
Abstract
Targeted protein degradation represents an area of great interest, potentially offering improvements with respect to dosing, side effects, drug resistance, and reaching “undruggable” proteins compared with traditional small-molecule therapeutics. A major challenge in the design and characterization of degraders acting as molecular glues is that binding of the molecule to the protein of interest (PoI) is not needed for efficient and selective protein degradation; instead, one needs to understand the interaction with the responsible ligase. Similarly, for proteasome targeting chimeras (PROTACs), understanding the binding characteristics of the PoI alone is not sufficient. Therefore, simultaneously assessing the binding to both PoI and the E3 ligase as well as the resulting degradation profile is of great value. The cellular thermal shift assay (CETSA) is an unbiased cell-based method, designed to investigate the interaction of compounds with their cellular protein targets by measuring compound-induced changes in protein thermal stability. In combination with mass spectrometry (MS), CETSA can simultaneously evaluate compound-induced changes in the stability of thousands of proteins. We have used CETSA MS to profile a number of protein degraders, including molecular glues (e.g., immunomodulatory drugs) and PROTACs, to understand mode of action and to deconvolute off-target effects in intact cells. Within the same experiment, we were able to monitor both target engagement by observing changes in protein thermal stability as well as efficacy by simultaneous assessment of protein abundances. This allowed us to correlate target engagement (i.e., binding to the PoI and ligases) and functional readout (i.e., degrader induced protein degradation).
Introduction
The mainstream in drug discovery is developing chemical modulators of various protein targets. Chemical modulators, typically small molecules that bind to an enzyme or receptor active site, function by locking their target protein in a state that augments or prevents it from performing its intended function. However, about 80% of the human proteome is estimated to lack favorable features for such drug development schemes (i.e., considered undruggable) 1 , and therefore, strategies for modulating the function of the bulk of the proteome is warranted. A possible approach to such undruggable proteins has been to regulate target protein abundance or availability, and this has been achieved in the preclinical setting by means of gene editing (CRISPR/Cas9), 2 blocking transcription,3,4 or selectively destroying target proteins. The latter approach gained substantial attention recently because of the increased understanding of the cellular protein disposal machinery via the ubiquitination–proteasome pathway.1,5–7 So far, only a handful of such modalities have reached clinical trials, albeit without any reports on the clinical outcome.
The ubiquitin proteasome system consists of a cascade of enzymes that catalyze the conjugation of the 76-amino-acid protein ubiquitin onto lysine residues of specific proteins, marking them for degradation by the large 26S proteasome complex. The molecular determinants of which proteins are targeted for ubiquitination are defined by a class of multi-subunit enzymes known as E3 ubiquitin ligases, which carry both the enzymes responsible for the ubiquitination and the nonenzymatic substrate recognition subunits to ensure a regulated breakdown of proteins. 6 It is now known that the specificity of the substrate recognition subunit can be modulated with small molecules, in which a notable example is the phthalimide class of compounds known as immunomodulatory drugs (IMiDs). First described in the 1950s and 1960s as a sedative, the IMiD thalidomide was later discovered to be a potent teratogen 8 that caused severe phocomelic side effects to newborns. However, since then, thalidomide and its close analogs have been repurposed as potent anticancer agents.9,10 In 2010, a major step toward understanding IMiD action was made in which cereblon (CRBN) was identified as a major target driving the thalidomide teratogenicity.11,12 Another major breakthrough in the understanding of IMiDs was the identification of the transcription factors Ikaros and Aiolos as proteins that were ubiquitinated upon IMiD treatment.13–16 More recently, casein kinase 1α (CK1α) was also identified as a protein selectively degraded by the IMiD (thalidomide analogue) lenalidomide.17,18 Finally, a number of proteome-wide studies extended the list of IMiDs-induced neosubstrates of CRBN.16,19,20
Although IMiDs have found clinical success, the applicability of the system is currently limited. The rational design of thalidomide analogs that target specific proteins for degradation would be difficult given the small structural determinant on the potential substrate, making it challenging to predict and exploit. A strategy to go around this problem was presented by Sakamoto and colleagues, 7 which used bifunctional molecules, in this case composed of a small molecule binding the target protein MetAP2 and a peptide that bound the SCF-β-TRCP E3 ubiquitin ligase. This bifunctional molecule was successful in bringing MetAP2 in close proximity to the E3 ubiquitin ligase, which resulted in increased MetAP2 ubiquitination and subsequent degradation. 7 These types of molecules were later denoted proteolysis targeting chimeras (PROTACs) and have come to adopt several E3 ubiquitin ligase binders, including IMiD molecules, but they also target a wide range of target proteins. An example of such a molecule is BSJ-03-204, which comprises pomalidomide as the CRBN-binding domain and palbociclib as the moiety binding the protein of interest (PoI). 21 The main PoIs, in this case CDK4 and CDK6, were ubiquitinated and degraded upon administration of BSJ-03-204 to live cells. 21
There are several validation methods for targeted protein degradation at different stages of the ubiquitin–proteasome pathway that can be used for PROTACs characterization, including cellular ubiquitination, target binding, and cellular degradation. 22 Such monitoring of protein degradation serves as a direct indication of successful binding, to not only the PoI but also to the responsible E3 ligase. However, the bifunctional nature of a PROTAC molecule could afford a broader target engagement profile than the sum of its components, as well as the opposite (i.e., that the different domains could sterically hinder specific interactions). It has been shown that, in addition to target engagement, for PROTACs the formation of a productive ternary complex is critical for effective protein degradation. 23 Remarkably, this is a profoundly different feature from the classical small molecule, in which a stronger affinity can drive the drug selectivity toward the target and is usually a primary aim in the design of new therapeutic compounds.
Using CRBN- and VHL-recruiting PROTACs that bind >50 kinases, Bondensson et al. 24 showed that only a subset of bound targets is degraded, pointing out the relevance of the additional factor in addition to target engagement. Examples of the importance of the cooperativity of the ternary complex formation are also given by work of the Gray group, in which the lack of KRAS oncoprotein degradation for the PROTAC based on the covalent KRAS inhibitor is reported to be due to the inability to induce KRAS polyubiquitination and subsequent degradation. 25 Similarly, the same group speculated that PROTAC failure to degrade tubulin may be due to the inability of those compounds to mediate the formation of a productive tubulin–PROTAC–CRBN ternary complex. 26
Ciulli et al. used thermodynamic techniques, including isothermal titration calorimetry, to quantify the stability and cooperativity of the binary and ternary complex for a series of PROTACs targeting BRD4/VHL and showed that more potent inhibitors do not necessarily generate more potent PROTACs. 27 Therefore, PROTACs with only modest target affinity can nonetheless induce potent target degradation because of positive cooperativity of the ternary complex. 28 It would therefore be interesting to not only elucidate the quantitative changes in the proteome after PROTAC administration to live cells but also to map the molecule’s high-affinity binding partners in both intact cells and lysates.
A crucial part of any drug development program is to ensure proper and specific target engagement, and several strategies can be employed for such investigations. 29 The cellular thermal shift assay (CETSA) was introduced in 2013 as a method to monitor target engagement in an intact cell environment. CETSA is based on the same principle as classical thermal shift assays, with the important addition that it can be employed in more complex biological systems such as living cells, tissues, and whole blood. 30 Importantly, the CETSA protocol can be carried out in both intact cells as well as lysates; in the latter we expect very little, if any, biological activity. Therefore, intact cell experiments allow both direct monitoring of ligand binding as well as downstream effects from such perturbations, whereas lysate-based experiments offer a cleaner picture of only the direct targets.
Originally, the method was used only to prove specific target engagement in cells, but in taking advantage of quantitative mass spectrometry (MS)–based proteomics for detection, CETSA MS allows for unbiased profiling of a molecule’s interactions and using intact cells as well as downstream effects on the global proteomic scale. This has turned out to be a powerful tool for target deconvolution after phenotypic screening campaigns as well as for the assessment of target specificity of a molecule.31,32
Herein, we set out to combine CETSA MS with quantitative proteomics to study both the target specificity as well as degradation specificity of several IMiDs, both in intact cells and cell lysates. We have carried out our study in intact cells of different origin: transformed human cells (K562); induced pluripotent stem cells (iPSC) and iPS-derived embryoid bodies (EBs). Lastly, we also compared the observed effects of IMiD treatment with the target engagement and degradation specificity of an IMiD-based PROTAC (BSJ-03-204) and its PoI ligand palbociclib.
Materials and Methods
Cells
K562 cells were obtained from ATCC and cultured following ATCC protocols. Cell cultures were kept subconfluent, and the cells were given fresh medium 1 d before harvesting. For the experiment, the cells were washed and detached with 1x TrypLE (Gibco, Life Technologies, Carlsbad, CA). The detached cells were diluted into Hank’s Balanced Salt solution (HBSS), pelleted by centrifugation (300 g, 3 min), washed with HBSS, pelleted again, and resuspended in the experimental buffer (20 mM HEPES, 138 mM NaCl, 5 mM KCl, 2 mM CaCl2, 1 mM MgCl2, pH 7.4) constituting the 2x cell suspension. For the lysate experiments, K562 cells were resuspended in the experimental buffer, and cells were lysed by three rounds of freeze-thawing. The lysate was clarified by centrifugation at 30 000 × g for 20 min and used immediately in the CETSA experiment constituting the 2x lysate. Reagents were purchased from Sigma unless otherwise noted.
Cellartis human iPSC line 22 (ChiPSC22) was obtained from Takara (Kyoto, Japan), expanded, and cultured using Cellartis DEF-CS Culture System following the Cellartis protocol.
EBs were obtained by seeding ChiPSC22 in in vitro differentiation medium (IVD) containing advanced RPMI 1640, B-27 50X, GlutaMAX 100X, PEST 10,000 units/mL of penicillin and 10,000 µg/mL of streptomycin (Gibco, ThermoFisher Scientific, Waltham, MA), and 5 µM of Y-27632 (Selleckchem, Houston, TX) at a density of 5 × 104 cells per well in a 96-well plate (nontreated, V bottom), centrifugation at 400 × g at room temperature for 5 min, followed by a 7 d incubation at 37 °C, 5% CO2, and ≥90% humidity. After 7 d, EBs were transferred into coated six-well plates (DEF-CS COAT, Takara) at a density of 30 EBs per well in IVD medium and harvested after 7 d.
Compounds
Thalidomide, lenalidomide, and pomalidomide were purchased from Sigma Aldrich as lyophilized powders and redissolved in DMSO to 30 mM stock solutions. BSJ-03-204 was purchased from Tocris (#6938) and redissolved in DMSO to a 10 mM stock solution.
Two-Dimensional CETSA MS Experiment
The K562 cell suspension was divided into five aliquots, and each was mixed with an equal volume of compound solution prepared at 2x final concentration in the experimental buffer (as above). The resulting final concentrations of compounds (thalidomide, lenalidomide, or pomalidomide) were 0.03, 0.3, 3, and 30 µM; 1% DMSO only was used as control. Incubations were performed for 60 min at 37 °C with end-over-end rotation. The treated cell suspensions were each divided into 10 aliquots that were all subjected to a heat challenge for 3 min, each at a different temperature between 44 and 62 °C. After heating, protein aggregates were pelleted by centrifugation at 20,000 × g for 20 min, and supernatants containing soluble fractions were kept for further analysis.
Compressed CETSA MS Experiment in Lysed iPSCs and EBs
iPSCs (ChiPSC22) and EBs were lysed by three rounds of freezing in liquid nitrogen and thawing in a water bath (20 °C). Cell debris was removed by centrifugation at 30,000 × g for 20 min. Clarified lysate was then divided into eight aliquots, and each was mixed with an equal volume of compound solution prepared at 2x final concentration in the experimental buffer (as above). The resulting final concentrations of pomalidomide were 0.02, 0.2, 2, 6, 20, 60, and 200 µM; 1% DMSO only was used as control. Incubation was performed for 15 min at room temperature. After incubation, samples were divided into 24 aliquots and subjected to a heat challenge for 3 min, two samples per temperature point, in a temperature range of 44 to 66 °C (in increments of 2 °C). After the heating step, samples corresponding to the same compound concentration were pooled together, and 16 resulting samples (eight concentration points in two replicates) were centrifuged for 20 min at 30,000 × g to pellet aggregated proteins. Supernatants containing soluble fractions were kept for further analysis.
Time-Course–Compressed CETSA MS Experiment in Intact iPSCs and EBs
iPSCs and EBs were incubated with 30 µM of pomalidomide or vehicle control (1% DMSO) in media (DEF-CS for iPSCs and advance RPMI and 50X B27 for EBs) for 15, 30 (EBs only), 60, 120, and 480 min at 37 °C, 5% CO2, and ≥90% humidity. At harvest, the cultures were washed in dPBS [−]CaCl2 [−]MgCl2, trypsinized with TrplE, washed in dPBS, centrifugated at 200 × g for 3 min, and resuspended in the experimental buffer (as above). All solutions used for harvest contained 30 µM pomalidomide or vehicle control, respectively. The cell suspensions were divided into 13 aliquots, of which 12 were subjected to a heat challenge for 3 min, each at a different temperature between 44 and 66 °C (increments of 2 °C), while one aliquot remained unheated. After heating, samples corresponding to the same treatment were pooled together. In all samples, protein aggregates were pelleted by centrifugation at 30,000 × g for 20 min, and supernatants containing soluble fractions were kept for further analysis.
LC-MS Sample Preparation and LC-MS/MS Analysis
The total protein concentration of the soluble fractions was measured by DC protein colorimetric assay (BioRad, Hercules, CA). The same protein amount from each sample was taken and subjected to reduction and denaturation with tris(2-carboxyethyl)phosphine (Bond-breaker, Thermo Scientific) and RapiGest SF (Waters, Milford, MA), followed by alkylation with chloroacetamide. Proteins were digested with Lys-C (Wako Chemicals, Richmond, VA) overnight and trypsin (Trypsin Gold, Promega, Madison, WI) for 6 h.
After complete digestion had been confirmed, samples were labeled with 10-plex Tandem Mass Tag reagents (TMT10, Thermo Scientific) according to the manufacturer’s protocol. Labeling was performed for 3 h at room temperature and ~30% acetonitrile in the buffer. Labeling reactions were quenched by the addition of hydroxylamine to the final concentration of 0.1%. For the two-dimensional (2D) CETSA MS experiment, concentration series from two consecutive temperatures were combined in one TMT10-plex sample. For the eight-concentration-point–compressed CETSA MS in lysed iPSCs, eight CETSA samples were combined with two nonheated samples (treated with highest concentration of compound and corresponding DMSO control) within the same TMT10-plex sample. For the time-course experiment, samples from the same time point were combined together (six CETSA samples + four nonheated samples).
The labeled samples were subsequently acidified and desalted using polymeric reversed phase (Strata X, Phenomenex or Oasis HLB, Waters). Liquid chromatography (LC)–MS grade liquids and low-protein binding tubes were used throughout the purification. Samples were dried using a centrifugal evaporator.
For each TMT10-plex set, the dried labeled samples were dissolved in 20 mM ammonium hydroxide (pH 10.8) and subjected to reversed-phase high-pH fractionation using an Agilent 1260 Bioinert HPLC system (Agilent Technologies, Santa Clara, CA) over a 1 × 150 mm C18 column (XBridge Peptide BEH C18, 300 Å, 3.5 µm particle size, Waters). Peptide elution was monitored by ultraviolet absorbance at 215 nm, and fractions were collected every 30 s into polypropylene plates. The 60 fractions covering the peptide elution range were evaporated to dryness, ready for LC-MS/MS analysis.
Thirty individual fractions were analyzed by high-resolution Q-Exactive HF or Q-Exactive HF-X mass spectrometers (Thermo Scientific) coupled to high-performance nano-LC systems (Evosep One, Evosep, or Ultimate 3000, Thermo Scientific).
MS/MS data were collected using higher-energy collisional dissociation, and full MS data were collected using a resolution of 120 K with an AGC target of 3e6 over the m/z range of 375 to 1650. The top 12 most abundant precursors were isolated using a 1.2 Da isolation window and fragmented at normalized collision energy values of 35. The MS/MS spectra (45 K resolution) were allowed a maximal injection time of 120 ms with an AGC target of 1e5 to avoid coalescence. Dynamic exclusion duration was 30 s.
Protein Identification and Quantitation
Protein identification was performed by a database search against 95,607 canonical and isoform human protein sequences in Uniprot (UP000005640, download date: February 21, 2019) using the Sequest HT algorithm as implemented in the ProteomeDiscoverer 2.2 software package. Data were recalibrated using the recalibration function in PD2.2, and final search tolerance setting included a mass accuracy of 10 ppm and 50 mDa for precursor and fragment ions, respectively. A maximum of two missed cleavage sites was allowed using fully tryptic cleavage enzyme specificity (K\, R\, no P). Oxidation of methionine, deamidation of asparagine and glutamine, and acetylation of protein N-termini were also allowed as variable modifications. Carbamidomethylation of Cys, TMT-modification of lysine, and peptide N-termini were set as static modifications.
Peptide-spectrum matches were filtered at a 1% false discovery rate level by employing target-decoy search strategy and percolator rescoring. 33
For quantification, a maximum co-isolation of 50% was allowed. Reporter ion integration was performed at 20 ppm tolerance, and the integration result was verified by manual inspection to ensure the tolerance setting was applicable. For individual spectra, an average reporter ion signal-to-noise ratio of ≥20 was required. Only unique peptide sequences were used for quantification.
Data Analysis
Protein intensities were normalized ensuring the same total ion current in each quantification channel. Intensity values were then log2-transformed and normalized between treatments and replicates, so the median protein intensity is the same in all treatments and replicates.
The fold-changes of any given protein across the concentration range is quantified by using the vehicle condition as the reference (i.e., a constant value of 1). Fold-changes are also transformed to log2 values, to achieve a normal distribution about 0.
To estimate effect size (amplitude) and P value (significance) of the protein hits in the eight-concentration-point compressed CETSA MS experiments, the individual protein concentration-response curve is fitted via nonlinear least squares algorithm using the following formula:
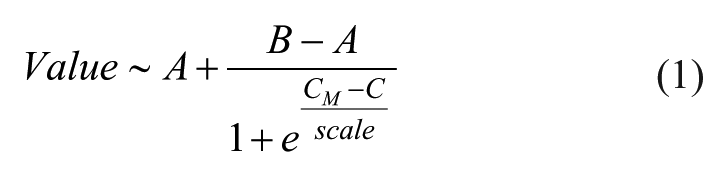
where
To estimate the effect size (amplitude) and P value (significance) of the protein hits in 2D CETSA MS experiments, protein log-fold changes over the temperature and concentration domain were fitted via the nonlinear least squares algorithm using the 2D surface equation formula:
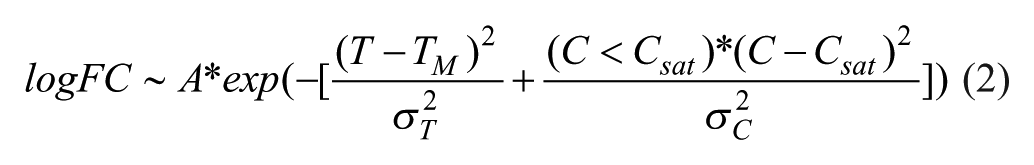
where
To estimate the significance of time-dependent compound-induced changes in either protein abundance or protein thermal stability, following the mixed effect, a linear model was built:
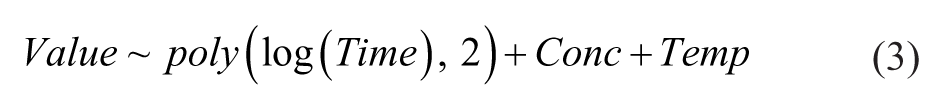
where poly(log(Time),2) is the second-order polynomic expression using the log2-transformed time value, Conc is the two-level factor equal either to 1 for pomalidomide-treated samples or to 0 for nontreated samples, and Temp is the two-level factor value equal either to CETSA or non-CETSA for heated and nonheated samples, respectively.
The significance of the effect for each protein was assessed using an analysis of variance (ANOVA) F test comparing the fit results with the trivial model fit. The Benjamini-Hochberg correction was applied to the F test–derived P values to adjust for multiple comparison.
Results
Profiling of the three classical IMiDs in intact K562 cells (
Fig. 1a
) was performed using a 2D thermal proteome profiling approach (2D CETSA MS). Here, each protein can be characterized by a heat map (
Fig. 1b
), in which the protein-fold change (relative to vehicle control for each temperature) is represented by the color. Alternatively, each protein can be characterized by the observed maximum/minimum log-fold change (amplitude) and significance derived from an ANOVA-based F test to present the results as volcano plots (
Fig. 1c
). Only a handful of ~7000 proteins quantified in the experiments were found to be significantly affected by the IMiD treatment. Among them, we were able to observe a clear temperature- and concentration-dependent stabilization of CRBN for all three compounds, confirming the binding to the endogenous target.11,34,35 Notably, for proteins ZFP91, IKZF1, and RNF166, the same concentration-dependent reduction of soluble protein amount was observed for all sample temperatures, suggesting compound-induced protein degradation rather than destabilization, which is well in line with previous findings.14,16,36 When the same experiment was performed for pomalidomide in lysed cells (
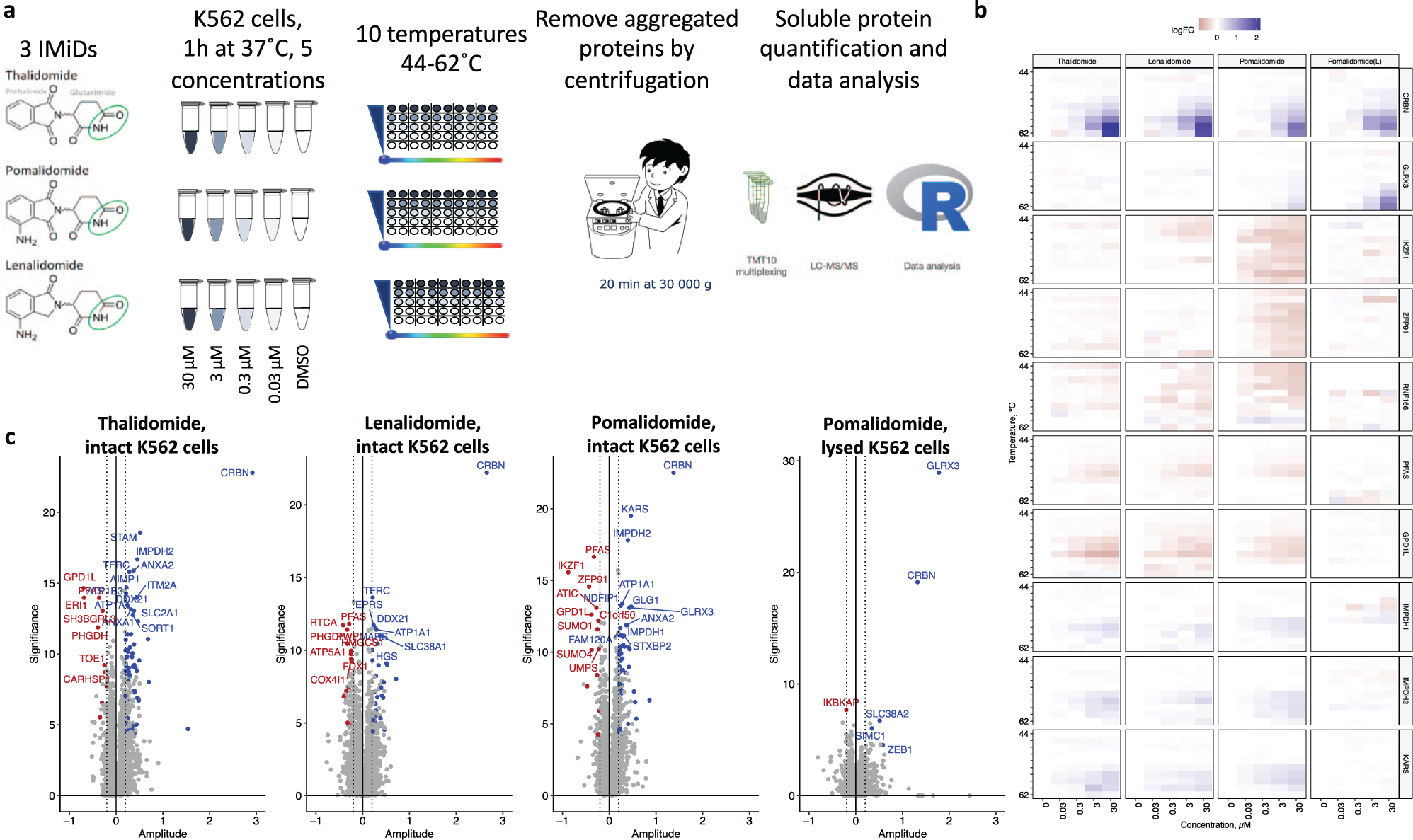
Two-dimensional (2D) cellular thermal shift assay (CETSA) mass spectrometry (MS) profiling of thalidomide, pomalidomide, and lenalidomide in intact and lysed K562 cells. (
The results presented above in the transformed human cell line K562 encouraged us to investigate target engagement and degradation profiles in other sample matrices. In addition to antineoplastic effects, IMiDs are also infamous for their teratogenic effects, which are best studied in immature cells. Human iPSCs, three-dimensional iPSC aggregates, EBs, in which cells spontaneously differentiate into a heterogenous mix of derivatives of the three germ layers, 37 are the golden standard in in vitro teratogenic assessment. iPSCs and EBs were treated with pomalidomide, and target engagement was assessed by CETSA MS, with quantitative proteomics applied to measure protein degradation ( Fig. 2a ).
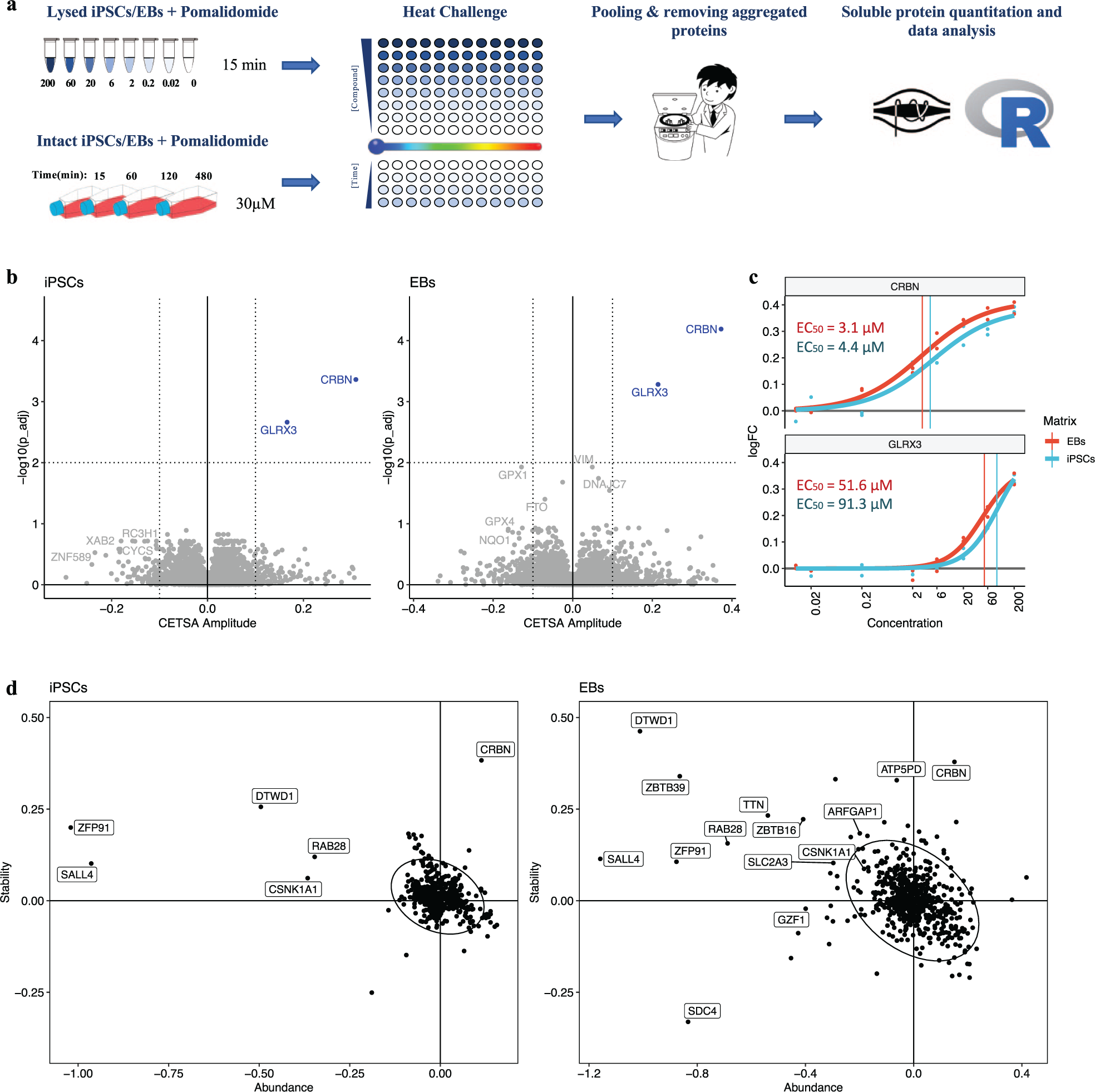
Compressed cellular thermal shift assay (CETSA) mass spectrometry (MS) profiling of pomalidomide in intact and lysed induced pluripotent stem cells (iPSCs) and iPSC-derived embryoid bodies (EBs). (
By applying the recently developed one-pot, or compressed, format,38–40 we have explored a wider concentration range (lysate experiment) as well as the time dependence of the binding and degradation profile (
Fig. 2
).
Figure 2b
shows the results of the eight-concentration pomalidomide profiling in lysed iPSCs and EBs. In this experiment 7593 protein groups were quantified. Each protein is represented by its amplitude (relative stability change) and significance (-log10–transformed P value). CRBN and GLRX3 were the only two proteins that showed a significant concentration-dependent change in thermal stability upon treating either lysed iPSCs or EBs with pomalidomide, supporting the observations in lysed K562 cells (
The previous experiments showed that a single concentration of pomalidomide of 20 to 30 µM allows for nearly complete stabilization of cellular targets of pomalidomide, without causing major off-target binding events. To study binding events over time, human iPSC and EBs were incubated with either 30 µM pomalidomide or vehicle control for 15 min, 30 min (EBs only), 1 h, 2 h, and 8 h. We combined the heat-and-pool (CETSA compressed) and nonheated (protein abundance) samples within one TMT-multiplexed sample, which allowed us to apply a mixed-effect linear model to the protein measurements to retrieve pomalidomide-induced abundance and stability change contributions into the measured pomalidomide-induced protein intensity changes ( Fig. 2d ).
In this experiment 7084 and 8011 protein groups were quantified in iPSCs and EBs, respectively. The higher number of detected protein groups in EBs correlates well with the increased cellular diversity from the three germ layers. In line with the 2D CETSA MS results in K562 cells, a clear degradation of the known targets ZFP91 and DTWD1 was observed after 1 h of incubation in both iPSCs and EBs cells ( Fig. 1b ). Ikaros protein was not identified in iPSCs and EBs, and according to the previously published gene expression data, it is not expressed in either iPSCs or EBs. 43 In addition, time-dependent degradation of SALL4, RAB28, and CSNK1A1 was observed in two cell systems. Profiling in EBs identified even more pomalidomide-induced CRBN neosubstrates, including ZBTB16, ZBTB39, and GZF1 ( Fig. 2d ), with monotonous time-dependent degradation profiles ( Fig. 3a ). Other proteins such as SDC4, SLC2A3, or TTN also showed significant alteration of protein abundance. However, the time-course profile of these proteins ( Fig. 3a ) show either maximal absolute fold change after just 15 min of incubation (SLC2A3, TTN) or nonmonotonous time dependence (SDC4). Such inconsistent time-dependent abundance changes suggest that these are not degradation events but rather protein solubility changes due to biological perturbations in the cells. These slightly noisier protein abundance and solubility changes are more pronounced in the EB data compared with iPSCs and might reflect the fact that EB samples are more heterogenous and prone to have more sample-to-sample variation.
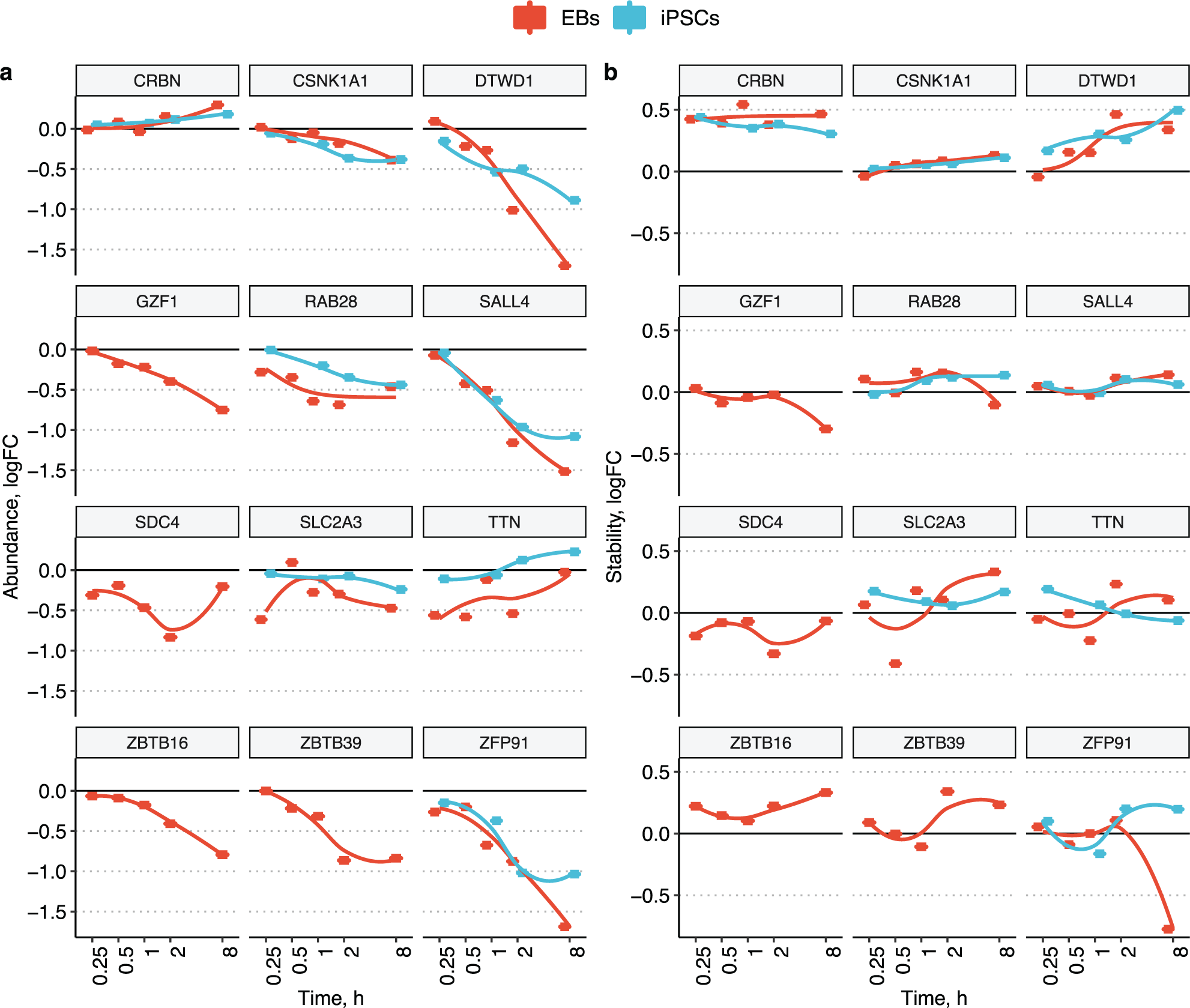
Time-dependent pomalidomide-induced protein abundance (
Stabilization of CRBN is observed at all time points studied (
Fig. 3b
). Moreover, a time-dependent increase in CRBN abundance can be also seen (
Fig. 3a
), likely caused by the inhibition of autoubiquitination. In addition, for most CRBN neosubstrates, degradation is accompanied by an increase in protein stability, which is most prominent for the DTWD1 (
We next explored the effect of the palbociclib and palbociclib-IMiD–based PROTAC BSJ-03-204 on the proteome of K562 cells. To evaluate the changes in compound-induced proteome stability, intact and lysed K562 cells were incubated with seven concentrations of either palbociclib or BSJ-03-204 (0.02, 0.067, 0.2, 0.67, 2, 6.7, and 20 µM) for 1 h and 15 min, respectively. The same samples used for intact CETSA MS profiling of BSJ-03-204 were also subjected to soluble protein abundance measurements without any heat treatment applied either directly (0, 0.67 µM) or after an additional 5 h of incubation (0, 0.2, 0.67, and 2 µM) to trace slower-acting drug-induced protein degradation.
In this experiment, heat-treated (CETSA) samples and nonheated samples were measured as different TMT-multiplexed samples. In the heat-treated samples 5122 and 5209 protein groups were quantified in lysed and intact K562 cells, respectively. The binding profile of the PROTAC molecule BSJ-03-204 in the lysed K562 cells combines pomalidomide binder (CRBN) and palbociclib (CDK4, CDK6, PIP4K2A, PIP4K2C, CSNK2A2, PLK1, etc.) signatures ( Fig. 4a ). Ferrochelatase (FECH), a common kinase-inhibitor off-target,44,45 was found to be more stabilized by BSJ-03-204 as compared with palbociclib alone ( Fig. 4b ).
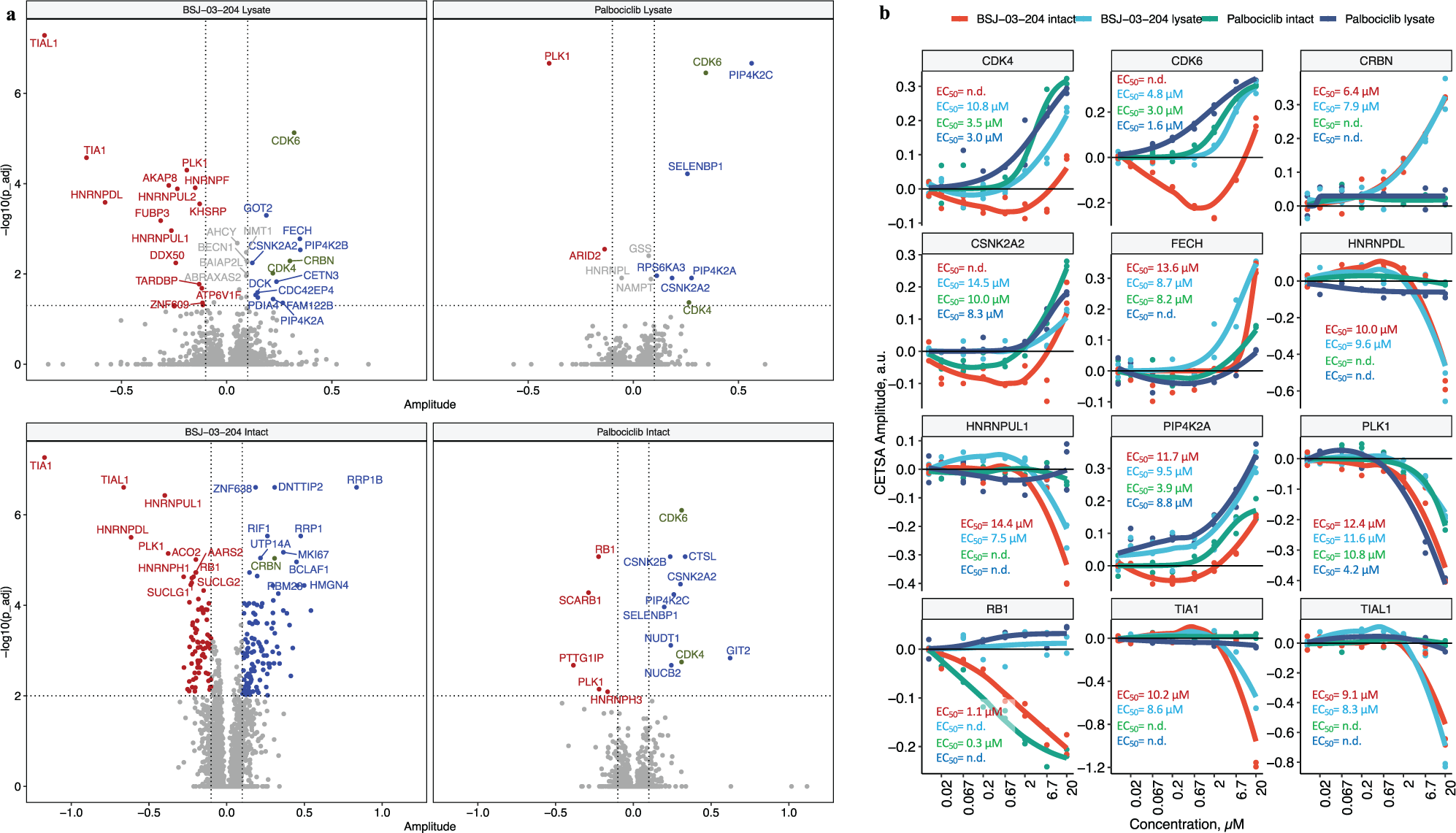
Compressed cellular thermal shift assay (CETSA) mass spectrometry (MS) profiling of palbociclib and palbociclib-based PROTAC molecule BSJ-03-204 in intact and lysed K562 cells. (
Although palbociclib stabilizes on-target (CDK4, CDK6) as well as off-target (PIP4K2C, CSNK2A2, etc.) kinases in intact cells, at first glance no cyclin-dependent kinases were found to be affected by BSJ-03-204 in intact cells ( Fig. 4a ). This is likely due to the compressed CETSA MS signal consisting of a combination of changes in protein abundance and changes in protein stability. In the intact cell experiment, BSJ-03-204 should induce both degradation of CDK4 and CDK6 as well as thermal stabilization (as is evident in the lysate experiment). At concentrations less than 6 µM, degradation is much more prominent and results in negative CETSA fold-change observed. However, when the concentration of BSJ-03-204 is high enough to ligate CDK4/6 and CRBN into separate binary complexes, ubiquitination of the PROTAC substrates is diminished and no degradation occurs (the so-called hook effect, previously described for several PROTACs 46 ), with a positive fold-change seen at 20 µM ( Fig. 4b ).
Both palbociclib and BSJ-03-204 treatment induce destabilization of RB1, which is a direct substrate of CDK4 and CDK6.47,48 Our data suggest that at least at early time points (1 h), the inhibition of CDKs by palbociclib, or degradation by BSJ-03-204, result in very similar concentration-dependent downstream events.
BSJ-03-204 has additional hits in both lysate and intact cells, which could not be explained from the binding profiles of either palbociclib of pomalidomide. TIAL1, TIA1, PLK1, HNRNPUL1, and HNRNPDL were identified as hits in both lysate and intact cell samples, which suggests that these are direct binders to the PROTAC molecule. However, hits that occur only in intact cells may constitute pathway effects induced by the biological effects of CDK4/6 degradation and/or inhibition. Among them, several proteins are involved in transcriptional regulation, RRP1B, RRP1, ZNF638, DNTTIP2, and RIF1.
When only protein abundance changes after incubation with BSJ-03-204 are considered (i.e., focusing on target protein degradation), we can observe a very clear compound profile ( Fig. 5a ). CDK4 and CDK6 showed a consistent, time-dependent degradation, whereas CRBN was rescued from degradation upon the treatment ( Fig. 5b ). In addition, time-dependent accumulation of KLHL8 was observed.
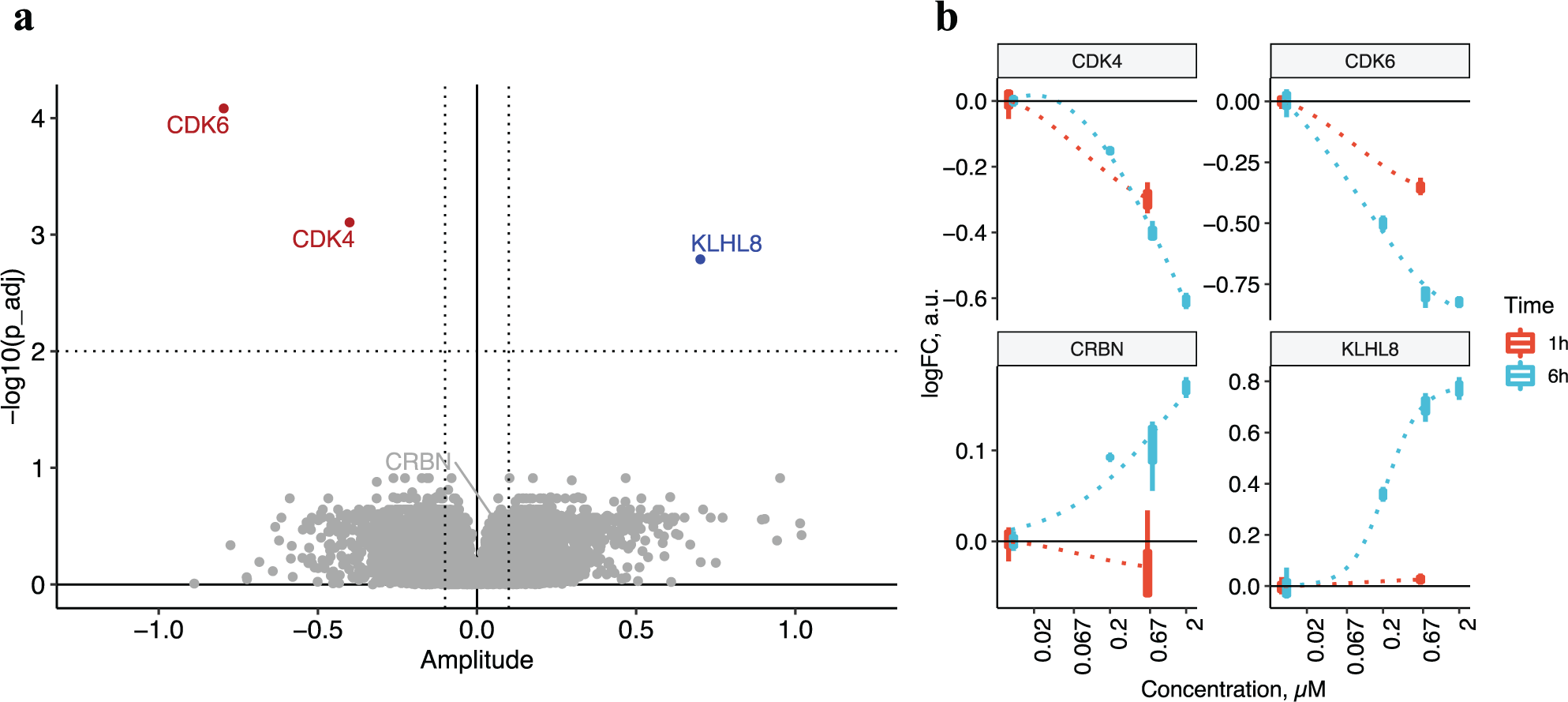
(
Discussion
In this study, we show the target deconvolution capabilities of CETSA MS with regard to delineating the target engagement profiles of the classical IMiDs: thalidomide, lenalidomide, and pomalidomide. Moreover, we designed our experiments in such a way that we also could simultaneously monitor both target engagement and pharmacologic effect (protein degradation), which resulted in a comprehensive profiling of the investigated IMiDs in relatively short and efficient LC-MS experiments. We took this concept further by applying the same experimental design on the IMiD-based PROTAC BSJ-03-204, enabling us to determine target engagement as well as degradation profiles of BSJ-03-204 in K562 cells.
Target deconvolution by CETSA MS revealed that all IMiDs are relatively selective drugs; CRBN stands out as the high-affinity target whose functional modulation is in turn responsible for downstream cellular effects ( Fig. 1 ). Other proteins displayed altered thermal stability in intact cells, but only CRBN and GLRX3 did so in lysate. A higher concentration of pomalidomide was needed to shift GLRX3 as compared with CRBN in both K562 and iPSC. In the lysate format, most of the biology is turned off; that is, thermal shifts are more likely to be caused by direct target-ligand interactions and not by signaling events. GLRX3 is an [2Fe-2S] iron-sulphur-complex binding chaperone, 49 which is interesting when considering another frequent off target for small-molecule drugs: FECH. FECH has also been reported to bind [2Fe-2S] iron-sulphur complexes. 50 We found that BSJ-03-204 stabilized FECH in both intact cells and lysate, and FECH has been reported by others as an off-target for many small molecules, including kinase inhibitors.40,44,45 Moreover, even higher pomalidomide concentrations altered the thermal stability of several other redox-associated proteins. This may be in line with previous reports on antioxidative activity of IMiDs, 41 and teratogenic mode of action, in which increased levels of reactive oxygen species were detected upon thalidomide treatment. 51
Three of the proteins that had affected thermal stability in response to IMiD treatment belonged to the inosine synthesis pathway: IMPDH1, IMPDH2, and PFAS. The two formers were the therapeutic targets of the immunosuppressant drug mycophenolic acid. 52 Our finding that the thermal stability of IMPDH1 and IMPDH2 was affected downstream of IMiD treatment might account for the immunomodulatory effects of IMiDs. In addition, IMPDH expression is increased in neoplasms, 52 which indicates that the antineoplastic effects of IMiDs could in part stem from the modulation of IMPDH function.
We were able to confirm many of the previously identified IMiD neosubstrates in our experiments with iPSCs, EBs, and K562 cells. However, not all known neosubstrates were detected in the cell types under investigation. Notably, Ikaros protein (IKZF1) was found to be degraded upon pomalidomide treatment in the immortalized cancer cell line K562 ( Fig. 1 ) but was not detected in either iPSC or EBs, confirming the absence of IKZF1 gene expression in iPSCs and EBs. 43 Aiolos (IKZF3) is a protein related to Ikaros and is also reported to be a neosubstrate,13,15 but Aiolos was not identified in any of our experiments, most likely because of the low protein expression levels.
The time-dependent degradation profile revealed degradation of the intended targets of BSJ-03-204: CDK4 and CDK6. CRBN, on the other hand, showed a slight increase in line with the notion that IMiDs inhibit CRBN’s autoubiquitination. BSJ-03-204 also had a large impact on the levels of Kelch-like protein 8 (KLHL8), whose levels increased over time. In the nematode (Caenorhabditis elegans), a paralog of human KLHL8 (KEL-8) has been reported to function as a substrate adaptor together with Culin 3 (CUL3) containing E3 ligase (BTB-CUL3-RBX1), which controls ubiquitination and degradation of the protein Rapsyn. 53 Our data suggest that CRBN in turn may regulate ubiquitination and degradation of KLHL8. However, we did not detect the suggested KHLH8 substrate Rapsyn in K562 cells.
When comparing the PROTAC BSJ-03-204 with pomalidomide alone (
In conclusion, we have used CETSA MS in combination with quantitative proteomics to simultaneously study target engagement between protein targets and small-molecule degraders as well as the resulting degradation of prey proteins. This has proven effective, as it allows us to keep track of both target engagement as well as the downstream efficacy.
Regardless of modality, novel targets identified by our approach need to be confirmed and validated, because target engagement and subsequent decreased protein levels do not necessarily have to be dependent on the ubiquitin-proteasome system. For instance, target engagement could result in downstream, secondary effects that regulate protein synthesis (transcription and translation) or proteasome-independent degradation. Such effects could be investigated further by the inclusion of pharmacologic proteasome inhibitors in the assay.
For the IMiDs, our straightforward approach allowed us to identify the same primary target as well as downstream preys, as has been described collectively during the past 20 y of IMiD research, although using a plethora of other techniques. For PROTACs, the formation of a productive ternary complex is required for effective protein degradation. With the experimental setup used in this study, we provide a tool for those wanting to distinguish between productive binding (i.e., those inducing degradation) and silent targets (i.e., those proteins that bind only the molecule). A detailed study of cooperativity between binary and ternary complex formation, as well as characterization of steric hindrances to form fully functional ternary complexes, offered by different PROTACS has been beyond the scope of this study and possibly also beyond reach for CETSA MS studies.
Supplemental Material
sj-pdf-1-jbx-10.1177_2472555220984372 – Supplemental material for A Tale of Two Tails: Efficient Profiling of Protein Degraders by Specific Functional and Target Engagement Readouts
Supplemental material, sj-pdf-1-jbx-10.1177_2472555220984372 for A Tale of Two Tails: Efficient Profiling of Protein Degraders by Specific Functional and Target Engagement Readouts by Alexey L. Chernobrovkin, Cindy Cázares-Körner, Tomas Friman, Isabel Martin Caballero, Daniele Amadio and Daniel Martinez Molina in SLAS Discovery
Footnotes
Supplemental material is available online with this article.
Declaration of Conflicting Interests
The authors declared the following potential conflicts of interest with respect to the research, authorship, and/or publication of this article: D.M.M. is a co-founder and shareholder of Pelago and co-inventor of patents originating from PCT/GB2012/050853. All authors are employees of Pelago Bioscience AB, Sweden.
Funding
The authors received no financial support for the research, authorship, and/or publication of this article.
References
Supplementary Material
Please find the following supplemental material available below.
For Open Access articles published under a Creative Commons License, all supplemental material carries the same license as the article it is associated with.
For non-Open Access articles published, all supplemental material carries a non-exclusive license, and permission requests for re-use of supplemental material or any part of supplemental material shall be sent directly to the copyright owner as specified in the copyright notice associated with the article.