
Abstract
Oncogenic forms of KRAS proteins are known to be drivers of pancreatic, colorectal, and lung cancers. The goal of this study is to identify chemical leads that inhibit oncogenic KRAS signaling. We first developed an isogenic panel of mouse embryonic fibroblast (MEF) cell lines that carry wild-type RAS, oncogenic KRAS, and oncogenic BRAF. We validated these cell lines by screening against a tool compound library of 1402 annotated inhibitors in an adenosine triphosphate (ATP)-based cell viability assay. Subsequently, this MEF panel was used to conduct a high-throughput phenotypic screen in a cell viability assay with a proprietary compound library. All 126 compounds that exhibited a selective activity against mutant KRAS were selected and prioritized based on their activities in secondary assays. Finally, five chemical clusters were chosen. They had specific activity against SW620 and LS513 over Colo320 colorectal cancer cell lines. In addition, they had no effects on BRAFV600E, MEK1, extracellular signal-regulated kinase 2 (ERK2), phosphoinositide 3-kinase alpha (PI3Kα), AKT1, or mammalian target of rapamycin (mTOR) as tested in in vitro enzymatic activity assays. Biophysical assays demonstrated that these compounds did not bind directly to KRAS. We further identified the mechanism of action and showed that three of them have CDK9 inhibitory activity. In conclusion, we have developed and validated an isogenic MEF panel that was used successfully to identify RAS oncogenic or wild-type allele-specific vulnerabilities. Furthermore, we identified sensitivity of oncogenic KRAS-expressing cells to CDK9 inhibitors, which warrants future studies of treating KRAS-driven cancers with CDK9 inhibitors.
Introduction
The RAS GTPase family consists of HRAS, NRAS, and KRAS, and they encode four RAS isoforms (with KRAS encoding two splice variants: KRAS-4A and KRAS-4B). All RAS isoforms function at the plasma membrane, and cycle between the active guanosine triphosphate (GTP)-bound state and the inactive guanosine diphosphate (GDP)-bound state. RAS acts as a GTP/GDP switch linking extracellular stimuli to intracellular signaling pathways, to regulate key cellular activities and maintain homeostasis. Two major downstream signaling pathways, RAF/MEK/ERK and phosphoinositide 3-kinase alpha (PI3K)/AKT/mammalian target of rapamycin (mTOR), control cell proliferation and maintain cell survival. 1 RAS was identified as an oncogene more than 40 years ago, and it is one of the most mutated genes in human cancer. A recent analysis estimated that about 19% of cancer patients carry a RAS mutation, with 75% of them being KRAS mutations. In particular, KRAS mutations predominate in pancreatic adenocarcinoma (88%), colorectal adenocarcinoma (50%), and lung adenocarcinoma (32%). 2 KRAS mutations are found in three hotspots: codons 12, 13, and 61, amino acids that are within the guanine nucleotide-binding interface. These missense mutations result in increased nucleotide exchange (GDP for GTP) and/or decreased GTP hydrolysis, and consequently hyperactive KRAS. Aberrant downstream signaling activation (e.g., RAF/MEK) leads to uncontrolled cell proliferation and ultimately tumor formation. 1 Given the prevalence of KRAS mutations in human cancers, KRAS has been an attractive target for drug development.
Despite significant efforts, directly targeting oncogenic KRAS has proven to be challenging. Targeting the KRAS guanine nucleotide-binding site seems unachievable, given the high binding affinity for GTP and GDP, and high cellular concentration of GTP. 3 Previous efforts to target KRAS posttranslational processing have also failed. 4 Currently, there are several compounds under clinical trials that were developed to covalently bind directly to KRASG12C, and they showed promising efficacy against tumors with KRASG12C. They were not, however, expected to have any effects on other oncogenic KRAS alleles.5,6 Alternatively, inhibitors that target major downstream signaling molecules (e.g., RAF and MEK) have been studied in clinical trials. RAF inhibitors were unfortunately shown to cause paradoxical activation in RAS mutant cells, and hence were not recommended for oncogenic RAS-driven cancers. 7 MEK inhibitor monotherapies also showed only limited response rates in a majority of the trials. 8 More importantly, these signaling pathways are required for normal cell survival and homeostasis, and as a result, inhibitors targeting these pathways potentially have small therapeutic windows, limiting their use. New approaches are being developed to identify new biological targets for treating oncogenic KRAS-driven cancers. 9
One of the new approaches is the synthetic lethal screen. It is a powerful tool for identifying previously unknown genotype-specific vulnerabilities. 10 For oncogenic KRAS, this approach allows us to identify signaling pathways or biological targets on which the mutated cells have acquired dependence. While inhibition of a synthetic lethal target does not cause detrimental effects on normal cells, it may be lethal to oncogenic KRAS-expressing cells. Two distinct approaches have been used to investigate oncogenic KRAS synthetic lethality. In one approach, RNA inteference (RNAi) or clustered regularly interspaced short palindromic repeat (CRISPR)-mediated screens have successfully identified multiple genes that are specifically required for oncogenic KRAS-expressing cell survival.11–16 However, the path to developing small molecules and ultimately showing the same synthetic lethality by these compounds is challenging. In another approach, a small-molecule compound library is used in phenotypic screens to directly identify compounds that have genotype-specific activity.17–19
In this study, we developed and validated a panel of isogenic MEFs consisting of cell lines that express wild-type (WT) RAS (KRAS-4B, HRAS, and NRAS) as well as cell lines that express oncogenic KRAS and BRAF alleles that are commonly found in human cancers (KRASG12C, KRASG12D, KRASG12V, KRASG13D, KRASQ61R, and BRAFV600E). We then used these cell lines to perform a high-throughput screening campaign with an adenosine triphosphate (ATP)-based cell viability assay to identify compounds that showed antiproliferative effect preferentially in oncogenic KRAS-expressing MEF cell lines over WT RAS-expressing cell lines. Secondary cell-based, biochemical, and biophysical assays were subsequently performed to identify the biological targets and the underlying molecular mechanisms of these selected compounds. Ultimately, we identified five chemical clusters, including one natural product, that show selective activity against oncogenic KRAS-expressing cells. Three of these five clusters were shown to demonstrate CDK9 inhibitory activity in an in vitro kinase assay and in KRASG12D-expressing MEFs, demonstrating the potential use of CDK9 inhibitors in treating cancers carrying oncogenic KRAS alleles.
Materials and Methods
Reagents and Cell Culture
Mouse embryonic fibroblasts (DU1473) null for both Hras and Nras were provided by M. Barbacid’s laboratory (CNIO, Madrid, Spain). 20 Cells were treated with 600 nM 4-hydroxy tamoxifen (Sigma-Aldrich, St. Louis, MO) for 11 d to eliminate the endogenous floxed Kras gene. The MEF panel was developed with cells lacking all endogenous Kras and were growth-arrested, transduced with lentiviral constructs expressing the WT RAS, mutant KRAS, or mutant BRAF allele. The peripheral blood mononuclear cells were purified from human blood samples (buffy coats) purchased at the Etablissement Français du Sang (Strasbourg, France). MRC5 and LS513 cells were obtained from the American Type Culture Collection (ATCC, Manassas, VA). Colo320 and SW620 were obtained from the DSMZ-German Collection of Microorganisms and Cell Cultures (Braunschweig, Germany) and the European Collection of Authenticated Cell Cultures (Salisbury, UK), respectively. The inhibitor library (cat. no. L1100) was purchased from Selleckchem (Houston, TX). The following antibodies were purchased from Cell Signaling Technology (Danvers, MA): phospho-Ser2 RNA polymerase II (cat. no. 13499) and total RNA polymerase II (cat. no. 14958).
Western Blots
Cells were lysed in Tris-based lysis buffer plus protease and phosphatase inhibitor cocktail (ThermoFisher). Fifteen to thirty micrograms of protein for each sample were then separated by sodium dodecyl sulfate–polyacrylamide gel electrophoresis (SDS-PAGE) using Invitrogen’s Bolt system (Invitrogen, Carlsbad, CA), transferred to polyvinylidene fluoride (PVDF) membranes (Bio-Rad, Hercules, CA), and blocked using Odyssey blocking buffer (LI-COR, Lincoln, NE). Membranes were incubated overnight with primary antibodies (1:1000) in Odyssey blocking buffer plus 0.2% Tween-20 (LI-COR), incubated with LI-COR IRDye 800CW or IRDye 680RD secondary antibodies at 1:15,000, and analyzed by the Odyssey imaging system (LI-COR).
ATP-Based Cell Viability Assay
Cell viability was measured by the CellTiter-Glo assay (Promega, Fitchburg, WI), following the provided protocol. Briefly, for the primary screen, assay-ready plates (384-well, cat. no. 781092; 1536-well, cat. no. 782092; both from Greiner Bio-One, Kremsmünster, Austria ) containing 150 nl or 25 nl of 10 mM compounds were prepared with an Echo acoustic dispenser (Labcyte, San Jose, CA). The final concentration was 5 µM. Cells were then dispensed in the assay plates with a Multidrop Combi Reagent dispenser (ThermoFisher). Cell culture media, cell plating densities, and cell line doubling times are listed in
For dose–response curve experiments, serial dilutions were made with an automatic multi-channel pipetting robot (CyBio, Jena, Germany), diluted on-line with the Multidrop Combi Reagent dispenser, and added to the assay plates with a V-prep station (Agilent Technologies, Santa Clara, CA). 72 h (MEFs) or 96 h (colorectal cancer cell lines) later, CellTiter-Glo reagent was added to the cells. After a 15 min incubation, luminescent signal was read with either the EnVision (PerkinElmer, Waltham, MA) or PHERAstar (BMG Labtech, Ortenberg, GA) plate reader.
Cellular Toxicity Assay
Cell toxicity was assessed with the 4-Plex Apoptosis Kit (Intellicyt, Ann Arbor, MI), which measures four parameters: cell viability (membrane integrity), caspase activity (caspase 3/7 substrate), annexin V binding (surface detection of phosphatidylserine), and mitochondrial damage (mitochondrial depolarization). One thousand cells were plated per well of a 384-well plate and were incubated with the tested compounds. Forty-eight hours later, 10 µl staining cocktail was added to the cells, and was incubated for 1 h at room temperature (RT). Data were then acquired using the iQue Screener (Intellicyt).
Kinase Profiling
Activities of up to 365 kinases were assessed with the Kinase Profiler service (Eurofins, Luxembourg). Briefly, the in vitro kinase assay measures enzymatic activity via substrate phosphorylation based on incorporation of radioactivity from γ-[32P]-ATP in the presence of the tested compounds.
Cell-Based Homogeneous Time-Resolved Fluorescence (HTRF) Assay
Phosphorylation levels of ERK (cat. no. 64AERPEH, Cisbio, Bedford, MA), AKT (cat. no. 64AKSPEG, Cisbio), and EGFR (cat. no. 64EG1PEG, Cisbio) were measured by the HTRF assay kit, following the provided protocol. Briefly, 15,000 cells were seeded for each well of a 384-well plate. On the next day, cells were treated with the tested compounds. Twenty-four hours after inhibitor treatments, cells were lysed with the provided lysis buffer, followed by addition of the cryptate-coupled antibody and the acceptor-coupled antibody. The lysate was incubated with the antibodies for 4 h, and the HTRF signal was then measured with the EnVision or PHERAstar plate reader.
Biochemical Assays
PI3K enzymatic assay: Human p110α with an N-terminal poly-His tag was co-expressed with a p85α subunit in an Sf9 baculovirus expression system, and the p110α/p85α heterodimers were purified by sequential Ni-NTA (nickel–nitrilotriacetic acid) and heparin chromatography. Lipid kinase activity assays were performed using the PI3K HTRF assay kit according to the manufacturer’s instructions (Merck, Kenilworth, NJ). Serial dilutions of inhibitors were preincubated for 15 min at RT with 10 μM PI(4,5)P2 substrate and 150 pM enzyme mixture before starting the reaction by the addition of 100 μM ATP. After 15 min of incubation at RT, the revelation mixture was added (Merck kit). The fluorescence intensity signals at 665 nm and 620 nm were recorded after overnight incubation at 4 °C. The results were expressed as the fluorescence signal ratio [(665 / 620) × 10,000].
ERK2 and AKT1 enzymatic assays: Human recombinant ERK2 protein was purchased from Carna Bioscience (Kobe, Japan), and hAKT1 from Life Technologies (Carlsbad, CA). Serial dilutions of compounds were preincubated with 2 nM ERK2 enzyme for 30 min at RT before starting the reaction by the addition of substrate mixture 50 µM ATP/1.5 µM FL-Peptide 8 (cat. no. 760352, Caliper Life Sciences, Hopkinton, MA) in reaction buffer A [100 mM Hepes pH 7.5, 10 mM MgCl2, 1 mM dithiothreitol (DTT), 0.004% Tween, and 0.003% Brij-35]. After 60 min at RT, the enzymatic reaction was stopped by the addition of 35 mM ethylenediaminetetraacetic acid (EDTA). For the AKT1 assay, serial dilutions of compounds were preincubated with 2.5 nM enzyme for 30 min at RT before starting the reaction by the addition of substrate mixture 50 µM ATP/2 µM FL-Peptide 6 (cat. no 760350, Caliper Life Sciences) in buffer A. After 45 min at RT, the enzymatic reaction was stopped by the addition of 35 mM EDTA. In both cases, the assay plates (384-well format) were then processed on a Caliper Labchip 3000 (Caliper Life Sciences) according to the manufacturer’s instructions for readout.
MEK1 enzymatic assay: Human MEK1 protein was purchased from Carna Bioscience. The enzymatic activity was monitored using ERK2 K54R-6His-tagged (ProQinase, Freiburg, Germany) as substrate and HTRF as readout. Serial dilutions of compounds were incubated for 80 min at RT with 75 nM ERK2, 50 µM ATP, and 1.2 nM enzyme in reaction buffer [50 mM Tris-HCl pH 7.5, 10 mM MgCl2, 1 mM egtazic acid (EGTA), 0.008% Brij-35, 5 mM β-glycerophosphate, and 1 mM DTT]. Then, the revelation mixture containing anti-6His-XL665 plus anti-phospho ERK antibodies (Cisbio) was added, and the fluorescence intensity signals at 665 nm and 620 nm were recorded after 2 h of incubation at RT.
mTOR enzymatic assay: Human mTOR protein was purchased from Life Technologies. The enzymatic activity was monitored using GFP-4E-BP1 as substrate in a time-resolved fluorescence energy transfer (TR-FRET) assay kit according to the manufacturer’s instructions (Life Technologies). Serial dilutions of compounds were incubated for 30 min at RT with 400 nM GFP-4E-BP1, 8 µM ATP, and 3 nM enzyme in reaction buffer (50 mM HEPES pH 7.5, 10 mM MnCl2, 1 mM EGTA, 0.5% glycerol, 0.01% NV10, and 2 mM DTT). Then, the revelation mixture containing anti-Tb-p4E-BP1 (pThr46) antibody was added, and the fluorescence intensity signals at 490 nm and 520 nm were recorded after 1 h of incubation at RT.
BRAF assay: Human BRAF (V600E) protein was purchased from ThermoFisher. The enzymatic activity was monitored using MEK1 inactive as substrate (purchased from ThermoFisher). Serial dilutions of compounds were incubated for 30 min at RT with 1.4 nM BRAF, 100 nM MEK1, and 50 µM ATP in reaction buffer [50 mM HEPES pH 7.5, 10 mM MgCl2, 1 mM EGTA, 10 mM β-glycerophosphate, 0.008% Brij-35, 1 mM DTT, and 0.1 mg/ml bovine serum albumin (BSA)]. Then, the enzymatic reaction was monitored using the ADP-Glo kit according to the manufacturer’s instructions (Promega).
Biophysical Assays
Surface plasmon resonance (SPR) was performed with MASS2 (Sierra Instruments, Monterey, CA) using KRASWT 1–169 and KRASG12D 1–169 as ligands, and the following experimental conditions: 50 mM HEPES, 150 mM NaCl, 1 mM MgCl2, pH 7.4, 0.03% P20, and 5 µM GppNHp for GppNHp-loaded forms or 10 µM GDP for GDP-loaded forms; for the streptavidin chip: immo = 3800–2500 RU, and theoretical Rmax = 50 RU.
Results
Validation of the Isogenic MEF Panel with Screening against a Tool Compound Library
To facilitate screening for compounds that can specifically target oncogenic KRAS, we developed a panel of isogenic MEF cell lines based on the “RAS-less” MEFs generated by the Barbacid group.
20
These “RAS-less” MEFs have both Hras and Nras ablated, as well as a conditional Kras knockout allele. On tamoxifen treatment, Kras is further removed, resulting in truly RAS-less MEFs (lacking HRAS, NRAS, and KRAS). At the same time, these MEFs cease proliferation, indicating that RAS is required for MEF proliferation. This nonproliferative state is reversible with ectopically expressed RAS introduced by viral transduction.
20
We subsequently generated a panel of isogenic MEF cell lines that ectopically express unique WT or oncogenic alleles of RAS complementary DNAs (cDNAs) (
To validate the MEF panel, specifically to determine if they can be used to correctly identify allele-specific vulnerabilities, we performed a high-throughput screen with a tool compound library in an ATP-based cell proliferation assay. This library contained 1402 well-annotated small-molecule inhibitors that target key cellular-signaling pathways. Figure 1 shows some of the results from this screen. We compared the sensitivities of the HRASWT-expressing and KRAS-4B-expressing MEFs to all inhibitors in the library. The majority of the inhibitors did not show specific activities against either MEF line ( Fig. 1A ).
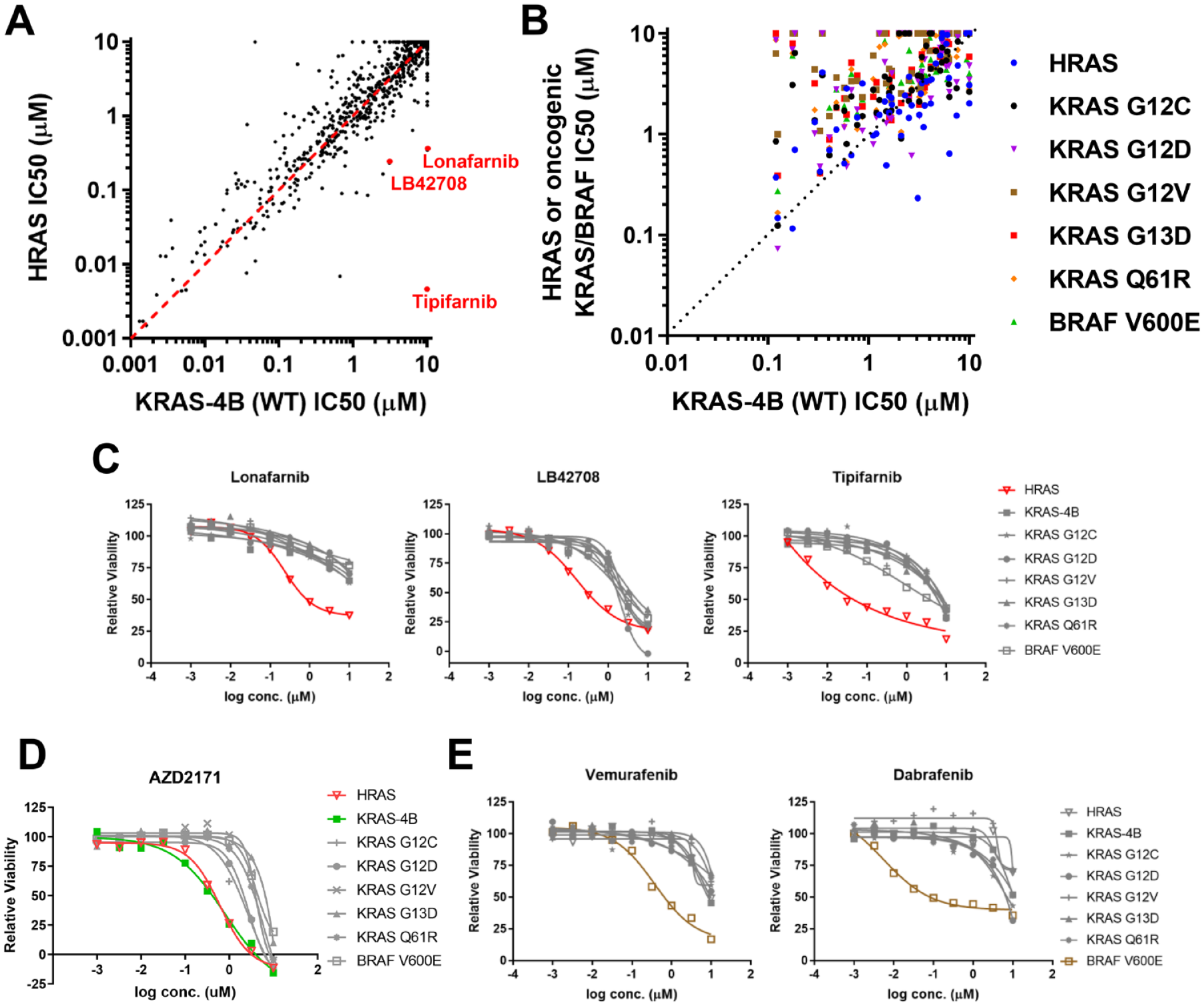
Validation of the isogenic mouse embryonic fibroblast (MEF) panel with a tool compound screen: (
RAS proteins function at the plasma membrane, and posttranslational lipid modification is required for membrane localization. Prenylation is one of these key modifications. Farnesylation of HRAS is required for its membrane localization, while KRAS-4B can be either farnesylated or geranylgeranylated. As a result, the HRASWT-expressing MEF is uniquely vulnerable to farnesyl-transferase inhibitors. As expected, HRASWT-expressing MEFs were more sensitive to farnesyl-transferase inhibitors (Tipifarnib, Lonafarnib, and LB42708) compared to KRAS-4B-expressing MEFs ( Fig. 1A ). Indeed, compared to other MEF cell lines in the panel, the potency of cell growth inhibition of these inhibitors was at least 10-fold higher in the HRASWT-expressing MEF line ( Fig. 1C ). We performed similar analysis comparing the sensitivities of the MEF cell lines to receptor tyrosine kinase (RTK) inhibitors. RTKs are upstream activators of RAS in the signaling cascade. As anticipated, HRASWT-expressing and KRAS-4B-expressing MEFs were more sensitive to the RTK inhibitors than other MEFs expressing oncogenic KRAS or BRAFV600E ( Fig. 1B ). To illustrate this, we showed the dose–response curves of AZD2171, a VEGFR/c-Kit/PDGFRβ inhibitor. The IC50 (half-maximal inhibitory concentration) values for the oncogenic KRAS- or BRAFV600E-expressing MEFs were at least three times larger than those of the RASWT-expressing MEFs ( Fig. 1D ). Finally, we showed that dabrafenib and vemurafenib, two US Food and Drug Administration (FDA)-approved drugs for BRAFV600E/K mutation–positive melanoma, indeed showed specific activity against BRAFV600E-expressing MEFs ( Fig. 1E ).
Collectively, these data demonstrated that the isogenic MEF panel has the expected biological properties. They can be used to identify compounds that can differentiate the HRASWT from the KRASWT allele. They can also be used to identify compounds that can differentiate the RASWT from oncogenic RAS or BRAF alleles. Hence, the MEF panel is an ideal tool for the phenotypic screen.
Phase 1: High-Throughput Screen with the Sanofi Compound Library
Following validation of the MEF panel, we initiated the screening campaign with the subset pair of MEF cell lines: HRASWT- and KRASG12D-expressing cell lines. We screened 923,000 compounds from the Sanofi compound library, including 6000 natural product compounds. An ATP-based cell proliferation assay was used to identify compounds that have specific activity against KRASG12D-expressing compared to HRASWT-expressing MEFs (
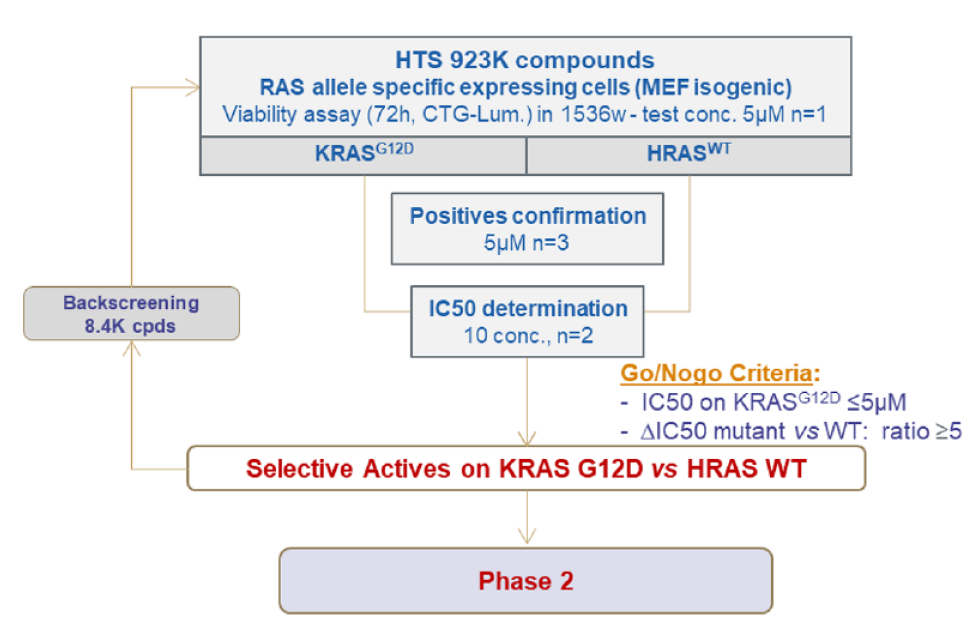
Phase 1: High-throughput screening of the proprietary compound library with the isogenic mouse embryonic fibroblast (MEF) panel. The isogenic MEF panel was used to conduct high-throughput screening with Sanofi’s compound library (923,000 compounds in the primary screen and 8376 compounds in the backscreen). In Phase 1, 696 active compounds were identified. 19 clusters and 8 singletons were discarded based on the biological profile and drug likeness. 126 compounds (28 clusters, 10 singletons, and 1 natural product) were ultimately selected for secondary assays for Phase 2.
Phase 2: Characterization of Specific Actives and Mechanism-of-Action Studies
KRAS-specific compounds selected from the phase 1 high-throughput screen were further examined in a series of cell-based, biochemical, and biophysical assays to identify potential biological targets and mechanisms of action (
Fig. 3
). Cell-based assays including pERK, pAKT, and pEGFR HTRF assays were performed to examine the effects of the compounds on RAS signaling pathways. Biochemical assays for BRAFV600E, MEK1, ERK2, PI3Kα, AKT1, and mTOR activities were performed to evaluate the effects of the selected compounds. To assess direct compound binding to KRAS, surface plasmon resonance (SPR) was used (data not shown). In addition, cell viability assays were performed with a panel of KRAS-dependent (SW620, KRASG12V and LS513, KRASG12D) and KRAS-independent (Colo320, RASWT) colorectal human cancer cell lines to confirm that KRAS-specific activity of the compounds is not limited to mouse fibroblasts (
Characterization of the Final Five Candidate Clusters.

Note: Data from the cell proliferation assay, kinase profiling (with up to 365 kinases), and toxicity assay (the cell viability values were shown) with quiescent PBMCs and normal human lung fibroblasts (MRC5) from the final five candidate series were summarized. IC50 values were determined from dose–response curves generated from at least two independent experiments. CRC, Colorectal cancer; IC50, half-maximal inhibitory concentration; %Inh, percent inhibition; MEF, mouse embryonic fibroblast; ND, not determined; PBMCs, peripheral blood mononuclear cells.
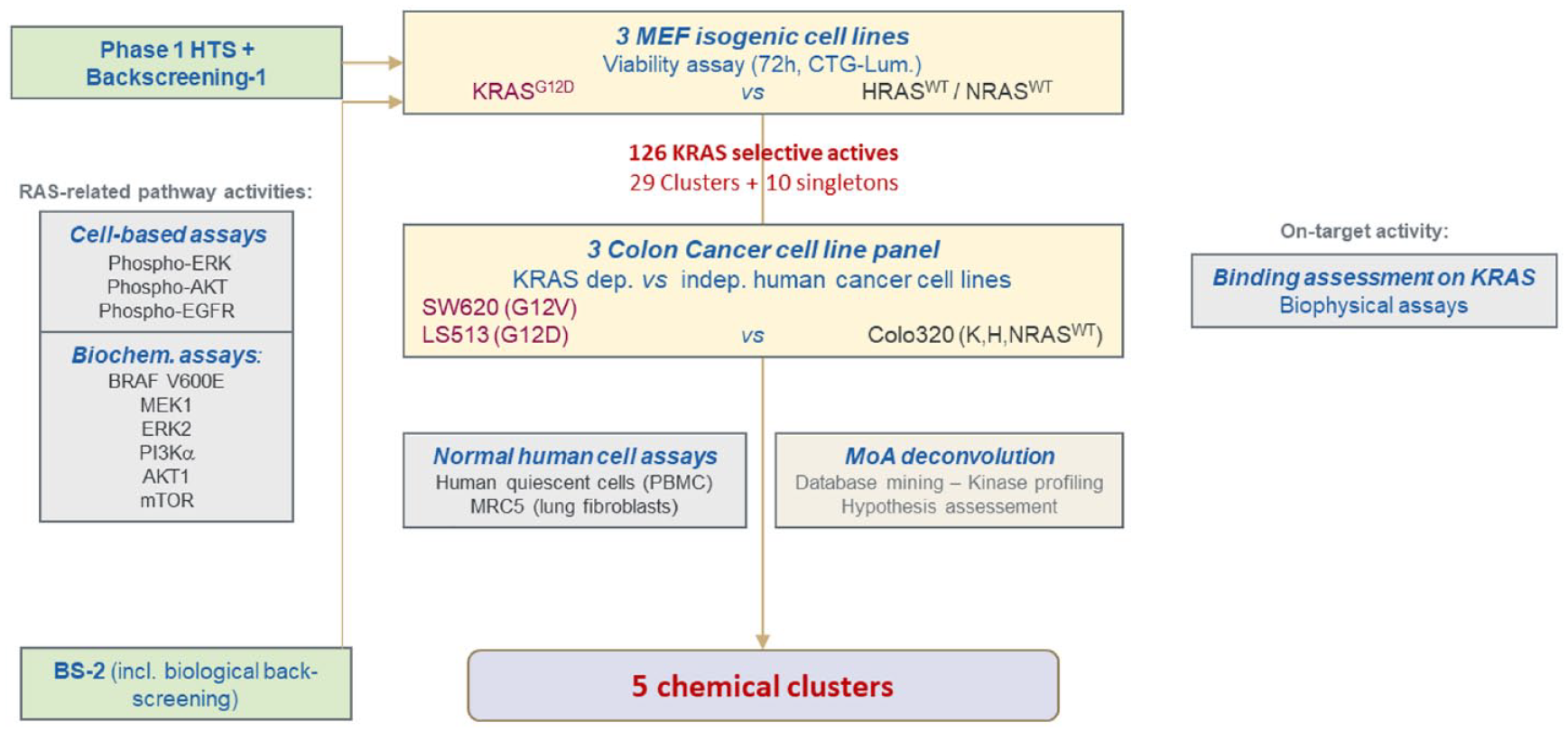
Phase 2: Characterization of the specific actives and mechanism-of-action studies. Specific actives (126 compounds) were further tested in a series of cell-based, biochemical, and biophysical assays to identify potential mechanisms of action. Cell viability assays using KRAS-dependent and -independent human colorectal cancer cell lines were performed. Cell-based assays [pERK, pAKT, and pEGFR homogeneous time-resolved fluorescence (HTRF)] as well as biochemical assays for BRAFV600E, MEK1, ERK2, phosphoinositide 3-kinase alpha (PI3Kα), AKT1, and mammalian target of rapamycin (mTOR) activities were performed to assess RAS-related pathway activities. Biophysical assays were also performed to assess direct compound KRASWT and KRASG12D binding.
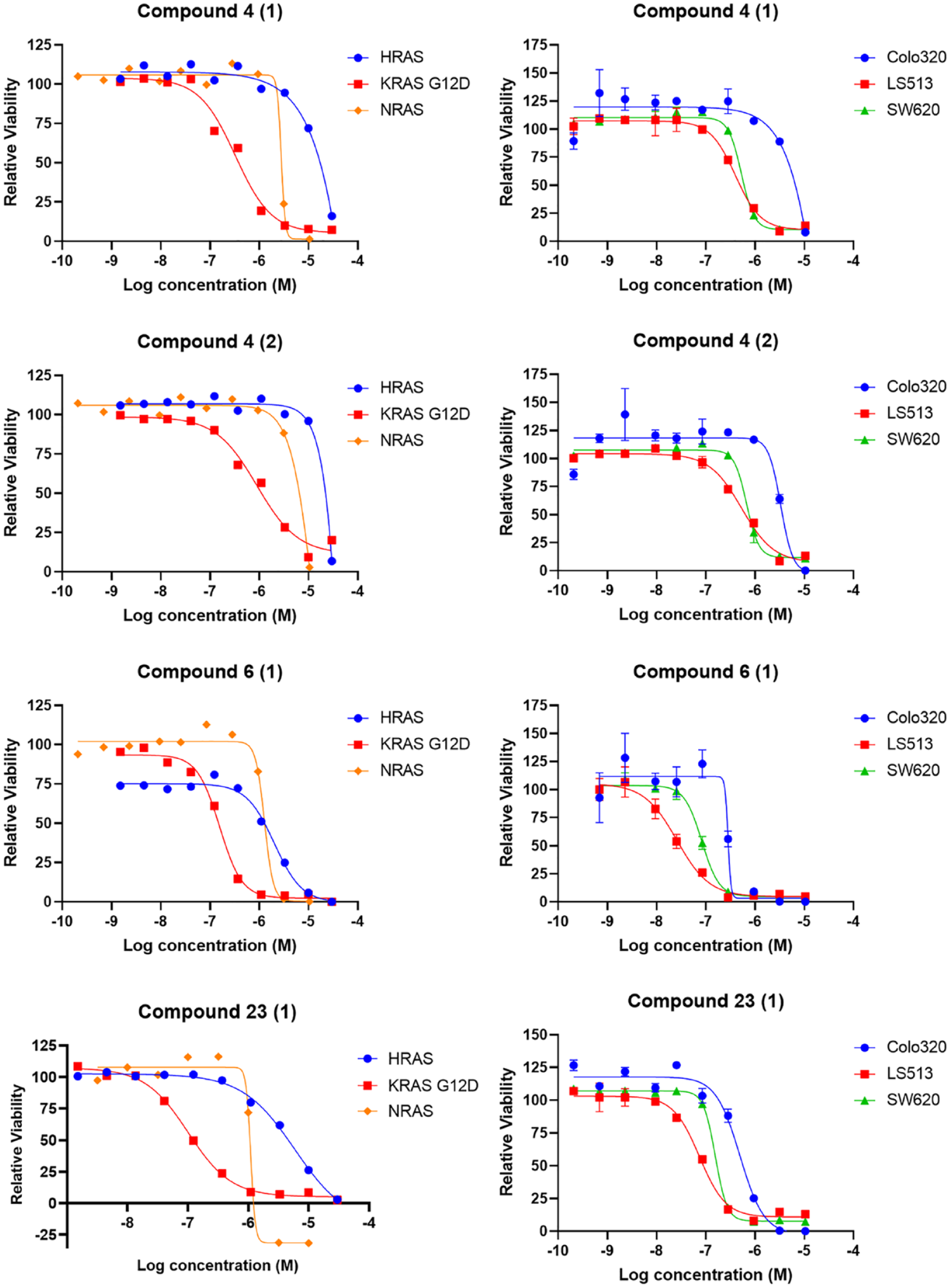
Dose–response curves of selected compounds in the cell viability assay: Mouse embryonic fibroblasts (MEFs) (72 h treatments) and colorectal cancer cell lines (96 h treatments) were treated with the indicated compounds and examined with the CellTiter-Glo assay. Data were normalized to DMSO-treated cells. Each experiment was performed at least twice, and the error bars represent the standard deviations.
Compounds from these five clusters were shown to have no specific binding to KRAS in the biophysical SPR assay. They did not have consistent effects on both ERK and AKT pathways in the cell-based HTRF assays, and showed weak or no activity in the biochemical assays (
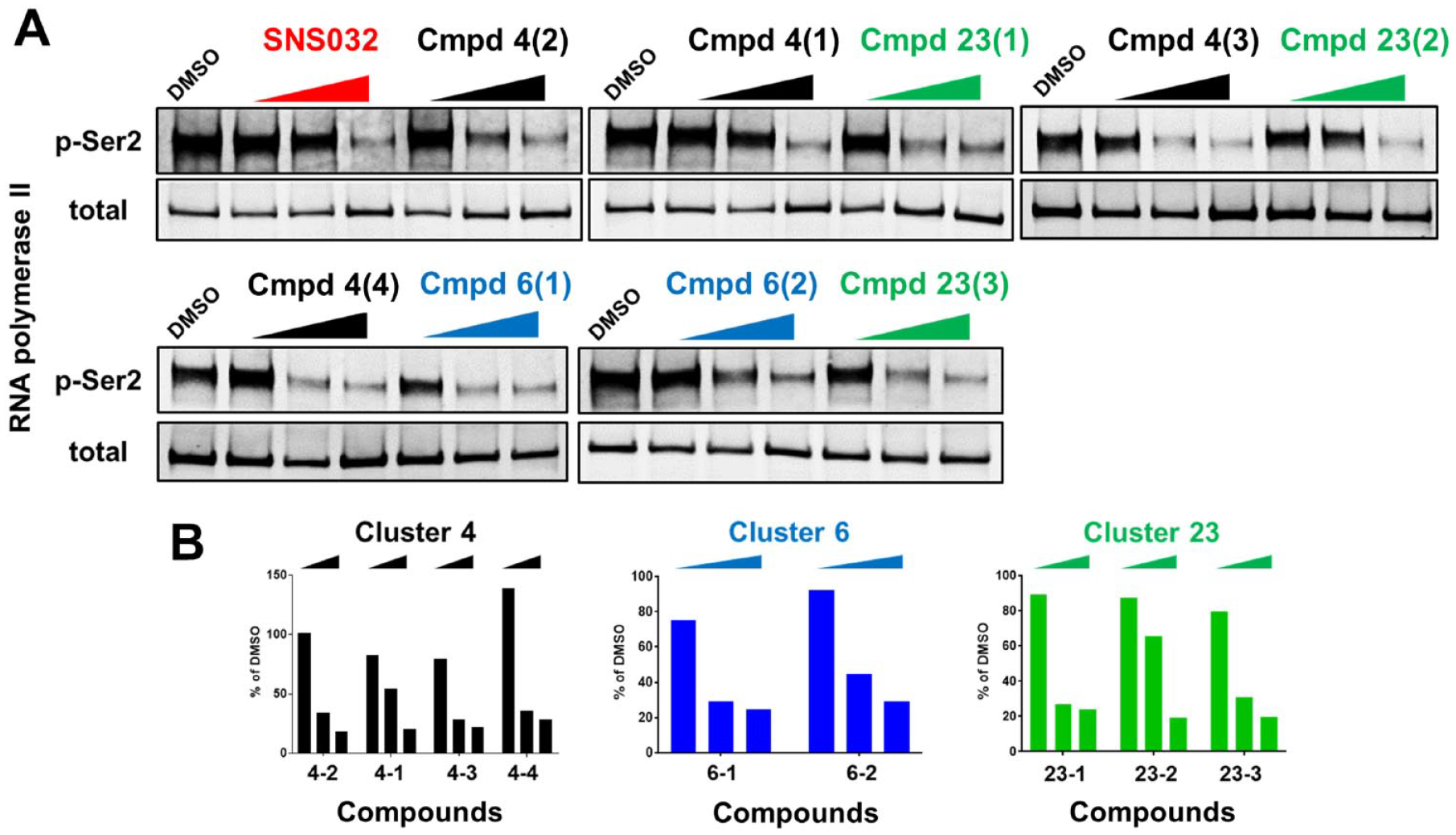
Three candidate chemical clusters demonstrated CDK9 inhibitory activity: KRASG12D-expressing mouse embryonic fibroblasts (MEFs) were treated with the indicated compounds for 6 h. Multiple doses (0.1 µM, 1 µM, and 10 µM) and multiple compounds from the same series were tested. (
Discussion
To explore the potential to identify new therapeutic opportunities, we developed and validated a KRAS-specific screening capability based on isogenic MEF cell lines and carried out an extensive high-throughput screen on a large, chemically diverse screening library. RAS is one of the first identified and the most frequently mutated oncogenes. Gain-of-function RAS mutations are found in almost 20% of all human cancers. In particular, KRAS mutations are most predominant among the three RAS isoforms. Unfortunately, it has proven challenging to develop targeted therapeutics for oncogenic KRAS. The aim of this screening campaign was to identify lead compounds that have selective activity against oncogenic KRAS-expressing cells. We first developed and validated a panel of isogenic MEFs that express WT KRAS, oncogenic KRAS, or oncogenic BRAF allele. We then screened almost 1 million compounds with this MEF panel in a phenotypic high-throughput screen. This was followed by targeted cell-based, biochemical, and biophysical assays to further characterize the selected compounds. We ultimately identified compounds from five different chemical clusters, including one natural product. Surprisingly, three of these five clusters demonstrated CDK9 inhibitory activity, revealing the critical role of CDK9 activity in oncogenic KRAS-expressing cell proliferation.
Phenotypic screens were previously used to identify potential therapeutics for oncogenic RAS-driven cancers. One of the first of these screens was performed by the Stockwell group. They identified a novel compound, named Erastin, by screening 23,550 compounds for their ability to cause selective lethality in genetically engineered human tumor cells that express HRASG12V. They showed that Erastin causes non-apoptotic cell death only in HRASG12V-expressing cells. 18 In a second screen, MEFs derived from a transgenic mouse model that expresses KRASG12D were used to screen >50,000 compounds. A class of compounds that cause oxidative stress and non-apoptotic cell death specifically in KRASG12D-expressing cells was identified. 19 Subsequently, two more HRASG12V-selective compounds with nanomolar potencies were also identified in another phenotypic screen with 303,282 compounds. 17 The biological targets of these selective compounds were not identified in these screens, however, highlighting a weakness of phenotypic screens. Renewed efforts to screen a larger panel of cell lines and to apply new technologies in mechanism-of-action deconvolution may prove to be key in tackling the challenge of targeting KRAS. A recent screen of 280,000 small molecules with a panel of 10 KRAS-dependent and –independent human cancer cell lines in both two-dimensional (2D) and three-dimensional (3D) cultures has led to the identification of the pyrimidine biosynthetic enzyme DHODH as a synthetic lethal vulnerability of oncogenic KRAS-expressing cells. 21 This previously unknown vulnerability was also identified in our current screen, and can potentially be exploited to develop new strategies targeting KRAS-mutant cancers.
In this study, we developed an improved screening process by using a panel of isogenic MEFs as well as screening a substantially larger compound library. Considering the large number of compounds, we initially focused on two MEF cell lines (HRASWT and KRASG12D). We reasoned that this comparison will allow us to identify compounds targeting specifically oncogenic KRAS instead of WT HRAS and WT KRAS. Compounds that showed KRASG12D-selective activity were subsequently used in a screen against the NRASWT MEF cell line. In addition, a panel of KRAS-dependent and –independent colorectal cancer cell lines (SW620, LS513, and Colo320) was used to confirm that the specific activity of the selected compounds is not limited to MEFs. It is worth noting that this MEF panel can be used to identify potential oncogenic allelic differences in future screens.
While phenotypic screens allow us to identify specific active compounds without prior knowledge of the molecular mechanisms, the process to determine the mechanism has proven to be challenging. To facilitate the mechanism-of-action deconvolution, we first performed cell-based and biochemical assays related to RAS signaling. Biophysical assays were also used to assess direct compound binding to KRAS. None of the compounds from the final five chemical clusters showed specific binding, highlighting the ongoing challenge of finding direct KRAS binders largely due to the lack of drug-binding pockets on the KRAS surface. The lack of consistent effects on RAS-related signaling pathways or signaling molecules tested makes these unlikely targets of the compounds. Hence, a comprehensive kinase profiling of these compounds was subsequently performed to identify the potential biological targets. Surprisingly, compounds from three of the final five clusters were shown to have CDK9 inhibitory activity. The CDK9 inhibitory activity was further demonstrated in KRASG12D-expressing MEFs.
CDK9 functions to regulate RNA polymerase II–directed transcription. It belongs to a multiprotein complex that is the main component of the positive transcription elongation factor b (P-TEFb) complex. Therefore, CDK9 inhibition prevents RNA polymerase II–directed transcription, resulting in global messenger RNA (mRNA) reduction. It was previously shown that in neuroendocrine cells, thyrotropin-releasing hormone activation of the MEK1/ERK signaling pathway upregulates nuclear CDK9 and promotes dimerization of cyclin T1 and CDK9, which subsequently increases P-TEFb activity and transcription of immediate early genes like c-fos and jun-B. 23 Its role in KRAS-dependent oncogenesis was not examined, however. While our screen was in progress, CDK9 was identified as a KRAS synthetic lethal target. Der’s group used a MYC degradation screen in KRAS-mutant pancreatic ductal adenocarcinoma (PDAC) cell lines to discover the role of CDK9 in stabilizing MYC, which was shown to be critical in promoting the growth of oncogenic KRAS PDAC. This study provided the molecular mechanisms underlying the vulnerability of oncogenic KRAS-expressing cells to CDK9 inhibition. 24 Our data obtained from an orthogonal approach support future studies on the potential use of CDK9 inhibitors for treating KRAS-driven cancers.
Pan-CDK inhibitors that exhibit potent CDK9 inhibition were evaluated in multiple clinical trials, but in most cases, significant adverse effects forced premature trial terminations. 25 The recent development of CDK9-selective inhibitors like BAY1143572 and AZD4573 may overcome some of the toxicities caused by the nonspecific inhibition of other CDKs,26,27 although results from clinical trials remain inconclusive. A BAY1143572 phase I trial was terminated prematurely due to severe adverse effects, and the AZD4573 phase I trial is ongoing (NCT03263637). The vulnerability of oncogenic KRAS-expressing cells to CDK9 inhibition may widen the therapeutic windows for the selective CDK9 inhibitors, potentially making them more clinically tractable.
In summary, we developed and validated the use of an isogenic MEF panel for phenotypic screens. We screened almost a million compounds with a pair of isogenic MEF cell lines and a panel of three KRAS-dependent and –independent colorectal cancer cell lines in a phenotypic synthetic lethal screen. We performed cell-based, biochemical, and biophysical assays as well as protein kinase profiling to characterize the candidate compounds. While we did not identify any direct KRAS binders, we identified CDK9 inhibition as the underlying mechanism of differential compound sensitivities between WT and oncogenic KRAS-expressing cells. This novel vulnerability of KRAS-mutant cells identified by this screen, and corroborated by another independent study, 24 warrants future genomics [small interfering RNA (siRNA) and CRISPR] as well as in vivo studies, and provides a potential new strategy to develop therapeutics for KRAS-driven cancers.
Supplemental Material
sj-pdf-1-jbx-10.1177_24725552211008853 – Supplemental material for Sensitivity of Oncogenic KRAS-Expressing Cells to CDK9 Inhibition
Supplemental material, sj-pdf-1-jbx-10.1177_24725552211008853 for Sensitivity of Oncogenic KRAS-Expressing Cells to CDK9 Inhibition by Lick Pui Lai, Viviane Brel, Kanika Sharma, Julia Frappier, Nadia Le-Henanf, Bertrand Vivet, Nicolas Muzet, Emilie Schell, Renaud Morales, Eamonn Rooney, Nicolas Basse, Ming Yi, Frederic Lacroix, Matthew Holderfield, Walter Englaro, Christophe Marcireau, Laurent Debussche, Dwight V. Nissley and Frank McCormick in SLAS Discovery
Footnotes
Acknowledgements
The authors would like to thank Mariano Barbacid for providing the MEF cell line, and our colleagues from the RAS Initiative and Sanofi for their ongoing support.
Supplemental material is available online with this article.
Declaration of Conflicting Interests
The authors declared the following potential conflicts of interest with respect to the research, authorship, and/or publication of this article: V.B., J.F., N.L., B.V., N.M., E.S., R.M., E.R., N.B., F.L., W.E., C.M., and L.D. are full-time Sanofi employees. F.M. is a consultant for the following companies: Amgen, Pfizer Inc., Daiichi Sankyo, Ideaya, PMV, and Quanta Therapeutics. F.M. is a consultant and co-founder with ownership interest including stock options of BridgeBio Pharma, Inc., and has ownership interest including stock options in Olema Pharmaceuticals, Opna, Kura, Wellspring, Avidity, and Quadriga. F.M. is scientific director of the NCI RAS Initiative at the Frederick National Laboratory for Cancer Research/Leidos Biomedical Research, Inc.
Funding
The authors disclosed receipt of the following financial support for the research, authorship, and/or publication of this article: This project has been funded in part with federal funds from the National Cancer Institute, National Institutes of Health, under contract no. 75N91019D00024. The content of this publication does not necessarily reflect the views or policies of the US Department of Health and Human Services, nor does mention of trade names, commercial products, or organizations imply endorsement by the US government.
References
Supplementary Material
Please find the following supplemental material available below.
For Open Access articles published under a Creative Commons License, all supplemental material carries the same license as the article it is associated with.
For non-Open Access articles published, all supplemental material carries a non-exclusive license, and permission requests for re-use of supplemental material or any part of supplemental material shall be sent directly to the copyright owner as specified in the copyright notice associated with the article.