
Abstract
Tay–Sachs disease is an inherited lysosomal storage disease resulting from mutations in the lysosomal enzyme, β-hexosaminidase A, and leads to excessive accumulation of GM2 ganglioside. Tay–Sachs patients with the infantile form do not live beyond 2–4 years of age due to rapid, progressive neurodegeneration. Enzyme replacement therapy is not a therapeutic option due to its inability to cross the blood–brain barrier. As an alternative, small molecules identified from high-throughput screening could provide leads suitable for chemical optimization to target the central nervous system. We developed a new high-throughput phenotypic assay utilizing infantile Tay–Sachs patient cells based on disrupted lysosomal calcium signaling as a monitor of diseased phenotype. The assay was validated in a pilot screen on a collection of Food and Drug Administration-approved drugs to identify compounds that could reverse or attenuate the disease. Pyrimethamine, a known pharmacological chaperone of β-hexosaminidase A, was identified from the primary screen. The mechanism of action of pyrimethamine in reversing the defective lysosomal phenotype was by improving autophagy. This new high-throughput screening assay in patient cells will enable the screening of larger chemical compound collections. Importantly, this approach could lead to identification of new molecular targets previously unknown to impact the disease and accelerate the discovery of new treatments for Tay–Sachs disease.
Introduction
Lysosomal storage disorders (LSDs) are a group of inheritable metabolic diseases with deficiencies in lysosomal proteins, including hydrolytic enzymes within the glycosphingolipid biosynthetic and metabolic pathways.1,2 As a consequence, nondegraded substrates accumulate, producing compromised function of lysosomes and other cellular organelles. Two-thirds of LSDs have central nervous system involvement associated with progressive and severe neurodegeneration. Tay–Sachs disease (TSD) is a GM2 gangliosidosis arising from mutations in the HEXA gene to produce a deficiency of the lysosomal enzyme, β-hexosaminidase A (HEXA). 3 The loss of enzyme activity leads to accumulation of GM2 ganglioside in neuronal cells and rapid, progressive neurodegeneration. 4 The three forms of TSD, infantile, juvenile, and adult, are classified on the severity and disease onset and correlated with the level of residual mutant β-hexosaminidase enzyme activity and rate of accumulating GM2 gangliosides. 4 The infantile form is the most severe, with little or no enzyme activity. Onset occurs between 3 and 6 months with rapid, progressive symptoms. This debilitating neurodegenerative form is fatal, and patients typically do not live beyond 2 years.5,6 Enzyme replacement is not a viable strategy to treat TSD due to the inability of the enzyme to cross the blood barrier and reach the target neuronal cell types. At present, no effective treatment options exist for TSD patients.
The application of high-throughput screening (HTS) approaches to identify small-molecule therapeutics for LSDs, including TSD, have previously utilized target-based assays designed to identify either chaperones to restore defective enzyme activity or compounds to clear accumulating substrates resulting from decreased enzyme activity. Chaperones or protein stabilizers of HEXA in TSD7,8 and glucocerebrosidase in Gaucher disease9,10 have been identified in enzyme-based HTS assays. The use of screening collections of Food and Drug Administration (FDA)-approved drugs identified pyrimethamine and ambroxol as pharmacological chaperones and facilitated advances into clinical trials to assess therapeutic efficacy in GM2 gangliosidosis and neuronopathic Gaucher disease patients, respectively.11,12
The use of phenotypic assays on patient-derived cells offers an alternative to target-based strategies and has potential to discover new and novel therapeutics.13,14 The availability of LSD patient-derived cells provides a unique opportunity for developing phenotypic-based screens to identify compounds that can either attenuate the underlying pathophysiology or restore function to a normal state and support identification of a new class of disease-modifying therapeutics.
Disruption of calcium homeostasis, in addition to abnormal lysosomal function, is observed in several neuronopathic LSDs—independent of the mutations associated with each disease and accumulated substrates as a result of impaired enzyme activity.15–18 Defective lysosomal calcium signaling may be a core defective mechanism shared among LSDs. Vesicle membrane fusion and transport in the endosomal/lysosomal system is a calcium-dependent process. Reduction of calcium from acidic lysosomal stores disrupts normal endosome/lysosome function and has been suggested to contribute to impairing autophagy and the ability of the cell to clear accumulating substrates. 15 Targeting restoration of abnormal lysosomal calcium signaling is a novel approach for therapeutic intervention in LSDs.
Disruption of calcium homeostasis in lysosomal stores and lysosomal dysfunction are measurable, quantitative hallmarks of LSDs.19,20 HTS assays based on measuring lysosomal calcium in LSDs have not been previously explored. With the goal to discover small-molecule therapeutics for treating neuronopathic LSDs, we developed a new phenotypic assay on LSD patient-derived cells. As a proof of concept, we evaluated TSD patient-derived cells to determine whether reduced calcium release from lysosomal acidic stores could be measured in a 384-well format and if the disease phenotype could be exploited as the foundation for developing HTS to drive discovery of small-molecule therapeutics for the treatment of TSD.
Materials and Methods
Chemicals
Gly-Phe-β-napthylamide (GPN) (cat. 14634), 2-aminoethyl diphenylborinate (2-APB) (cat. 64970), and ionomycin (cat. 11932) were from Cayman Chemical Co. (Ann Arbor, MI); pyrimethamine (cat. P-7771) was purchased from Sigma (St. Louis, MO). 4-Methylumbelliferyl-2-acetamido-2-deoxy-6-sulfate-β-
Cell Lines and Culture
Human fibroblast cell lines from Tay–Sachs patients (GM00221, GM00502, GM11853) and a healthy normal patient (GM05659) were from Coriell Institute for Medical Research (Camden, NJ); GM00221 donor is homozygous for a 4-base-pair insertion at nucleotide 1278 in exon 11 of the HEXA gene (c.1278insTATC), which leads to a premature termination signal. GM00502 donor is a compound heterozygote with one allele having a 4-base-pair insertion at nucleotide 1278 in exon 11 of the HEXA gene and the second allele having a G>C splice mutation in intron 12 (IVS12+1G>C). GM11853 donor is homozygous for a 4-base-pair duplication in exon 11 of the HEXA gene (1274_1277 dupTATC), which leads to a premature termination signal. Cell lines were grown in MEM media (Cellgro, cat. 10-010-CV, Manassas, VA) supplemented with 10% fetal bovine serum (Gemini Bio, cat. 900-108, West Sacramento, CA). Cells were maintained at <80% confluence under standard incubator conditions (5% CO2, 37 °C, and 75% humidity). Cell viability and cell number count were measured with CellTiter-Glo.
β-Hexosaminidase A Assay
Lysates were prepared from wild-type (WT) and Tay–Sachs patient fibroblasts. Cells were harvested in cold phosphate-buffered saline (PBS) with cOmplete protease inhibitor (Roche Applied Sciences, Penzburg, Germany, cat. 04-693-159-001, Mannheim, Germany) and pelleted at 2500 rpm for 5 min at 4 °C. Pellets were retained on dry ice for 10 min and then resuspended in 10 mM citrate buffer and 0.5% Triton (pH 4.2) with cOmplete, followed by centrifugation at 14,000 rpm for 10 min at 4 °C. β-Hexosaminidase A activity was measured by the release of 4-methylumbelliferyl fluorophore from 4-MUGS. Reactions were incubated for 1 h at 37 °C in a final volume of 100 µL with varying concentrations (300–1500 μM) of 4-MUGS. Reactions were terminated by the addition of 13 mM glycine/83 mM carbonate (pH 10.8) stop buffer. Fluorescent product formation was detected on a Clariostar fluorescence plate reader (BMG LabTech, Cary, NC) with excitation 364 nm and emission 450 nm.
Lysotracker Staining
Cells were seeded on glass coverslips and cultured overnight. Cells were live stained with 50 nM Lysotracker-Red DND-99 (LifeTech, cat. L7528, Carlsbad, CA) in media at 37 °C for 60 min, followed by two washes with Hanks Balanced Salt Solution (HBSS; Cellgro, cat. 21-022-CV). Nuclei were stained with NucBlue Live Cell Stain (LifeTech, cat. R37605). Cells were imaged on a Leica EL6000 DMI3000 confocal microscope system, Leica SP5 Microscope with a TCS confocal system using a 63× objective (NucBlue: excitation 405 nm, emission 488 nm; Lysotracker: excitation 532 nm, emission 635 nm).
Lysosomal Calcium Assay
Tay–Sachs fibroblasts were plated at 800 cells/well in 384-well black μClear plates (Greiner, cat. 781091) with a MultiFlo dispenser (BioTek, Winooski, VT) and cultured for 24 h at 37 °C, 5% CO2. Compounds were added to cells under sterile conditions and continued in culture for 72 h. On the day of assay, culture media was completely removed and replaced with 20 µL of dye loading buffer, 4.8 μM Fluo-8 AM (cat. 21083), and 1× Screen Quest Calcium Assay Buffer (cat. 36301) (both from AAT Bioquest, Sunnyvale, CA) in calcium-free HBSS containing 20 mM HEPES (pH 7.4) and incubated for 30 min at 37 °C, followed by room temperature for 30 min. Changes in fluorescence intensity are monitored on a Hamamatsu FDSS µCell fluorescence kinetic plate reader (excitation 480 nm, emission 540 nm) reading at 1 Hz for a 30 s baseline and followed by addition of 2-APB (25 μM final concentration). After 15 min, GPN (50 μM final concentration) was added and the change in fluorescence was continually recorded for an additional 10 min. The data from each well were normalized and expressed as a ratio by dividing the fluorescence over the entire real-time reading by the initial basal fluorescence recorded prior to GPN addition.
Prestwick Chemical Collection High-Throughput Screen
The Prestwick collection used in the pilot screen consists of 1200 FDA-approved drugs. Compound stocks (10 mM in 100% DMSO) in 96-well storage plates were quad-mapped into 384-well polypropylene plates and diluted in MEM (no serum) to prepare 7× stocks (140 µM) prior to addition to TSD fibroblasts (GM00221), with a final screening concentration of 20 µM in 0.20% DMSO. Cells are plated with the MultiFlo dispenser (800 cells/well in 30 µL of complete growth media) in 384-well black μClear plates (Greiner, cat. 781091) and cultured for 24 h at 37 °C, 5% CO2. Compounds (5 µL) are then added to cells under sterile conditions and continued in culture for 72 h at 37 °C, 5% CO2. On the day of the assay, the media is completely removed and replaced with dye load buffer as described above. Results were calculated as percent activation = [(test compound – median low control)/(median high control – median low control) × 100] using 0.2% DMSO-treated TSD fibroblasts as the low control (32 wells/plate) and 0.2% DMSO-treated WT cells as the high control (32 wells/plate). The Z′ and signal-to-background ratio (S/B) were calculated using the high-control and low-control wells. 21 In each case, a Z′ value greater than 0.5 was required for a plate to be considered valid. Hits were identified using a standard deviation-based hit threshold where a hit = 3× standard deviation (3SD) from the mean of all samples. Hits were confirmed in triplicate from 10 mM DMSO compound collection stocks and counterscreened on WT cells to determine selectivity for TSD versus normal patient cells. Selected hit compounds were analyzed in 10-point dose–response curves using fresh powder samples purchased from Sigma.
Western Blot Analysis
Cells were harvested in cold PBS with cOmplete protease inhibitor and pelleted at 2500g for 5 min at 4 °C. Pellets were lysed in cold RIPA buffer (50 mM Tris-HCl [pH 8.0], 150 mM NaCl, 1% NP-40, 0.5% sodium deoxycholate, and 0.1% sodium dodecyl sulfate [SDS] with cOmplete protease inhibitor) for 30 min on ice and then centrifuged at 8000g for 10 min at 4°C. Protein concentrations were determined using Bio-Rad protein reagent (Bio-Rad, Hercules, CA, cat. 500-0006). SDS–polyacrylamide gel electrophoresis (SDS-PAGE) was run using 12% Tris-glycine polyacrylamide gels (Nu-Sep, Germantown, MD, cat. NN10-012) in Tris-glycine-SDS buffer and proteins were transferred to Immobilon PSQ PVDF membranes (Millipore, Burlington, MA, cat. ISEQ10100). Membranes were blocked in Odyssey blocking buffer (LI-COR, Lincoln, NE, cat. 927-50100)/TBS (1:1) for 1 h at room temperature and then incubated in LC3II primary antibody (Novus, Centennial, CO, cat. NB100-2220) (1:1000, overnight at 4 °C), followed by IRDye 680RD donkey anti-rabbit secondary antibody (LI-COR, cat. 926-68073) (1:20,000, 1 h at room temperature). Membranes were then incubated with a β-actin primary antibody (GeneTex Inc., Irving, CA, cat. GTX26276) (1:5000, overnight at 4 °C), followed by IRDye 800RD goat anti-mouse secondary antibody (LI-COR, cat. 926-32210) (1:10,000, 1 h at room temperature). Signal intensities were measured with an Odyssey Infrared Imager (LI-COR) and analyzed with Image Studio Lite version 5.2.5 software (LI-COR). The LC3II signal intensity was normalized to actin and expressed as a ratio.
Data Analysis
Data were analyzed using GraphPad Prism 6.0 software (San Diego, CA) and are presented as mean ± standard error of the mean (SEM). EC50 values were calculated by fitting the dose–response curves with a four-parameter logistic regression. For the statistical analysis, two group comparisons were analyzed using unpaired, two-tailed Student t test, and differences were considered significant if p < 0.05. Primary HTS data were analyzed with Dotmatics Software in Studies and visualized in Vortex (Dotmatics Ltd., UK).
Results
TSD Patient-Derived HTS Assay Principle and Development
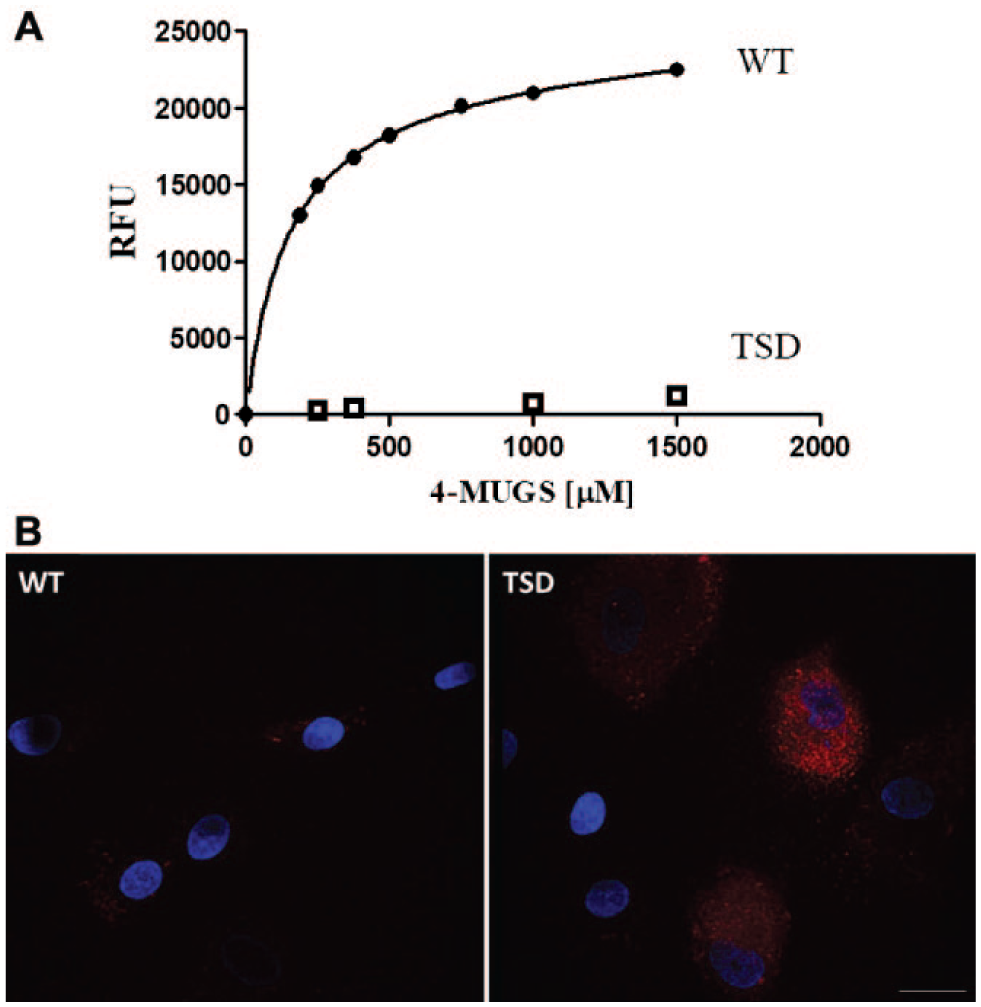
Calcium release from lysosomal stores can be induced by the addition of GPN, a substrate of cathepsin C, which upon hydrolysis produces osmotic lysis of lysosomes and release of calcium from acidic stores into the cytosol.20,22 The intracellular calcium response can be detected with a membrane-permeable, calcium-sensitive indicator, Fluo-8 AM. Measures of lysosomal calcium release from Gaucher disease and Niemann–Pick type C patient-derived cells have been previously reported; however, these studies utilized conventional fluorescence microscopy methods,19,20 which are not suitable for an HTS assay. With the goal to establish a phenotypic HTS in TSD patient-derived cells, we developed a 384-well-format cell-based fluorescence assay for measuring GPN-induced lysosomal calcium release. We obtained three infantile TSD patient fibroblast cell lines (GM00221, GM00502, GM11853) and a normal age-matched control (GM05659, WT). We first confirmed that the patient cell lines had a deficiency in HEXA activity, and lysosomal HEXA activity was measured in lysates prepared from TSD and normal, WT fibroblasts with the fluorogenic substrate 4-MUGS ( Fig. 1A ). In comparison with fibroblasts from a normal, WT patient, loss of enzyme activity was observed in the TSD patient cell lines, consistent with the mutation producing the lack of α-subunit expression. Lysosomal enlargement, a hallmark of LSD patient-derived cell lines, can be measured with Lysotracker, a fluorescent dye that preferentially accumulates in cellular acidic compartments.23,24 TSD patient cells, compared with WT cells, showed higher intensity of Lysotracker staining, indicative of lysosome enlargement ( Fig. 1B ). These studies confirmed the disease phenotype in TSD patient cell lines and validated their selection for developing the HTS assay.
(
We next compared calcium release from lysosomal stores from TSD and WT patient fibroblasts in response to acute addition of GPN to determine if reduced calcium signaling could be detected in TSD patient-derived cells and whether this difference was robust to support development of a 384-well-format HTS cell-based assay. To functionally isolate calcium release from lysosomes, the IP3 receptor antagonist 2-APB was added prior to acute GPN addition to block release from endoplasmic reticulum (ER) calcium stores.
20
High-throughput capacity was enabled with the FDSS µCell fluorescence kinetic plate reader. Kinetic tracings of the calcium responses are shown in
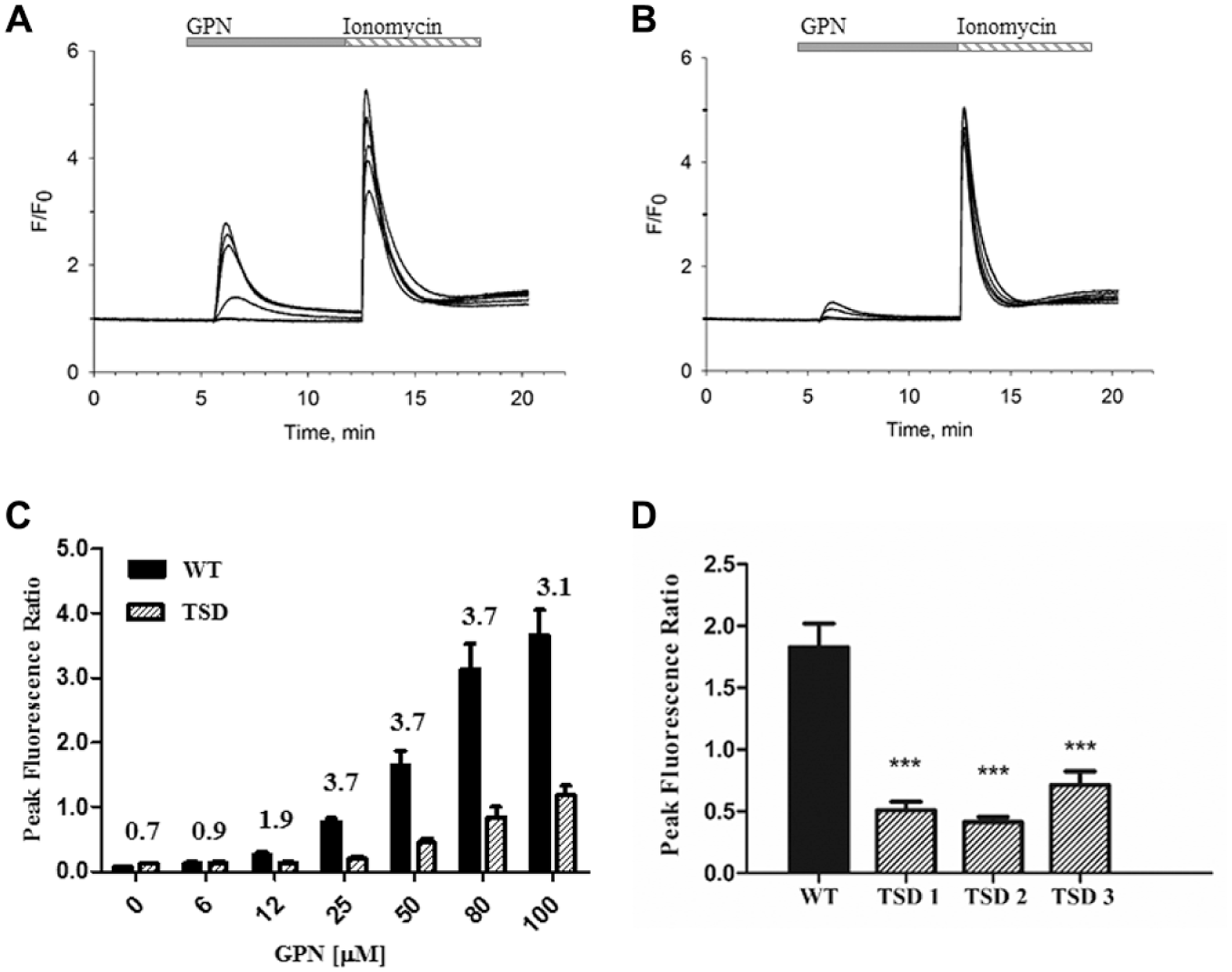
Development of a lysosomal calcium release assay in TSD patient-derived fibroblasts. Representative kinetic tracings from WT (
We determined the feasibility of using the reduction in lysosomal calcium release in TSD patient-derived fibroblasts as a phenotype to identify compounds with activity to restore lysosomal calcium signaling as a readout of disease-modifying activity. Parameters, including cell dependence, DMSO tolerance, and stability to cell passage, were determined to support the development of a screening protocol (data not shown). The response was stable up to 0.2% DMSO, with higher 0.4%– 1% DMSO significantly reducing the assay window. We selected a compound treatment interval of 72 h based on an expectation that active compounds would likely require downstream changes. We determined cell numbers/well after 72 h in culture postseeding to ensure that cell numbers were identical between WT and TSD patient-derived cell lines over the time interval and the observed differences in calcium response were not a consequence of different growth between normal and diseased cell types (data not shown). In parallel studies, we measured cell viability in both WT and TSD fibroblast lines after acute GPN addition under conditions used for the lysosomal calcium assay, thereby confirming that the addition of the lysosome osmotic agent produced no cell death and the response (i.e., change in fluorescence intensity) measured was not an artifact of cytotoxicity (data not shown). The timeline of the screening assay protocol is shown in Figure 3A .
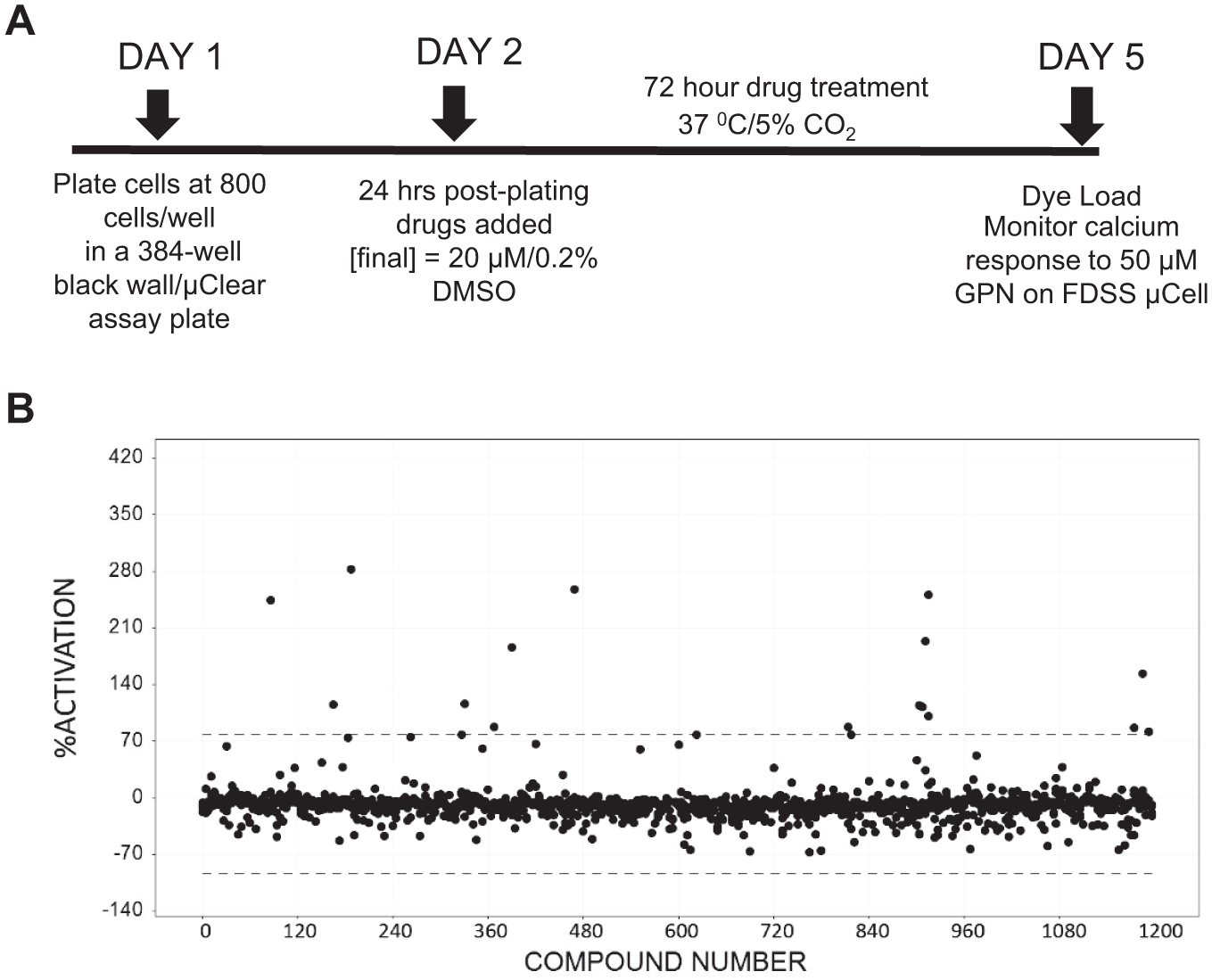
(
Pilot HTS with Prestwick Screening Collection
We performed pilot HTS on 1200 FDA-approved drugs (Prestwick collection) to determine whether small molecules could be identified with activity to restore lysosomal calcium signaling in TSD patient-derived cells. The pilot screen was performed at a single 20 μM compound concentration. A scatterplot of the primary HTS is shown in Figure 3B . The Z factor was calculated using vehicle-treated WT cells as the high control and vehicle-treated TSD cells as the low control. The screen performed with an overall mean Z′ factor of 0.56 ± 0.017. A value of ⩾0.5 is considered to have an assay window and acceptable variability for HTS assays. 21 Hits were identified using a standard deviation-based hit threshold (hit = signal mean plus 3SD in the compound data). The pilot screen identified 12 compounds as hits and delivered a 1% primary hit rate using the cutoff criterion. For confirmation, hit compounds from the primary screen were retested in triplicate at 20 μM and counterscreened on WT patient fibroblasts to assess selectivity for the diseased phenotype. Nine hits identified from the primary screen were confirmed with four compounds showing selectivity for TSD patient cells compared with WT cells. The hits were further confirmed from freshly prepared powder stocks, and dose–response curves were performed to determine functional potency for increasing lysosomal calcium from basal levels on TSD and WT cell lines ( Fig. 4 ).
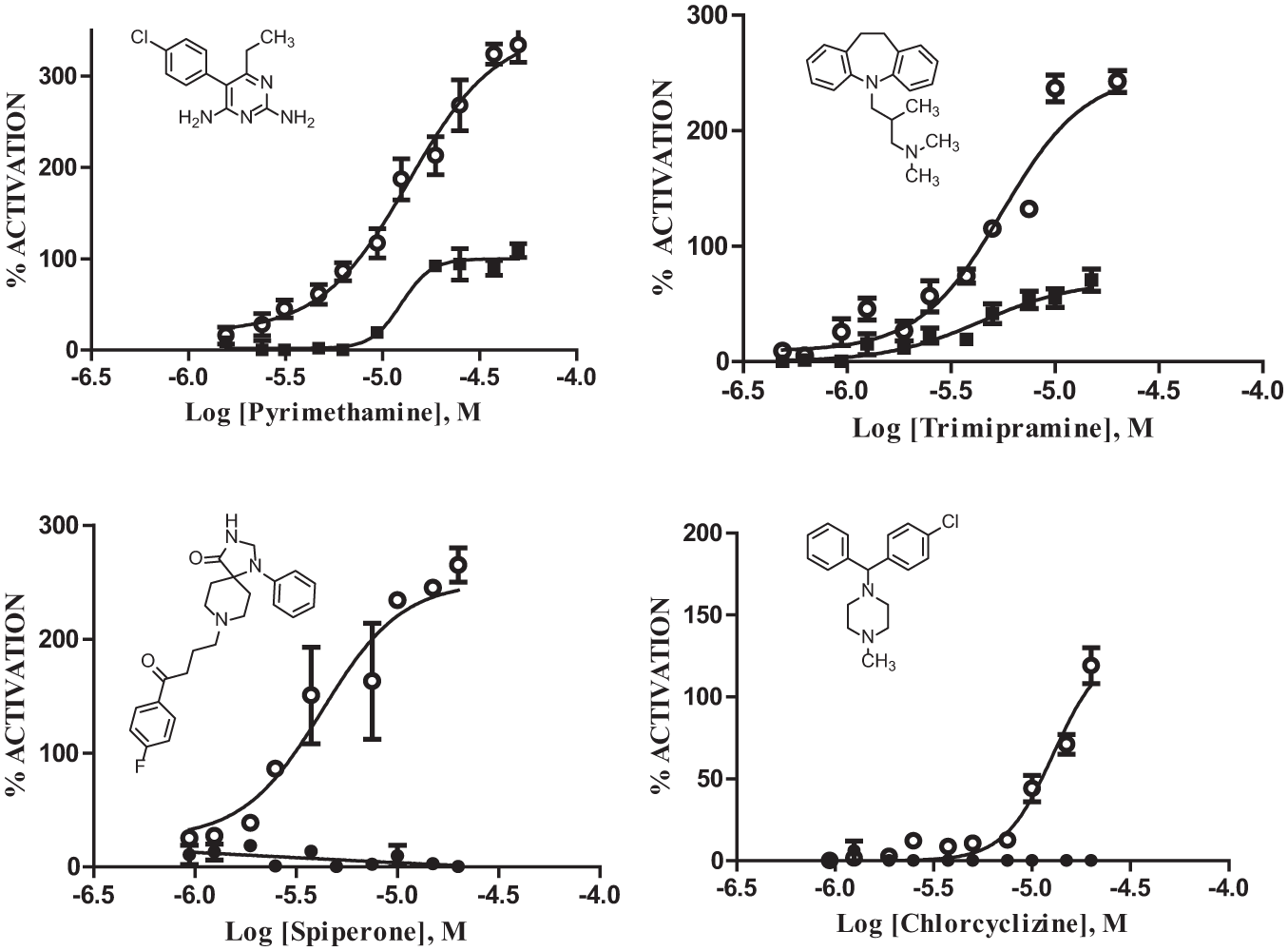
Dose–response analysis of selected hits in lysosomal calcium assay on WT and TSD patient cell lines. Percent activation of compounds on TSD and WT patient cell lines was calculated from the cell type-specific basal response measured in the absence of drug for TSD (open circles) and WT (closed circles). Data are mean ± SEM of triplicates for each concentration. EC50 values and percent activation: pyrimethamine TSD = 14 μM, 335%, WT = 12 μM, 100%; trimipramine TSD = 5.6 μM, 240%, WT = 4.4 μM, 70%; spiperone TSD = 4.3 μM, 250%, inactive on WT; chlorocyclizine TSD = 12.8 μM, 130%, inactive on WT.
Summary of Hits Identified in Prestwick Screen
The hits identified from the pilot screen represent diverse structures and biological activities for known targets. The four drugs selected for further evaluation for measuring functional potency exhibited modest micromolar potency in the phenotypic assay, with maximal response ranging from 130% to 335% activation of the TSD basal response to GPN measured in the absence of drug treatment ( Fig. 4 ). Pyrimethamine increased the GPN-induced calcium response in TSD cells with an EC50 of 14 µM and 335% activation over vehicle-treated TSD cells. Trimipramine, a tricyclic antidepressant, produced an EC50 of 5.6 μM and 240% activation over the basal GPN response in vehicle-treated TSD cells. Pyrimethamine and trimipramine showed activity (100% and 70% activation, respectively) on WT cells, but with a much lower maximal activation compared with TSD cells. Spiperone, a dopamine D2 receptor antagonist and typical antipsychotic, produced an EC50 of 4.3 μM and 250% activation over non-drug-treated TSD cells. Chlorcyclizine, an H1 histamine receptor antagonist, produced a small 130% activation over the basal GPN response in vehicle-treated TSD cells with an EC50 of 12.8 μM. Spiperone and chlorcyclizne were inactive on WT cells and displayed selective activity for TSD cells.
Pyrimethamine is a known pharmacological chaperone of HEXA. This activity was identified from a target-based HTS assay on purified enzyme to detect competitive inhibitors within the National Institute of Neurological Disorders and Stroke (NINDS) collection of known FDA-approved drugs.7,8 Pyrimethamine was further shown to function as a pharmacological chaperone and enhanced mutant enzyme activity in fibroblasts from late-onset forms of TSD patients. 25 We would not have expected pyrimethamine to act as a chaperone in the TSD patient-derived cell line utilized in our phenotypic HTS, as the mutation incorporates a premature termination stop codon, thereby disrupting expression of a full-length, functional β-hexosaminidase α-subunit. Therefore, pyrimethamine acting as a chaperone could not explain the mechanism of action in our phenotypic screen. Pyrimethamine is an antimalarial drug and potent inhibitor of dihydrofolate reductase. In melanoma cell lines, pyrimethamine was shown to be an autophagy modulator. 26 To further understand the mechanism of action of pyrimethamine to reverse the lysosomal disease phenotype in TSD patient cells, we evaluated the activity of pyrimethamine to modulate autophagy in TSD and WT patient cell lines.
Monitoring Autophagy in TSD and WT Patient Cells
Assessment of autophagy can be monitored by measuring the conversion of cytosolic microtubule-associated protein light chain 3 (LC3I) to the lipid-modified, membrane-bound form (LC3II) as a protein marker on autophagosomes. Autophagy is a dynamic process whereby autophagosomes fuse with lysosomes to allow degradation and clearance of organelles and proteins. This process can be inhibited with bafliomycin A1, an inhibitor of the vacular-type H+-translocating ATPase, which blocks autophagosome/lysosome fusion. This allows the assessment of the level of autophagic flux as the degradation pathway is inhibited and the LC3II expression levels detected under these conditions reflect the amount that would have been degraded by autophagy over the treatment period.
We measured LC3II expression levels by Western blot analysis comparing WT and TSD patient fibroblast cell lines ( Fig. 5A ). Similar basal levels of LC3II expression were detected in vehicle-treated WT and TSD patient fibroblasts. Upon bafliomycin A1 treatment, levels of LC3II expression increased in both WT and TSD cells; however, the level of LC3II in the TSD cells was 32% lower compared with that in WT patient cells ( Fig. 5B ). This suggests that autophagy is defective in the TSD patient cells due to either a reduction in the number of autophagosomes produced or the amount of autophagic flux. We next determined if pyrimethamine could reverse the autophagy defects in TSD cells. LC3II expression was higher after treatment with pyrimethamine plus balfiomycin in both TSD and WT patient cells compared with bafliomycin A1 treatment alone. A 49% increase in LC3II expression in TSD patient cells with pyrimethamine in the presence of balfiomycin was observed compared with bafliomycin alone. In WT cells, a 21% increase in LC3II expression was observed with pyrimethamine in combination with balfliomycin compared with bafliomycin alone ( Fig. 5B ).
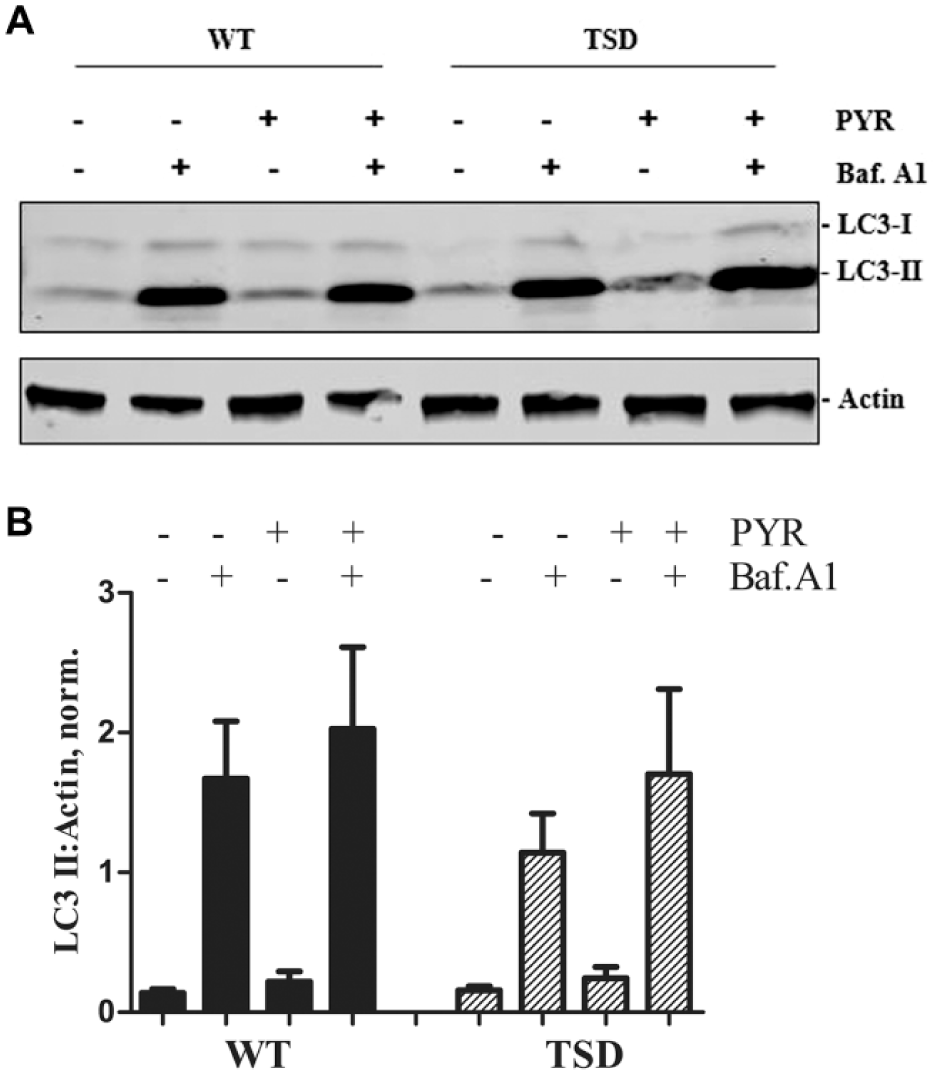
Autophagy detection in WT and TSD fibroblasts. (
Discussion
Here we describe a patient-derived cell-based phenotypic HTS assay to identify small molecules as disease-modifying therapeutics for TSD disease. We developed this novel assay based on a deficit in lysosomal calcium release as a monitor of the diseased state. The pilot HTS on a collection of known FDA-approved drugs demonstrated the ability to identify compounds that could improve lysosomal function in the diseased state and showed selectivity for the diseased state over WT, normal patient cells.
Pyrimethamine, an antiparasitic drug, has previously been shown to be a pharmacological chaperone for HEXA. We found pyrimethamine to be active in the primary screen and confirmed that it had modest functional potency in restoring lysosomal calcium release in TSD patient cells. Since pyrimethamine was previously reported to modulate autophagy in other cell types, 26 we evaluated the compound’s activity on autophagy in TSD and WT patient fibroblasts. Impairment in autophagy has been implicated as an underlying mechanism in a number of LSDs.27,28 Functional lysosomes are critical for autophagy as they fuse with autophagosomes to deliver cellular material for digestion by lysosomal enzymes. Disruption of the autophagy/lysosomal pathway reduces cellular capacity to remove accumulating nondegraded material impacting normal autophagy flux. In TSD patient cells, pyrimethamine was effective in improving autophagic flux. Additional studies are required to determine the mechanism for modulation of autophagy in TSD patient cells and to understand the target engaged by pyrimethamine within the autophagy/lysosomal pathway. The mTor inhibitors rapamycin and Torin 1 did not show improvement of lysosomal calcium release in TSD patient-derived cells as measured by the FDSS assay (data not shown). Profiling additional autophagy modulators with known mechanisms of action on TSD patient-derived cells could provide insight into pyrimethamine’s mechanism of action.
In addition to pyrimethamine, the TSD phenotypic screen identified three drugs from a collection of FDA-approved drugs, revealing new and previously unknown pharmacological activities to improve the diseased phenotype in TSD patient cells. The functional potency on TSD patient cells is modest; however, the drugs can be further utilized as probes to understand disease biology and facilitate the identification of new targets for drug discovery.
The use of patient-derived cells in high-throughput phenotypic screening assays has several advantages for drug discovery in comparison with target-based assays. The screen is performed in a disease cellular environment containing the defective biochemical and signaling pathways downstream from the known genetic defect. The assay is unbiased for compound mechanisms of action. Importantly, this approach could lead to the identification of new molecular targets previously not considered to be disease modifying. Our phenotypic screening approach may have a broader applicability to other LSD diseases. The phenotypic assay we developed on TSD patient cells was based on a lysosomal deficit that has been reported in other LSD diseases. Dysregulation and deficits in calcium signaling have been found in patient-derived cells from other LSDs, including Niemann–Pick type C and Gaucher disease.19,20 Active compounds identified from our screen may have broader therapeutic utility in treating other LSDs targeting restoration of deficiency in lysosomal calcium signaling. The phenotypic HTS assay platform may be applicable to other LSDs, and we are currently expanding screening on patient-derived cells from these diseases.
Based on the pilot HTS, the assay was validated and can support the screening of large chemically diverse libraries to identify novel pharmacophores to support hit-to-lead chemistry efforts. Establishing this new phenotypic assay platform is a significant step for advancing drug discovery for TSD with the potential to identify novel targets for targeting drug intervention.
Footnotes
Declaration of Conflicting Interests
The authors declared no potential conflicts of interest with respect to the research, authorship, and/or publication of this article.
Funding
The authors disclosed receipt of the following financial support for the research, authorship, and/or publication of this article: This work was supported by a research grant from the University of Pennsylvania Orphan Disease Center (MDBR-16-115-TSA).