaced by compound-containing activation buffer, and leucine-triggered currents were recorded in a single run.
Data Analysis
Currents (I) were recorded and analyzed using Surfe 2 r-specific software packages (SurfControl and DataControl, Nanion Technologies). B0AT1 currents were induced and recorded after application of activating buffers, and current integrals (area under the curve) were calculated. In general, high controls (leucine activation) were repeated three times to ensure stable signals, after compound addition and incubation leucine currents were triggered once. The current integrals were then calculated and used for data analysis. The Michaelis constant (KM) was calculated using the Michaelis–Menten equation. The signal-to-background (S/B) ratio was quantified as follows: S/B = mean (high control)/mean (low control). Inhibition of B0AT1 was calculated as percent inhibition (% inh) and determined as follows: % inh = 100*(1 – Iinhibition/Icontrol). For the comparison of the different amino acids (see Fig. 2A ), the currents obtained with the parental membranes were subtracted from the currents of B0AT1 membranes to calculate the corrected values. The difference currents were normalized relative to Ileucine: Inormalized = (IB0AT1 – Iparental)/Ileucine*100. IC50 was quantified using the four-parameter logistic model and fitted using Sigma Plot 13. All errors are quoted as standard deviation values.
Results
Detection of Electrical Currents of B0AT1 Using an SSM-Based Electrophysiology Setup
B0AT1 utilizes energy stored in the transmembrane potential and the Na+ gradient for transport of neutral amino acids across membranes, resulting in a net movement of ionic charge. 31 With regard to the electrogenic properties of B0AT1, 31 transporter activity was assessed by an SSM-based electrophysiology technique. CHO cells overexpressing human B0AT1 and collectrin were used to prepare membrane fragments. The membrane vesicles were placed atop an SSM-coated gold electrode in each cavity of a 96-well sensor plate. The prepared sensor plate was inserted into the measuring chamber of the workstation, and buffers were rapidly applied via the automatic perfusion system according to the defined solution exchange protocol. After examination of several different assay setups, the final sequence of a complete multistep analysis used for all further experiments, unless mentioned otherwise, comprising six individual, successive activations (I–VI), and currents were recorded for 5 s each ( Fig. 1A ). In the first two measurements (I and II), only nonactivating buffer (without leucine) was applied during the complete analysis to ensure that the starting conditions were predefined and reproducible, as well as to generate a stable baseline. Synchronous activation of B0AT1 transporters was induced upon short application (150 ms) of activation buffer containing leucine, resulting in transient inward-directed electric currents (III–V). Replacement of the activation buffer by nonactivating solutions initiated a reverse current and slow return of the signal to the baseline.
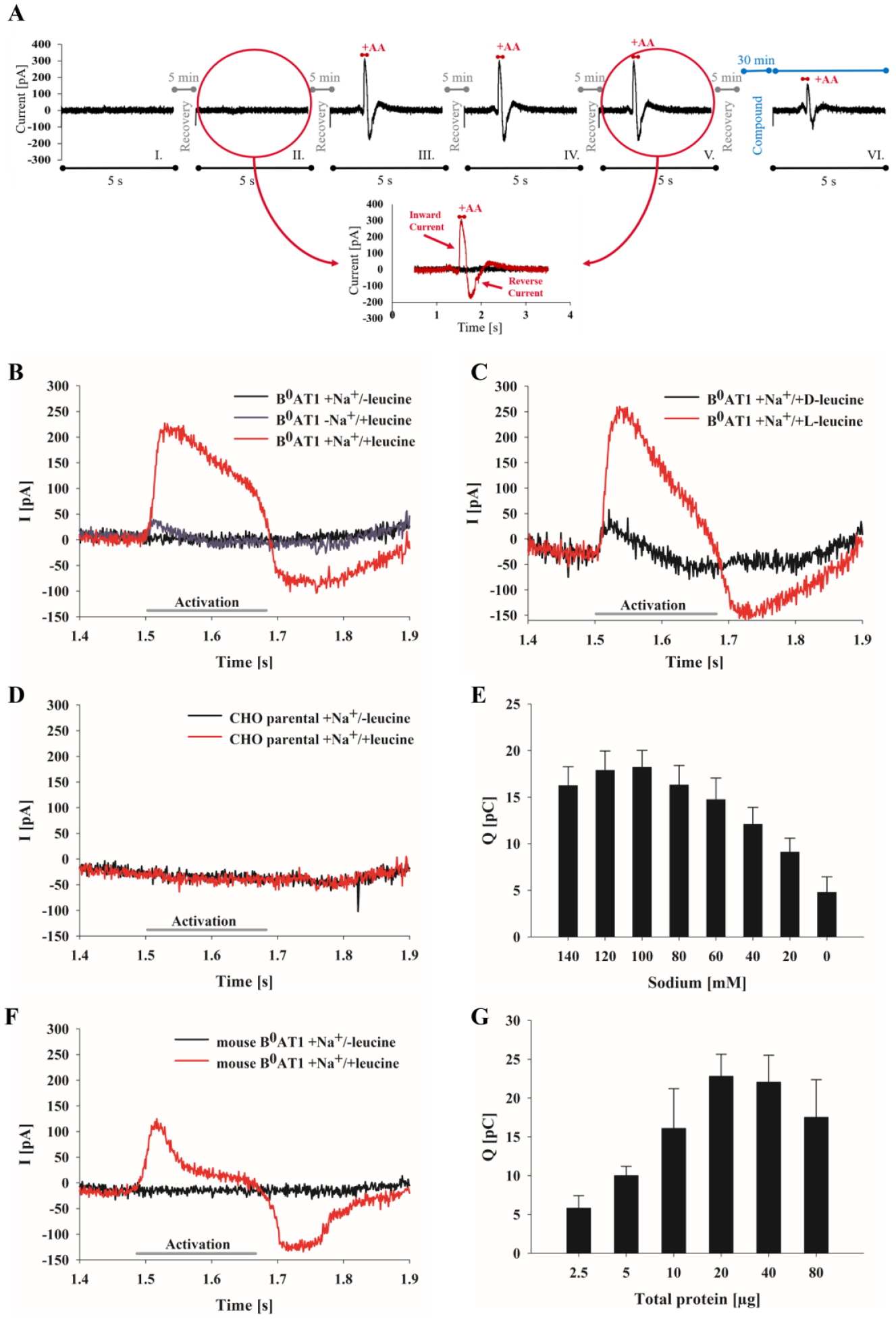
SSM-based analysis of B0AT1 transporter activity using CHO membrane preparations. (
Triggering of transient currents on sensors with membranes containing B0AT1 was dependent on substrate (leucine) availability (
Fig. 1B
, black and red curve). In control experiments in which the assay buffer had choline substituted for sodium (
Fig. 1B
, dark-blue curve), contained the stereoisomeric
In a hit-to-lead optimization campaign, potential hits are also subjected to analysis in nonhuman species for subsequent in vivo validation. For this purpose, the mouse variant of B0AT1 was expressed in CHO cells, membrane fragments were prepared, and SSM-based analysis was performed analogously to the human variant. Activation of mouse B0AT1 transporters with leucine induced fast, transient inward-directed currents ( Fig. 1F ). The transport kinetics of human and mouse B0AT1 differed noticeably after they reached peak maxima. The mouse B0AT1 responses declined more rapidly and exhibited a distinctive peak shoulder that could reflect responses that were characterized by faster kinetics with smaller amplitudes indicating a mixture of substrate binding and transport events. In contrast, the broad current peak seen for the human B0AT1 current peak shows the typical shape of transport processes. Although human and mouse B0AT1 share high amino acid sequence homology, small sequence variations can cause structural differences that would be sufficient to vary transport kinetics.
Titration of the Optimal Amount of Protein for Adsorption to the SSM
The quality of the electrophysiological analysis depends on the condition of the prepared sensor. Using too much or too little of the membrane preparations may result in the formation of a perforated layer or a thick membrane multilayer, respectively. Both conditions can considerably decrease sensor quality and result in increased sensitivity to mechanical and chemical disturbances during measurement, inappropriate capacitance and conductance values of the SSM, high background currents, or inappropriate S/B ratios.10,14 To guarantee sensitivity and reproducibility of the method, the optimal quantity of membranes containing B0AT1 that are immobilized on the SSM sensor must be determined for each membrane batch. For membrane preparations, the total protein concentration of the sample material can be used as a benchmark. Increasing protein amounts were applied to the sensors and the resulting B0AT1 signals were compared. The B0AT1 current amplitudes increased proportionally to the total protein amount ( Fig. 1G ). The highest current amplitudes were obtained with total protein amounts between 20 and 40 µg per sensor for the selected B0AT1 membrane batch. For subsequent measurements, a total protein amount of 20 µg was chosen to obtain optimized sensitivity and avoid waste of sample material.
Monitoring Substrate Specificity of B0AT1 Membrane Preparations
Studies applying two-electron voltage clamp (TEVC)23,31 analysis or transport assays using membrane potential-dependent fluorophores 32 determined that neutral amino acids are the preferred substrates for B0AT1. The SSM-based technology was also utilized to examine the transport selectivity of B0AT1. Amino acids were applied to B0AT1 membranes as well as parental membranes. The B0AT1 membranes’ current responses were monitored, adjusted by parental currents, and the difference currents were normalized to leucine-dependent currents ( Fig. 2A ). Large inward currents were induced by amino acids with hydrophobic (methionine, leucine [ Fig. 2B ], isoleucine) and polar (cysteine, glutamine [ Fig. 2C ]) side chains, coinciding with previous results.22,23,31,32 Amino acids with charged side chains, like arginine, histidine, and lysine, as well as glutamic and aspartic acid, induced no significant inward currents ( Fig. 2A ). The recorded responses after the addition of arginine ( Fig. 2D ) or glutamic acid ( Fig. 2E ) potentially represent artifact currents, because they appeared independently of B0AT1 expression and were insensitive to the tool compound N-(2,3-dimethylbenzofuran-5-yl)acetamide (DMBF-NAc; in-house identifier LB4895). Other transporter species or an unspecific interaction of amino acids with the lipid head groups of the SSM or the membrane fragments may have been the source for these interfering signals. Analysis and comparison of current responses from B0AT1 membranes with parental membrane vesicles can prevent misinterpretation of signals and identification of artifacts, as shown, for example, for arginine in Figure 2D .
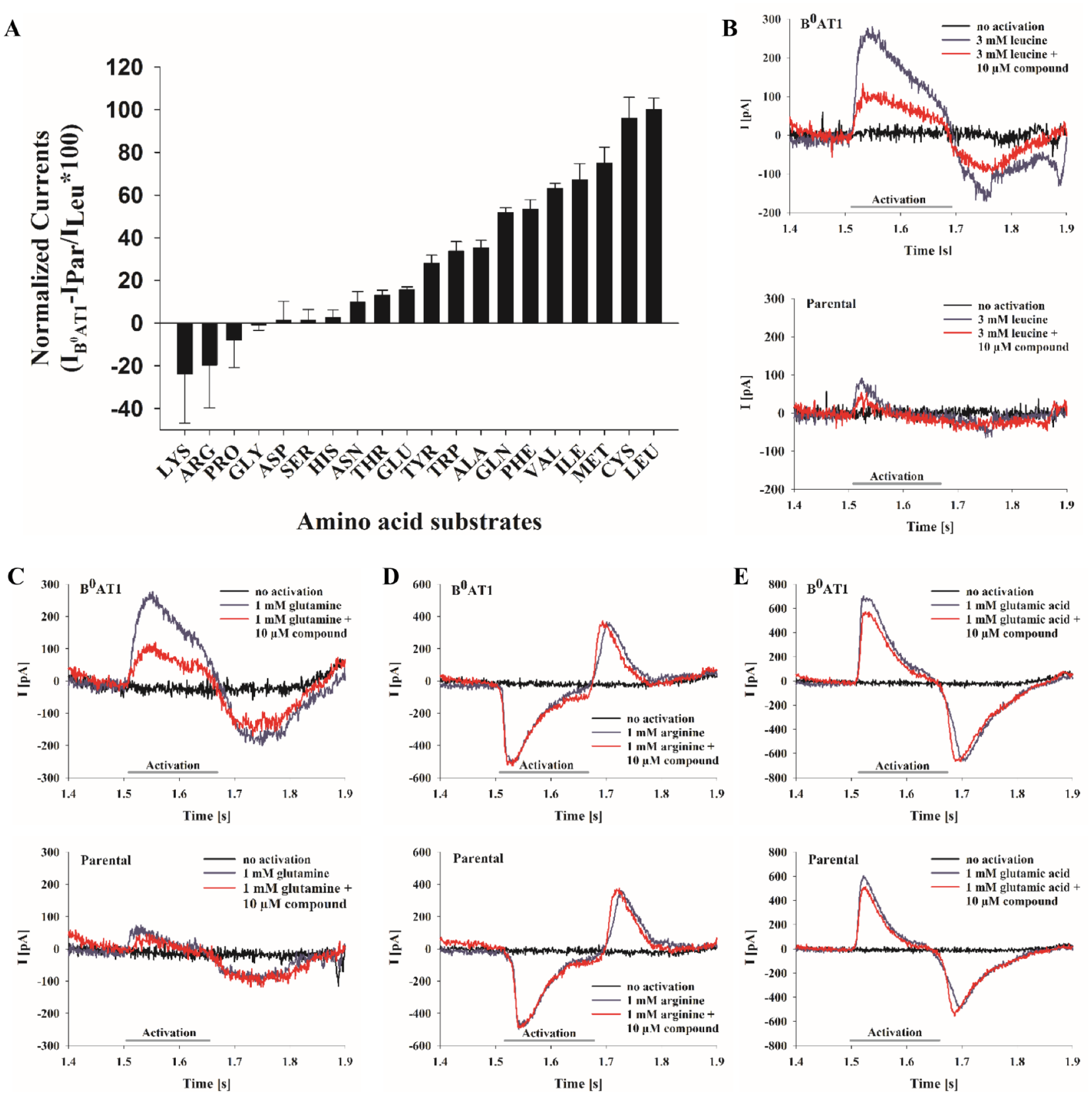
Amino acid specificity of B0AT1-dependent currents. (
Determination of B0AT1 Leucine Affinity
The apparent KM value of B0AT1 for leucine-mediated currents was determined by applying activating buffers having increasing leucine concentrations (0–100 mM) to the membrane preparations in the sensor plate. Data points used for determination of these kinetic parameters were obtained in one activation sequence, and increasing substrate concentrations were applied using the same sensor. Based on these measurements, an apparent KM value for leucine of 1.73 ± 0.31 mM was calculated for human B0AT1 ( Fig. 3A ). Parallel analysis of mouse B0AT1 samples showed a similar KM value of 1.59 ± 0.35 mM ( Fig. 3B ). Previous studies involving different experimental setups generally reported lower KM values compared with those obtained in the present study ( Table 1 ). Previous TEVC studies demonstrated that transport of amino acids via B0AT1 is supported by the membrane potential,23,31,33 and that KM values increased at more positive holding potentials.23,33 This observation could explain the higher KM values obtained with the SSM-based method, as the technique operates without active control of the membrane potential and involves only ionic conductivity of the membrane vesicles and concentration gradients, mainly the leucine gradient, to drive transport processes.
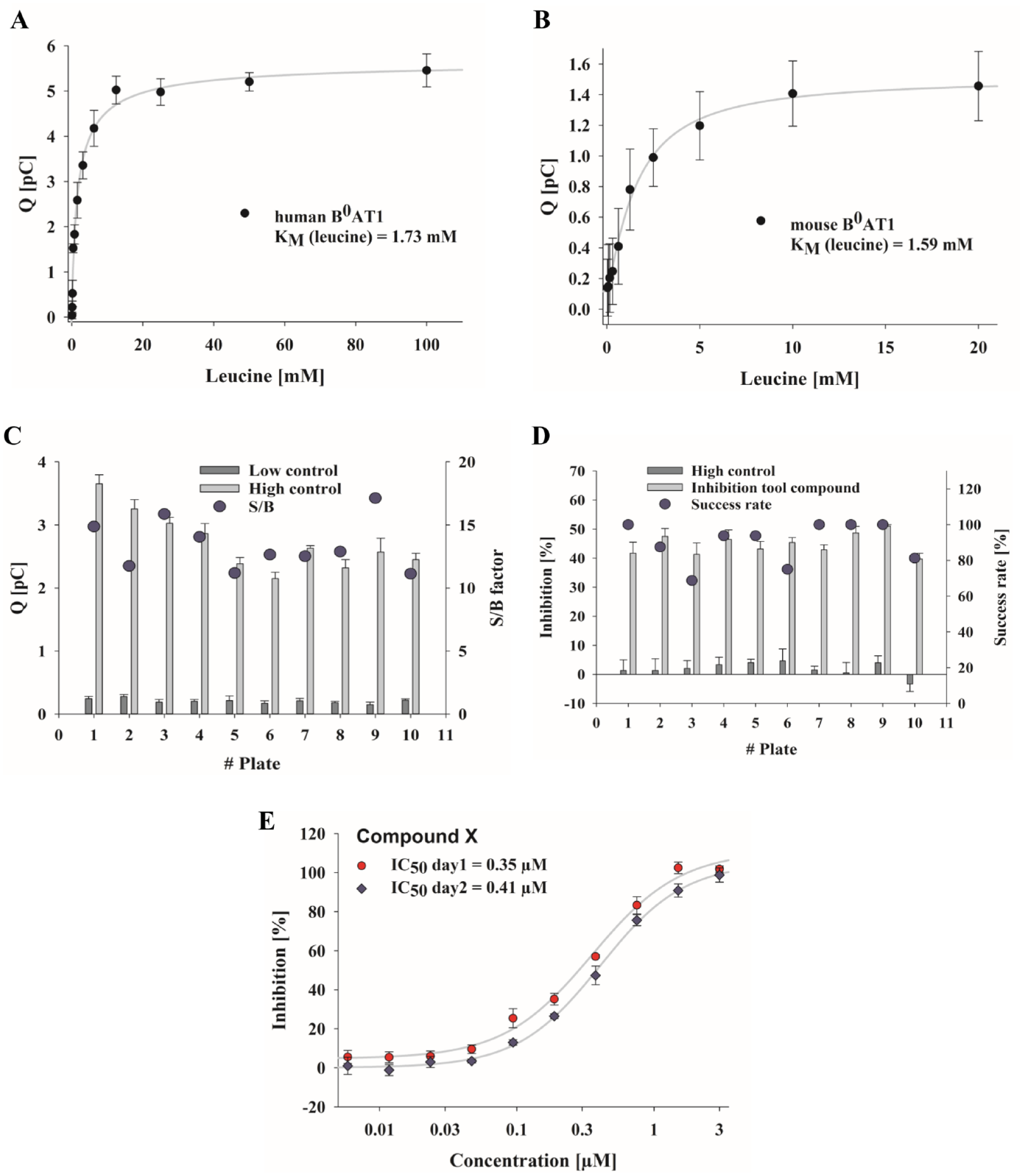
Kinetic parameters of B0AT1-dependent currents and stability and reproducibility of B0AT1 currents using SSM-based assay. Apparent leucine affinity of B0AT1 measured by application of activating buffers containing increasing concentrations of leucine (up to 100 mM). KM for (
Comparison of KM Values for Human and Mouse B0AT1 Determined Using Different Assay Platforms.

KM values determined by SSM-based electrophysiology (this study) were compared with those determined using other analytical approaches, as reported in the indicated studies.
Current Stability and Reproducibility of the SSM-Based Assay
Valid, robust, and reproducible assays are essential for pharmacological HTS and profiling campaigns. Thus, electrophysiological assays must generate stable and reproducible currents and have low background signals and sufficiently high current amplitudes during transporter activation to obtain adequate S/B ratios. To examine the stability of SSM-based currents, successive measurements of multiple assay plates were carried out. All wells represented stable and low background currents (low controls), adequate current amplitudes (high controls) after leucine-dependent activation, and conclusive S/B ratios (larger than 10), which is sufficient for biological assays ( Fig. 3C ). For further evaluation of assay robustness, the inhibitory activity of DMBF-NAc was repeatedly analyzed by applying the half-maximal inhibitory concentration of 10 µM, which was known from the MS assay and confirmed with dose–response measurements later in this study ( Fig. 4C ). The analysis showed reproducible current inhibition (of about 50%) within the same plate and between multiple plates ( Fig. 3D ). Furthermore, the SSM-based assay achieved a success rate, determined as the number of successfully measured wells per plate, of between 70% and 100% (90% ± 9.9% on average), which is acceptable for screening purposes ( Fig. 3D ).
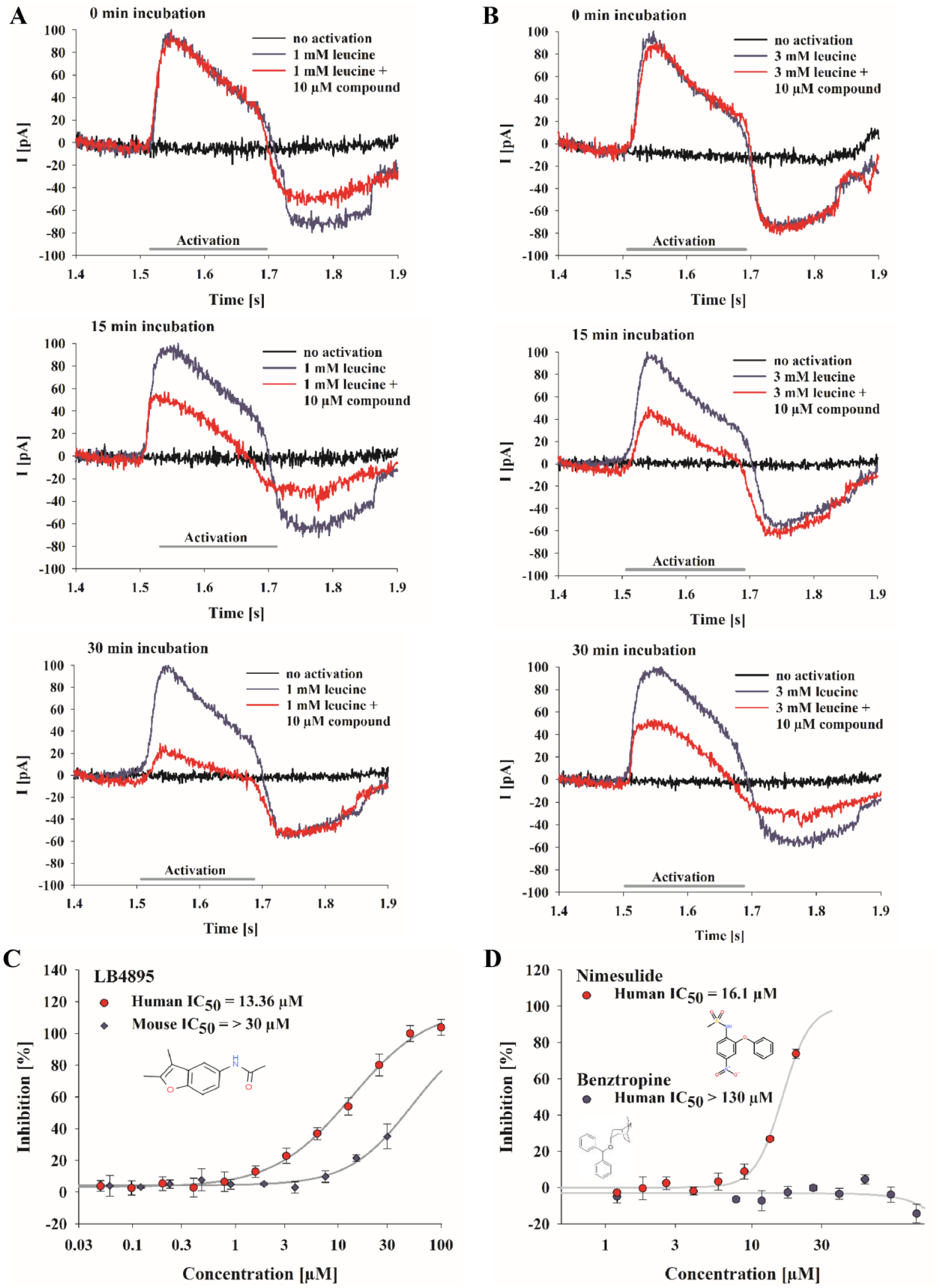
SSM-based analysis of the inhibitory effect of DMBF-NAc on B0AT1 as a tool compound. For characterization of the effect of DMBF-NAc (in-house identifier LB4895) on leucine transport, B0AT1 membranes were preincubated with 10 µM compound for 0, 15, and 30 min (from top to bottom). Transporter responses were triggered by replacing buffers containing 10 µM compound, and 1 mM (
IC50 results recorded during assay development and screening were also tested for reproducibility. The percentage inhibition of the dose–response curves was calculated as means from three or four independent recordings per assay per day, and at least two assay days were performed for each individual compound. Figure 3E shows the repetition of dose–response analysis for one of the profiled compounds. The deviation in IC50 values between the two measurements was sufficiently small and confirmed the suitability of the assay for screening purposes.
Validation of the SSM-Based Assay Using Established B0AT1 Inhibitors
The aim of this study was to test the feasibility of the SSM-based assay in a drug discovery context. The results described above demonstrate that robust B0AT1-specific currents are detectable using SSM-based technology. For further validation of this technique, pharmacological B0AT1 modulators were initially selected for SSM-based electrophysiology analysis.
The heterocyclic compound DMBF-NAc ( Fig. 4C ) was identified as a potent inhibitor of B0AT1 transport activity by an in-house HTS using a fluorescence-based membrane potential assay and was additionally confirmed by an MS-based amino acid uptake assay (data not shown). The inhibitory effect of this substance on B0AT1 was also analyzed with the established SSM-based assay. Sensors were coated with B0AT1 membranes and activations I–V ( Fig. 1A ) were performed as described. Subsequently, nonactivating buffer containing 10 µM DMBF-NAc was applied to the sensors and incubated for 0, 15, and 30 min. Upon replacing the buffer with activating solution containing 10 µM DMBF-NAc and 1 or 3 mM leucine, B0AT1-dependent leucine transport was induced ( Fig. 4A,B ). Prolonged incubation with DMBF-NAc measurably diminished the amplitude of the B0AT1-dependent currents ( Fig. 4A ). After 30 min of incubation, a nearly complete block of the currents was obtained. This incubation time was used for all further compound measurements. The higher concentration of leucine, that is, 3 mM versus 1 mM ( Fig. 4A,B ), clearly reduced the inhibitory effect of DMBF-NAc on B0AT1-mediated currents. The substrate concentration-dependent inhibition of currents indicates a putative competitive block of B0AT1 by DMBF-NAc, and the long incubation time could be explained by binding of the compound to the transporter on the trans-side. Performing dose–response experiments, this benzofuran-derivate inhibited leucine transport with an IC50 of 12.9 ± 1.2 µM ( Fig. 4C , showing an exemplary IC50), which is consistent with the IC50 of 12.2 ± 0.3 µM determined in an MS-based amino acid uptake assay (data not shown). In contrast, DMBF-NAc is only a weak inhibitor of the mouse variant of B0AT1 ( Fig. 4C ) and achieved only partial inhibition even at the highest compound concentration tested (30 µM).
To date, only limited data concerning potent inhibitors of B0AT1 are available. The anti-inflammatory drug nimesulide was identified as a promising B0AT1 inhibitor (IC50 = 23 µM) in a [3H]glutamine transport assay using B0AT1-reconstituted proteoliposomes, although high inhibitor concentrations (1 mM) were required for maximum inhibition.
34
Results from the SSM-based assay performed in this study also suggested a potentially inhibitory effect of nimesulide on B0AT1 with a calculated IC50 of 12.4 ± 4.3 µM (
Fig. 4D
, showing an exemplary IC50). However, the dose–response measurements for nimesulide failed to reach maximum inhibition and concentrations above 30 µM precipitated in the assay buffer. In a cell-based, radioactive uptake (
Recently, membrane potential-sensing fluorescent assays (FLIPR) were developed for the identification of novel inhibitors of B0AT1.32,35 Benztropine was one of the substances identified and further characterized by the FLIPR assay. 32 Using the SSM-based assay, the sensitivity of B0AT1 against benztropine could not be reproduced, even at high compound concentrations (up to 130 µM) ( Fig. 4D ). Off-target or toxic effects might exert influence on the potency of the inhibitor, manipulating the results of indirect fluorescence-based assays. The recently published inhibitors of the second generation, which exhibit a potency of around 1–15 µM, could be more promising tool compounds compared with nimesulide and benztropine. 35
Validation of In-House Compounds with SSM-Based Electrophysiology
Confirmation of the inhibitory effect of DMBF-NAc on human B0AT1, as well as proven specificity, sensitivity, and reproducibility of the assay, demonstrated the applicability of the method for identification of pharmaceutic agents that can modulate B0AT1 activity. Moreover, the SSM-based instrument allows automated and simultaneous processing of 96 samples, which considerably reduces the assay time compared with devices that lack robotic parallel processing and recording of sensors. Under these conditions, the method was used as a secondary assay within the B0AT1 screening cascade. Active compounds that were preselected using a liquid chromatography (LC)/MS-based HTS amino acid uptake assay 36 were tested with the validated SSM-based assay for confirmation of compound potency. In the first round of analysis, the substances were tested at a single dose at 10 µM (data not shown) using human and mouse B0AT1 membranes. Subsequent, active compounds (>30% inhibition) were selected and tested in dose–response experiments. The potencies of selected compounds from several chemically diverse clusters are summarized in Table 2 and Figure 5A–D . In general, the SSM-based measurements largely confirmed the MS-based results, in that the rank order of the compound potencies determined in both assays correlated approximately, but the potency to inhibit B0AT1 was decreased by a factor of 3 on average relative to the MS results. Correlation of IC50 results from the SSM- and LC/MS-based HTS illustrate these discrepancies ( Fig. 5E ). Different assay principles could contribute to this observed shift in potency. The LC/MS-based uptake assay is a cellular assay; thus, transporter activity is embedded in physiological processes and supported by an existing membrane potential. In contrast, the SSM-based assay involves membrane preparations that lack active control of the membrane potential and has the substrate gradient as the sole driving force. The LC/MS-based uptake assay performs endpoint measurements of substrate accumulation within the cell and is more susceptible to nonspecific or cytotoxic effects. Moreover, different expression systems (i.e., MDCK vs CHO) could affect the assay results. Importantly, the SSM-based method confirmed the most potent compound and series ( Table 2 , cluster A, compound 1), which was comparable to MS outcomes regarding sensitivity. The screen also identified two false-positive compounds ( Table 2 , compounds 7 and 8, cluster C). Both compounds were previously identified as active hits in the LC/MS-based primary screen. Through use of the SSM-based method, both substances were not active in inhibiting human B0AT1 and achieved only high IC50 values above 10 µM for mouse B0AT1. The inactivity of these two compounds was additionally confirmed in transwell–transporter assays, as well as in in vivo validation studies (data not shown).
IC50 Values for Selected Compounds Determined from SSM-Based Leucine Transport (This Study) and LC/MS-Based Isoleucine Uptake Assays.

Chemically related compounds are grouped into clusters (A–D). IC50 values were calculated from dose–response experiments using CHO membrane preparations in the SSM-based assay or with a cell-based (MDCK) isoleucine uptake assay.
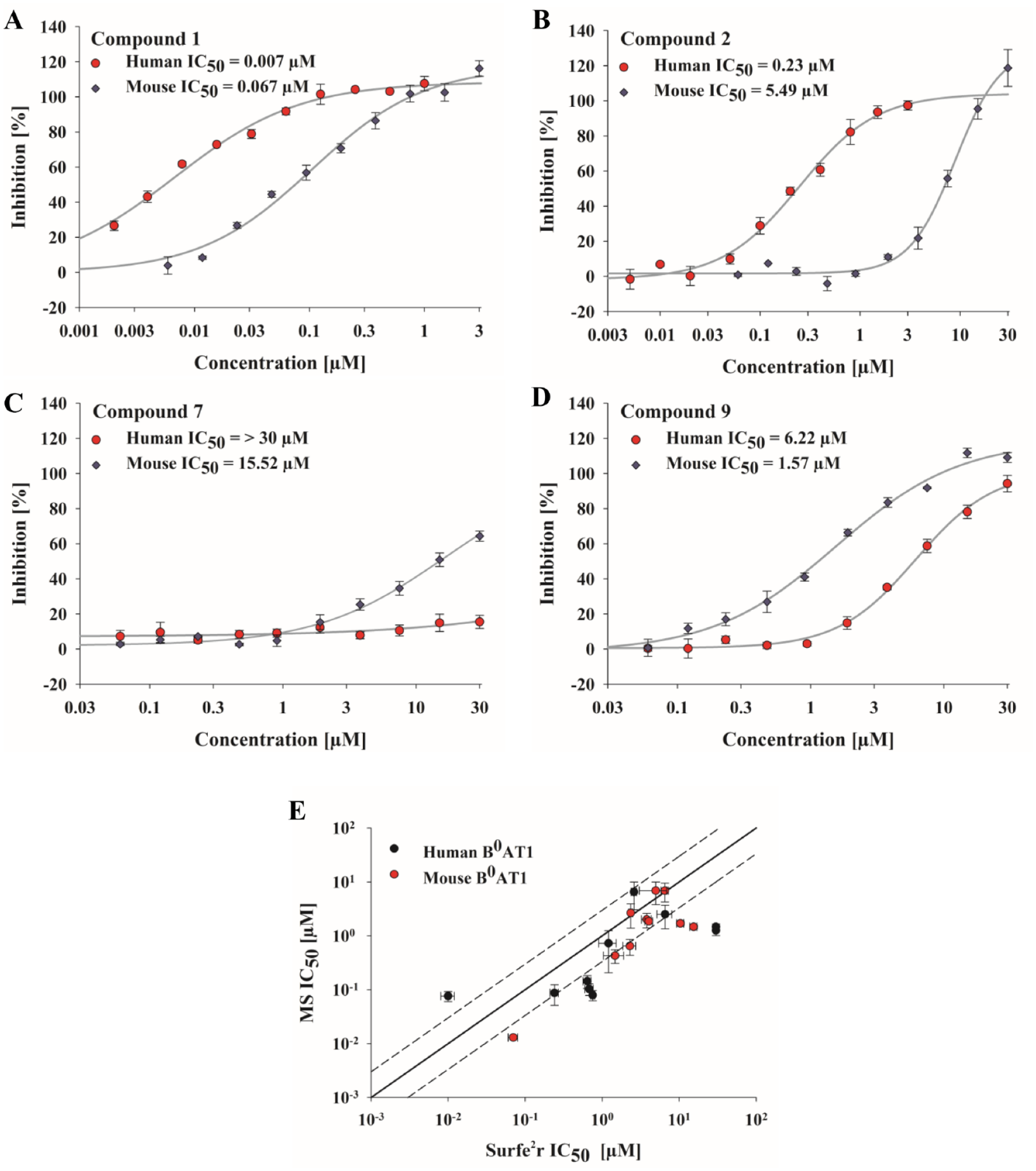
Dose-dependent activity of selected pharmacological agents on B0AT1. (
IC50 values determined for mouse and human B0AT1 isoforms exhibited noticeable differences for many compounds, and the overall compound potency was much lower for mouse B0AT1, independent of the applied assay technique. Alternative binding sites or varying binding affinities and kinetics of the compounds might explain this species-dependent difference in inhibitory potency. Such species-specific discrepancies have also been observed for ion channels, such as the sodium channels Nav1.8 37 and Nav1.7.38,39
Discussion
In this study, the suitability of the SSM-based approach for drug development was investigated using membrane preparations of B0AT1-overexpressing CHO cells. As a proof of concept, recording of leucine-dependent real-time currents was achieved only in B0AT1-containing membranes, demonstrating the specificity of the induced responses. Further electrophysiological characterization of the SSM-based technique in terms of substrate specificity, dependence on cosubstrates, and kinetic properties of B0AT1 correlated well with data obtained with classical TEVC analysis.23,31 In general, a higher KM value was obtained with the SSM-based approach compared with other methods. Possible reasons for this discrepancy include the diverse assay setups (TEVC, radioactive uptake assay, fluorescence-based protocols, different cells) and the reduced cellular context of the SSM-based technique, including the absence of a membrane potential such that the substrate gradient is the only remaining driving force.23,33
The current generation of SSM-based workstations promises sensitive, automated, and accelerated electrophysiology-based transporter analysis and could thus represent a useful complement for large-scale compound screenings. The analysis time for one 96-well plate with the applied B0AT1 protocol is around 1.25 h, including repeated measurement of low and high controls, addition and incubation of compounds, and final recording of leucine-triggered currents. The success rate per plate, defined as the number of evaluable wells per plate, was 90% on average, which minimizes the loss of data points and avoids the need for multiple repetitions of measurements. The preparation time for membrane material from cells depends on the applied protocol, but membranes can be isolated in large batches and stored for months without loss of activity, thus dispensing with the need for ongoing cell culture. The technical setup of the current SSM-based instrument allows automated preparation of the SSM, including the target-containing membrane vesicles on the sensor by the device itself, thus ensuring consistent quality of the SSM. Together, analysis of up to six sensor plates per 8 h workday can be carried out for the described B0AT1 protocol to allow direct, medium- to high-throughput electrophysiology-based measurements and compound screening.
In the course of assay development, the anti-inflammatory drug nimesulide and anticholinergic/antihistamine agent benztropine were used for assay validation. Both compounds were previously identified as potential inhibitors for B0AT1 using a [3H]glutamine uptake assay 34 or a FLIPR assay. 32 In contrast, using the SSM-based protocol nimesulide failed to reach maximum inhibition, and benztropine does not inhibit B0AT1 currents in dose–response experiments. This result is consistent with a study by Cheng et al. involving a radioactive uptake assay that showed that nimesulide was significantly less active. 32 The three studies differ in the applied screening method and expression system and can suffer from low sensitivity and potential nonspecific effects, but the divergent results emphasize the importance of hit validation using appropriate, sensitive, and direct assay technologies in the course of drug development projects. This importance is further highlighted by a recent pharmacological screen for new inhibitors of B0AT1 in which more than 3400 compounds were tested using two indirect cell-based assays. 36 In this screen, a FLIPR-based membrane potential assay achieved a hit rate of 7%, whereas an LC/MS-based uptake assay using stable isotope-labeled compounds had a hit rate of 1.5%, and the intersection of the hit rates was 0.6%.
For identification and development of new leads to modulate B0AT1 activity, an LC/MS-based uptake assay was chosen for initial screening of large compound libraries. For evaluation of the collected hits, the SSM-based assay was established as a secondary screen, detecting the transporter activity directly, reliably, and for the first time with acceptable throughput in electrophysiology-based measurements. Use of the SSM-based technique in the screening cascade for B0AT1 confirmed potent hits from the primary LC/MS-based screen and, more importantly, also detected false-positive compounds, demonstrating the usefulness of this direct screening method for hit validation and suggesting the advantage of a combination of both assays within one pharmaceutical screening campaign relative to a screening tree that includes only indirect assay systems. Hits identified in both assays can be selected for further characterization and testing within the screening campaign. A similar approach was successfully used for the pharmacological characterization of single compounds targeting nicotinic acetylcholine receptors.40,41
In summary, the current study demonstrates the successful implementation of SSM-based electrophysiology technology as a secondary screen for hit validation in a B0AT1 screening campaign. Through the direct measurement of electrical currents, SSM-based electrophysiological approaches are well suited for the analysis of versatile transport proteins. Thus, the SSM-based method represents a valuable medium-throughput technology for drug development in this target class.
Footnotes
Acknowledgements
The authors would like to thank Birgit Kaiser and her team for providing cell batches, Dieter Schmoll for providing the cell line, as well as Adam Belanger, Felix Baerenz, Alleyn Plowright, and John Macor for their constructive criticism and support.
Declaration of Conflicting Interests
The authors declared the following potential conflicts of interest with respect to the research, authorship, and/or publication of this article: All authors are Sanofi employees and may hold company shares and/or stock options.
Funding
The authors disclosed receipt of the following financial support for the research, authorship, and/or publication of this article: This work was fully funded by Sanofi.