
Abstract
Targeted protein degradation (TPD) is a recent strategy, utilizing the cell’s proteostasis machinery to deplete specific proteins. This represents a paradigm shift in early drug discovery, away from occupancy-driven to event-driven mechanisms.
Recent efforts have focused on the development of proteolysis-targeting chimeras (PROTACs). These heterobifunctional molecules combine a target-specific binding moiety linked to an E3 ligase ligand and trigger selective ubiquitination of the target protein, marking it for proteasomal degradation. While these molecules can be highly efficacious, they generally have unfavorable physicochemical properties due to their large size.
In contrast, smaller molecules that induce degradation could represent an attractive, simple option to overcoming the limitations of both traditional modulators and PROTACs. These molecules may have a range of mechanisms: recruitment of an E3 ligase (molecular glues), introduction of hydrophobic areas, or inducing local unfolding, each of which triggers degradation.
We recently completed a high-throughput screen of 111,000 compounds in a cellular HiBiT assay in an effort to identify such molecules. Preliminary analysis indicates that we have been able to identify alternative small-molecule degraders. We highlight methods for triage, characterization, selectivity, and mode of action. In summary, we believe that these types of small-molecule degraders, which may possibly have more acceptable physicochemical properties than the inherently larger heterobifunctional molecules, are an exciting approach for inducing TPD, and we illustrate that a general screening approach can be successful in identifying useful start points for developing such molecules.
Introduction
Recent studies have suggested that up to 93% of the human proteome may be undruggable by conventional drug discovery efforts. 1 Historically, drug discovery has focused on targeting binding pockets or regions on target proteins with a range of modalities in order to modulate the biological function of that target protein. For example, traditional small molecules, antibodies, anticalins, peptides, and protein therapeutics bind to their target proteins and either inhibit a catalytic function or prevent or disrupt other essential biological interactions. Recently, further modalities have also been applied to prevent (e.g., using antisense oligonucleotides) or induce (e.g., using modified RNA) the expression of the target protein in the disease setting. Another modality that has seen extensive interest recently is targeted protein degradation (TPD), 2 whereby the target protein is reduced in concentration or completely removed from the cell due to the introduction of an active molecule. Typically, this involves appropriation of the cell’s internal protein removal systems, and some have suggested that this approach may allow for the treatment of conditions where target proteins have previously not responded well to small-molecule inhibitor intervention. There are two different mechanisms by which cells degrade proteins: the lysosomal and proteasomal pathways ( Fig. 1 ). To date, most work on targeted degradation has focused on engaging the ubiquitin proteasomal system (UPS). In this process, modification of target proteins by the addition of ubiquitin chains is integral for subsequent degradation. The small protein, ubiquitin, is first exchanged from an E1 ubiquitin-activating enzyme to an E2-conjugating enzyme, followed by ubiquitination of the protein by an E3 ubiquitin ligase. 3 Following several rounds of catalysis, the resulting polyubiquitin chain attaches to the protein signals for degradation via the 26S proteasome. In this perspective, we introduce the different ways targeted protein TPD degradation may be mediated with small molecules, comment on their utility, and suggest ways to utilize screening to identify further, novel ligands.
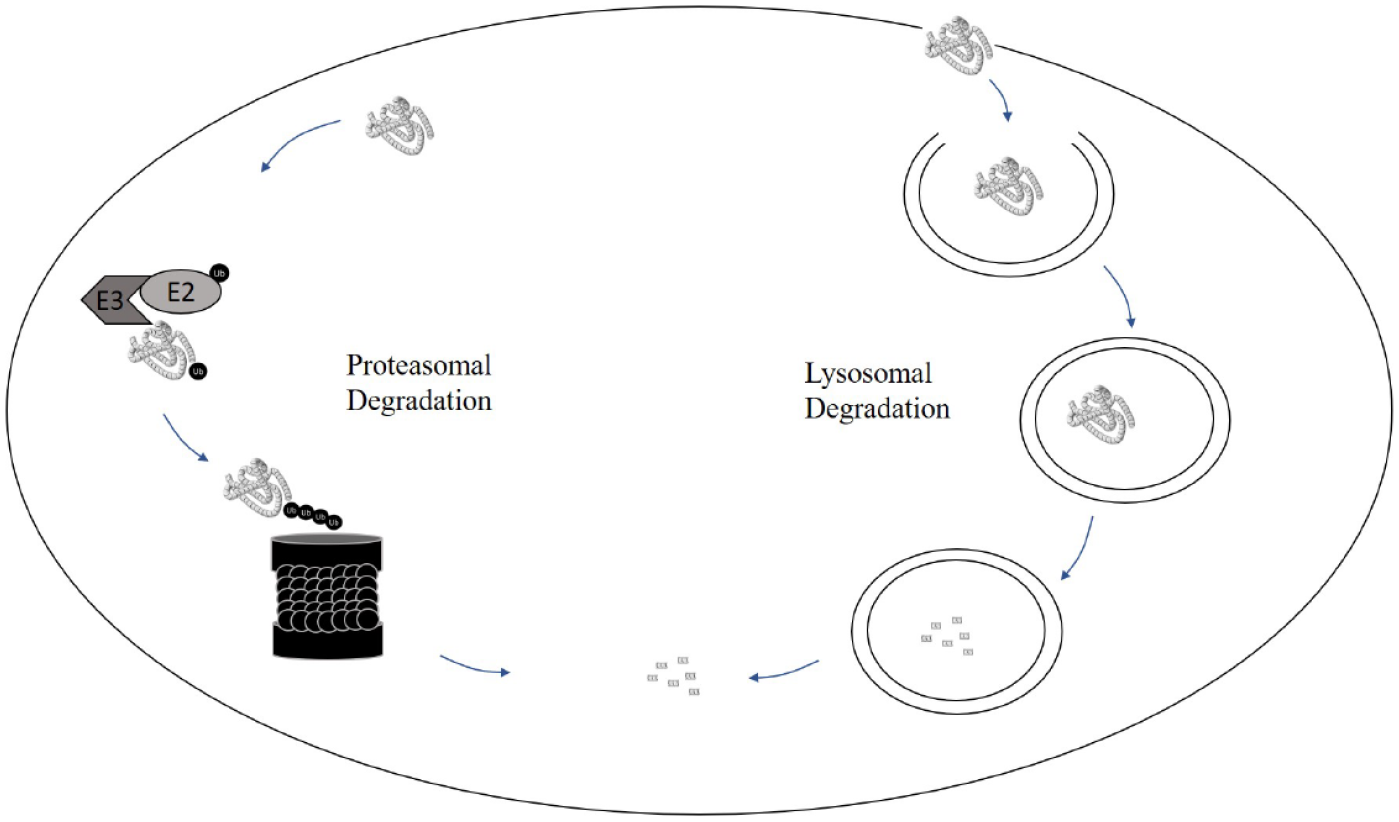
Mechanisms by which cells degrade proteins: the proteasomal and lysosomal pathways.
PROTACs
Perhaps, of the different types of chemical modalities that elicit target protein degradation, proteolysis-targeting chimeras (PROTACs) are the most well known. 4 PROTACs are heterobifunctional molecules containing two small-molecule ligands joined together by a linker ( Fig. 2 ). One of the small-molecule ligands is designed to bind to the target protein in the cell while the other binds with high affinity to an E3 ligase. By binding selectively to the target protein of interest and concomitantly binding to a specific E3 ligase, PROTACs are able to form a ternary complex, thereby maintaining the target protein and the E3 ligase in close proximity. Through the process described above, the E3 ligase links a polyubiquitin chain to the target protein and signals it for proteasomal degradation. Due to the bifunctional nature of these molecules and hence the necessity for combining ligands for both the target of interest and the desired E3 ligase, the physicochemical properties of PROTACs rarely conform to the well-known guidelines known as the rule-of-five. 5 While the physicochemical properties of these molecules are not ideal for oral administration, there are two PROTACs, both developed by Arvinas, currently undergoing clinical trials aimed at demonstrating therapeutic degradation of the androgen receptor (AR) and estrogen receptor (ER). 6 ARV-110 and ARV-471 are believed to target the Von Hippel–Lindau (VHL) E3 ligase. Although more than 600 different E3 ligases have been identified, 7 only for a small number of these proteins have suitable corresponding ligands been designed or discovered. This currently reduces the choice of E3 ligases utilized for PROTAC development, but also offers a wide range of further opportunity to access alternative E3 ligases, which may offer different tissue expression, and so offer additional therapeutic benefit. Alongside VHL, the most common examples of targeted E3 ligases include mouse double minute 2 (MDM2) and cereblon (CRBN). Since the first PROTAC, reported by the Crews and Deshaies laboratory in 2001, 8 which induced degradation of methionine aminopeptidase 2 (MetAP2) by recruitment of the SCFβ-TRCP E3 ligase, PROTACs for many diverse proteins have been reported, including classes such as kinases, BET proteins, and nuclear receptors.9,10 It is clear that PROTACs promise to have a major impact on the discovery of new medicines, with consideration of developing PROTACs now forming a part of many lead generation strategies. However, there are still some important aspects that will need to be addressed before we see PROTACs becoming commonplace in the pharmacy. Understanding which proteins are amenable and which may be refractory to the plethora of different E3 ligases is a key consideration, which may require a large matrix-based approach to define target and disease context sensitivity. Refining our ability to design linkers to allow the efficient formation of the functional ternary complex is also paramount. Not only does the linker allow correct positioning of the target protein and E3 ligase ligands, but also it has a major influence on the molecular properties of the resulting PROTAC, including cellular uptake, stability, and solubility. Finally, we await the results of the first clinical trials to really understand the efficacy of PROTACs in the in vivo disease setting. If the high cellular potency translates to well-tolerated clinical doses, protein reduction may truly become a highly effective strategy offering real benefit to patients.
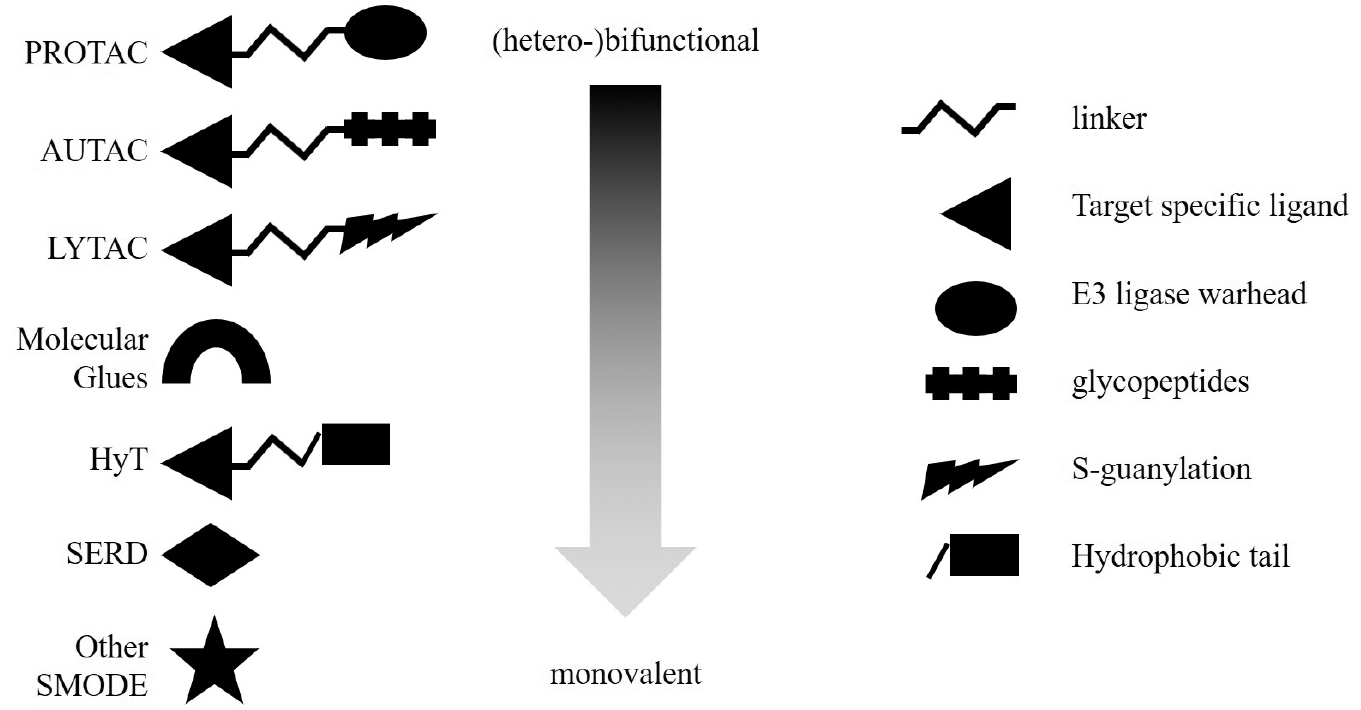
An illustration depicting the composition of various types of molecules that may induce TPD. The heterobifunctional molecules contain a target-specific ligand, a linker moiety, and an additional group that initiates the degradation process, usually by a further recognition process. The spectrum continues to different variations of bifunctional and monovalent types of degrader molecules.
AUTACs
Another approach that has been reported recently 11 is based on autophagy-targeting chimeras (AUTACs). These make use of the intracellular autophagic degradation pathway, where small, membrane-bound vesicles assemble and engulf material for degradation, via the lysosome. The composition of AUTACs links a specific ligand for a protein of interest to a guanine tag ( Fig. 2 ). The introduction of the guanine identifies the target for autophagy, and the presence of the guanine group has been shown to be sufficient to induce selective autophagy using halo-tagged introduction of S-guanylation into enhanced green fluorescent protein (EGFP) as a model system. Subsequently, AUTACs targeting MetAP2 and FK506 binding protein (FKBP12) were shown to be effective in degrading the respective target proteins. A potentially attractive advantage for AUTACs is their ability to degrade large and insoluble matter, for example, fragmented mitochondria, within the cell. Theoretically, at least, AUTACs may offer a mechanism to remove protein aggregates, which may prescribe their use for neurodegenerative diseases. While an exciting modality, a potential drawback with AUTACs is that they would not be expected to function within the cell nucleus, since the autophagic system is not active there. However, an AUTAC designed versus the BET family protein, BRD4, which shows nuclear localization, was also effective in reducing the BRD4 concentration, presumably due to the breakdown of the nuclear membrane during mitosis, resulting in the release of BRD4 into the cytoplasm.
LYTACs
So far, the approaches for TPD discussed above involve degradation of intracellular proteins. An extension of the approach to the AUTAC approach above explores degradation outside of the cell, again by engaging the lysosomal trafficking system. The so-called lysosome-targeting chimeras (LYTACs) are bifunctional molecules that bind the extracellular domain of a target protein and a cell surface lysosome-shuttling receptor in order to target the protein to the lysosome. 12 Initial LYTACs have employed antibodies linked to glycopeptide agonists of the cation-independent mannose-6-phosphate receptor (CI-M6PR). Proof of principle has been shown for LYTACs using cetuximab, an antibody targeting EGFR, combined with glycopolypeptides containing multiple serine-O-mannose-6-phosphonate (M6Pn) residues, where degradation levels of greater than 70% were observed. The approach also has been applied to PD-L1, where chaperone-mediated recycling has been shown to return the receptor to the cell surface. However, the results suggest that the system can prevent such recycling and lead to up to 50% degradation of PD-L1. As yet, there are no reported small-molecule-based LYTACs, and so the resulting molecules are extremely large, resulting from the linkage of the antibody to the M6Pn (often up to 90-mer). LYTACs face similar pharmacological challenges to those of PROTACs and AUTACs, and their utility may be improved if small-molecule ligands can replace the current antibody targeting strategy and/or the lysosome recruitment achieved using smaller ligands than the current glycopeptides. Of course, even then, the physicochemical properties may still pose a challenge for drug development, as the two ligand functional moieties will still ultimately need to be productively linked.
Molecular Glues
The term “molecular glues” generally describes small molecules that act by inducing or enhancing a protein–protein interaction (PPI). In the context of TPD, the term means ligands that, similarly to PROTACs, can facilitate close proximity of an E3 ligase and a protein-of-interest (POI), thereby inducing POI ubiquitination and subsequent degradation. 13 Probably the most prominent example of this class of degraders is thalidomide, a drug that was marketed drug for many decades before its mechanism of action was revealed, only a few years ago. Thalidomide was shown to bind to the CRL4CRBN E3 ligase and modulate substrate recognition, leading to the recruitment of neosubstrates to the E3 ligase complex. 14 Related molecules like ImiDs and cereblon E3 ligase-modulating drugs (CELMoDs) have since been identified and characterized and promise more flexibility and specificity of proteins targeted by this mechanism. 15 The anticancer agent indisulam was also only recently revealed to act as a molecular glue. Initially identified in a phenotypic screen for its antitumor activity, 16 Han et al. showed that it was the recruitment of the splicing factor RBM39 to the Cul4-DCAF15 E3 ligase complex that leads to its ubiquitination and subsequent degradation. 17 This, in turn, causes a multitude of pre-mRNA splicing aberrations.
It remains unclear how universal or limited this approach is with respect to target space and drug development, as similarly to PROTACs, so far it has only been applied to a limited number of different E3 ligases. An interesting variation of the molecular glue mechanism is the enhancement of natural E3 ligase–substrate interactions as recently described 18 for a mutant form of β-catenin binding to SCFβ-TrCP. In this case, ternary complex formation might be less of a challenge because an innate PPI rather than a new interaction surface is formed. den Besten and Lipford 19 have recently highlighted approaches correlating the drug sensitivity of clinical and preclinical drugs with the expression of ubiquitin ligase components as a way of searching for molecular glue behavior.
Mayor-Ruiz et al. recently described an elegant approach utilizing a hyponeddylated cell line to identify novel molecular glue-type degraders. 20 They profiled ~2000 known cytotoxic/cytostatic compounds on both an E2 mutated and a wild-type cell line and followed up on compounds with differential activity with a CRISPR-Cas9 screen focused on Cullin ring ligases and their regulators. They not only successfully validated their strategy by identifying a molecule that, similarly to indisulam, induced RBM39 degradation by associating with CRL4-DCAF15, but also found novel degraders promoting Cdk12-CyclinK recruitment to a CRL4B ligase complex.
Slabicki et al. applied a similar approach with a bioinformatics- based starting point probing data for a set of clinical/preclinical drug candidates for correlations of toxicity and E3 ligase expression. 21 Interestingly, they also identified a CDK12 inhibitor, CR8, as a selective cyclin K degrader and subsequently revealed that inhibitor binding turns CDK12 into a substrate receptor of the Cul4 E3 ligase complex. This leads to specific recruitment of cyclin K, which is ubiquitylated and consequently degraded. The authors of this study propose that these de novo interactions induced by target binding small molecules could be another strategy to develop a greater variety of molecular glue-type degraders.
SERDs/SARDs
Selective estrogen receptor degraders (SERDs) are a group of molecules that were initially identified and characterized as antagonists of estrogen receptor α (ERα). 22 Up to 75% of all breast cancers are ER positive, and drugs modulating the function of this nuclear hormone receptor are therefore the predominant treatment strategy for these cancers. Historically, the SERDs are part of a much larger classification of ERα effector molecules termed the selective estrogen receptor modulators (SERMs), which encompasses all of the various modalities. It took nearly 20 years to understand that SERDs, initially labeled as “downregulators,” are actually acting by induced degradation of ERα in cells, 23 and Wu et al. 24 finally showed that SERDs, upon binding, induce a conformational change in ERα that leads to increased external hydrophobicity and, consequently, identification by cellular degradation systems. In 2002, fulvestrant (Faslodex, AstraZeneca, Alderley Park, UK) was the first SERD to be approved by the U.S. Food and Drug Administration (FDA). 25 and several structurally diverse SERDs have been under clinical investigation since then. 26 Inspired by the success of SERDs, Bradbury et al.27,28 focused their attention on finding molecules that had a similar effect on the AR. However, AZD3514 was shown to decrease the synthesis of AR rather than inducing degradation, 29 making it a true downregulator rather than a degrader molecule.
Hydrophobic Tagging
Another related approach, possibly inspired by the hydrophobic nature of fulvestrant, which has been used to produce specific targeted degradation, is hydrophobic tagging. 30 This utilizes known high-affinity binders to a target protein with the addition of a chemical modification introducing a hydrophobic “degradation tail” to make a bifunctional molecule ( Fig. 2 ). Typically, these molecules contain an additional carbon linker that terminates in one of several different bulky side groups, such as adamantane or tert-butyl carbamate-protected arginine (Boc3Arg). The creation of these molecules was inspired by the recognition that surface-exposed hydrophobic patches of proteins may be recognized by the cell’s protein quality control machinery. These hydrophobic moieties may induce local regions of protein unfolding, significant protein structural changes, and ultimately protein destabilization. Alternatively, these groups may mimic hydrophobic patches of unfolded regions of proteins, triggering recognition by the unfolded protein response (UPR), leading to proteasomal degradation.
These mechanisms may be similar to the fulvestrant-induced surface hydrophobicity, leading to the degradation of ER (see the “SERDs/SARDs” section). Similar to the construction of PROTACs, hydrophobic tags (HyTs) can potentially be constructed using a toolbox approach, where known ligands for target proteins can be combined with different hydrophobic moieties to produce a range of putative degraders. The first HyT was targeted to both cytosolic and transmembrane HaloTag fusion proteins exploiting adamantyl–chloroalkane linked molecules. 31 The intractable target HRas was combined with the HaloTag developed by Promega (Madison, WI) and used in proof-of-concept (PoC) experiments for testing a range of distinct scaffold linkers with a broad selection of potential hydrophobic degrader moieties. A range of structurally distinct HyT degrader moieties were created to maximize their hydrophobicity (scores ranging from cLogP 3 to 5). They enabled a broad range of chemical diversity, included benzylic, ademantyl, tricyclic, and cyclohexyl bulky side groups that were linked to a varying range of HaloTag haloalkane reactive linker molecules.31,32
The chemical structures of example HyT degrader ligands are shown in Figure 1 of both Neklesa et al. 31 and Wang et al. 32 It was clearly demonstrated that the ademantyl fused molecule HyT13 had the ability to degrade the engineered HaloTag-HRAS (G12V) protein. 31 The HyT molecule was able to remain in mice for 24 h and be sufficient to inhibit the progression of HaloTag-HRAS (G12V) driven tumour growth by 80%. Additionally, association with HSP70 in HaloTag-HRAS (G12V) immuno-precipitation studies indicates HSP70’s possible involvement in recognition in adamantane-mediated degradation.33
Since the PoC of this approach promoting protein degradation, its utility has been exemplified by the identification of the first-in-class EZH2 selective degrader, which has been demonstrated to reduce EZH2 levels in cells. 34 This molecule has since demonstrated in vivo efficacy and shows promising therapeutic potential. Another successful example of the deployment of an adamantane-derived HyT was the degradation of the erythroblastosis oncogene B3 (ErbB3/HER3), a pseudokinase considered to be undruggable by conventional inhibition strategies, being a pseudokinase scaffold protein. 35 An ATP site-binding compound identified in a primary screen, optimized for affinity by addition of an acrylate group, was further modified by the introduction of an adamantyl moiety to create the HyTag degrader TX2-121-1. The degradation efficacy was, however, still found to be strongly dependent upon the covalent interaction.
HyTs based on the large Boc3Arg group are similarly described, with the first being exemplified to target gluthathione-S-transferase (GST) by attachment to ethacrynic acid (EA), which alkylates the active cystine in GST. 36 This compound demonstrated clear degradation with a 30%−80% removal of GST within 2–5 h. The major benefit of the HyT approach is that only a ligand for the target of interest needs to be discovered or developed. There is no requirement for a specific ligand targeting a designated E3 ligase, and therefore no necessity for ternary complex formation and issues with the notorious hook effect. However, the molecular characteristics that drive the efficacy of the linker chain combination with the hydrophobic moiety, in order to induce effective degradation, are not well understood. A further concern is raised by the hydrophobic nature of these large molecules and whether adequate selectivity can be sufficiently retained, driven by the molecular properties of the target-directed ligand functionality. Even if selectivity can be achieved, a further issue may be preferential binding of the hydrophobic component to serum proteins, which may limit bioavailability. It is likely that different classes of target protein will vary in their susceptibility to successful targeting by the hydrophobic tagging approach. Identifying and prioritizing those protein targets that are more sensitive to this approach will be important. For example, targeting proteins where significant ligand-induced conformational changes have been observed, suggesting that HyTags could induce exposure of degradation signaling motifs, may be prudent. Additionally, understanding how modification to the posttranslational state of the correctly folded target may occur, or interfering with requirements for certain PPIs, for example, with molecular chaperones, may represent another favorable strategy. 37
Unbiased Screening Approaches for Small-Molecule Protein Degraders
The modular character of bivalent molecules like PROTACs holds the tempting promise that a specific degrader molecule for any target protein could be built by simply attaching a target-specific ligand to an E3 ligase warhead. However, we so far have no evidence to suggest how universal this approach will be to both “to-be-degraded proteins” and different E3 ligase complexes. We will also need to explore how efficient ternary complex formation and/or off-target effects will hinder the development of efficacious and safe PROTAC drugs—a further challenge overcoming the issues presented by the often unfavorable physicochemical properties of these relatively large molecules. Understanding how to overcome these challenges will be critically important in allowing the systematic transition from optimized leads to clinically validated compounds.
We therefore propose that a more balanced approach to all possible degrader modalities be taken at the beginning of all hit-finding campaigns with the aim of complementing a potentially limited PROTAC angle on projects for which degradation is the preferred MoA. Interestingly, the most successful low-molecular-weight degraders known to date have been identified by chance, and the mechanism by which they act in cells has only been elucidated retrospectively. For example, several kinase inhibitors have been shown to bring about proteasomal degradation of their target kinases, or to interfere with interactions with molecular chaperones such as HSP-90 Cdc37. 12 As previously discussed, fulvestrant was first described as a “downregulator,” and only several years later was it shown to elicit this effect by locally destabilizing ER, inducing its degradation. In a similar serendipitous manner, the sulfonamide anticancer drug indisulam was very recently characterized as a “molecular glue,” having its effect by mediating the binding interaction between RBM39 and the E3 ligase DCAF15.
The history of thalidomide highlights the risk associated with a lack of mechanistic understanding, which would have potentially enabled an understanding of the teratogenic side effects ahead of treatment of pregnant women. In light of these examples, it is intriguing to speculate what proportion of previously identified binders and enzymatic inhibitors are actually acting by inducing degradation of their target in cells. Examples like the subset of BCL6 inhibitors from FP-based high-throughput screening (HTS) for binders not only show that this indeed seems feasible, but also highlight how a better mechanistic understanding could drive prioritization and optimization of chemistry efforts. 38
A high-throughput cellular assay that is able to detect all possible degradation MoAs ( Fig. 3 ) is therefore required to allow both profiling of hit outputs from biochemical screening campaigns and the de novo identification of cellular degraders from diversity libraries. While biochemical starting points provide initial knowledge on target engagement, screening of diversity libraries opens up unlimited opportunities for novel MoAs of small-molecule degraders (SMODEs) ( Fig. 2 ) and vastly expands the potential to discover degrader molecules with truly novel chemistry ( Table 1 ).
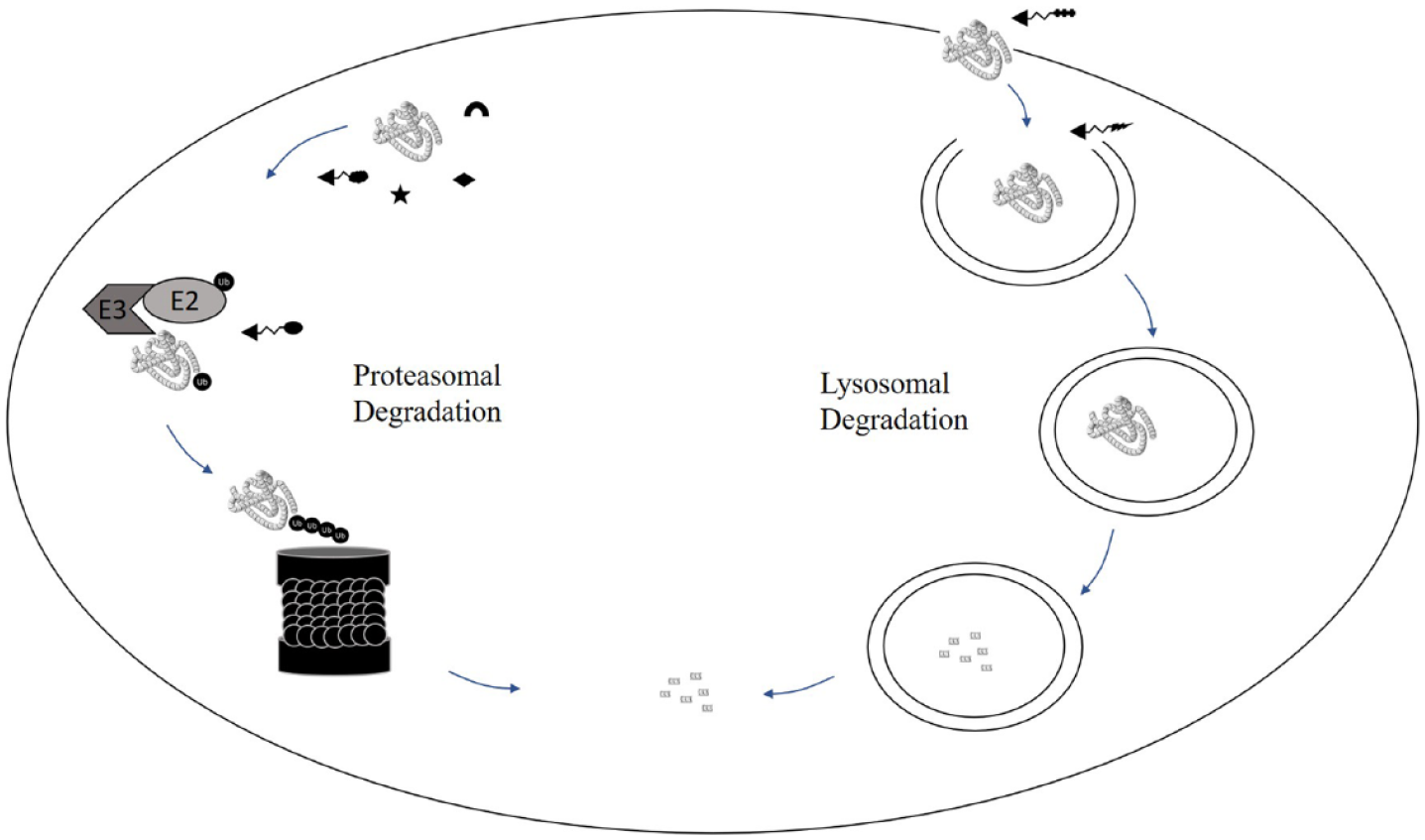
Schematic illustrating the likely sites of targeting for the different degrader modalities.
A Guide to Inform on a Degrader Strategy and When to Choose Different Degrader Modalities.
We have recently used Promega’s Nano-Glo HiBiT Lytic Detection System to build a high-throughput amenable assay to enable screening of larger numbers of compounds in an unbiased approach. CRISPR technology allows HiBiT tagging of endogenous proteins and offers an attractive alternative to immunofluorescent (IF)-based assay technologies that can be limited by the availability of specific antibodies or epitope masking upon small-molecule binding. Furthermore, the high reagent cost and multistep workflow of IF assays can make high-throughput applications and automation challenging. While the generation of CRISPR knock in (KI) HiBiT cell lines is still relatively time-consuming and the approach limits the initial characterization to only one cell line, we found that this assay technology is very sensitive and amenable to 1536-well format miniaturization and automation. This method enabled us to successfully screen a library of more than 110,000 compounds in less than 3 working days. Using a high-value oncology target as a PoC model system, we screened a diversity collection as well as a focused set of confirmed binders to the target previously identified in a 1.7-million-compound competitive binding screen.
Prior to the primary screen assay, robustness and reproducibility were evaluated by testing a smaller diversity subset on two separate occasions. Figure 4 shows data normalized to a target-specific PROTAC scale control plotted using a Bland–Altman analysis. A set of target-specific PROTACs (stars) served as standards to confirm the identification of putative degraders, and a set of known cytotoxic compounds (plus sign) were added to the validation experiment. The focused set of confirmed binders from the previous competitive binding HTS (triangles) were added to provide a greater likelihood of obtaining degradation alongside the likely lower probability for degradation from within the diversity set (circles). The assay proved to be well reproducible (95% confidence intervals ± 20%; see dotted lines in Fig. 4 ) and reliably identified known degraders as well as producing hits from the focused and diversity sets. The data also suggested that cytotoxic effects of compounds would indeed be a potential source for false positives and may significantly contaminate the output from larger screens, and we subsequently decided to add a cell viability assay to the first layer of the screening cascade. Furthermore, we included an artifact assay using a HiBiT-tagged control protein to assess whether compounds affected the detection system, for example, by directly inhibiting luciferase activity ( Fig. 5 , option 1). If specific antibodies are available, an IF assay at this stage could replace toxicity and artifact assays by providing an orthogonal readout and being amenable to multiplexing of several readouts ( Fig. 5 , option 2). This alternative approach could also include a specific selectivity readout, but care should be taken in evaluating potentially different sensitivities in this assay format.
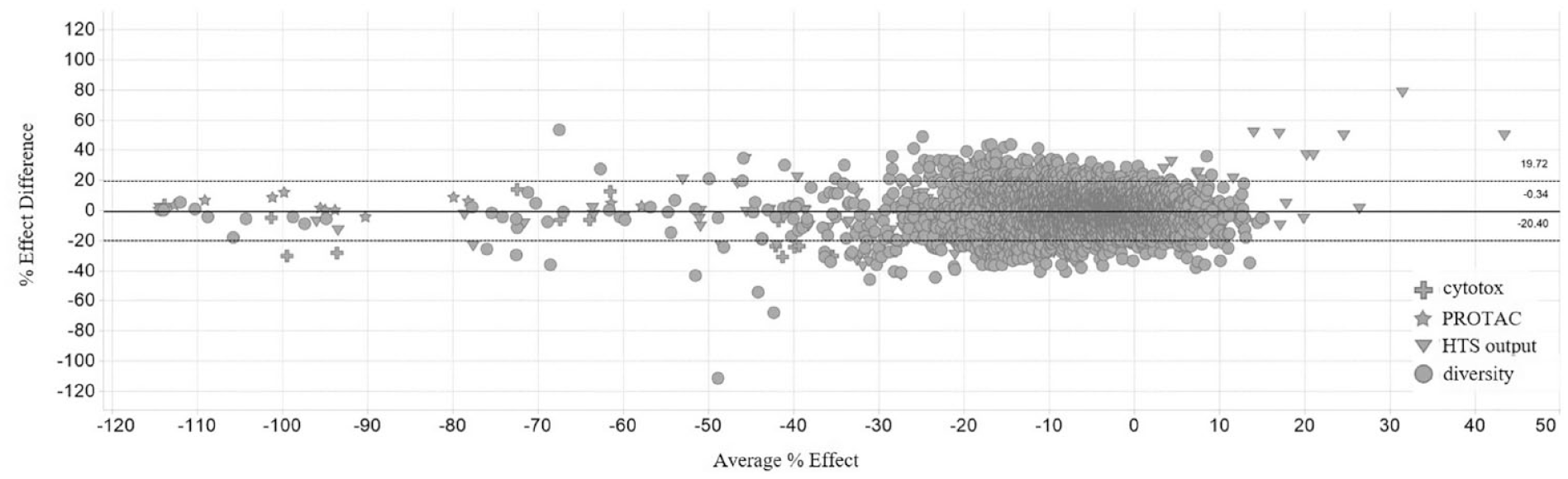
Bland–Altman plot visualizing the data for experiments performed on two separate occasions to assess assay reproducibility and robustness. Known cytotoxic compounds (plus) and target-specific PROTACs (star) are clearly identified as hit molecules. The focused set of confirmed binders to the target protein (triangle) and the validation diversity compounds (circle) were additionally profiled at this stage. The solid line indicates average, and the dotted lines describe 95% confidence intervals.
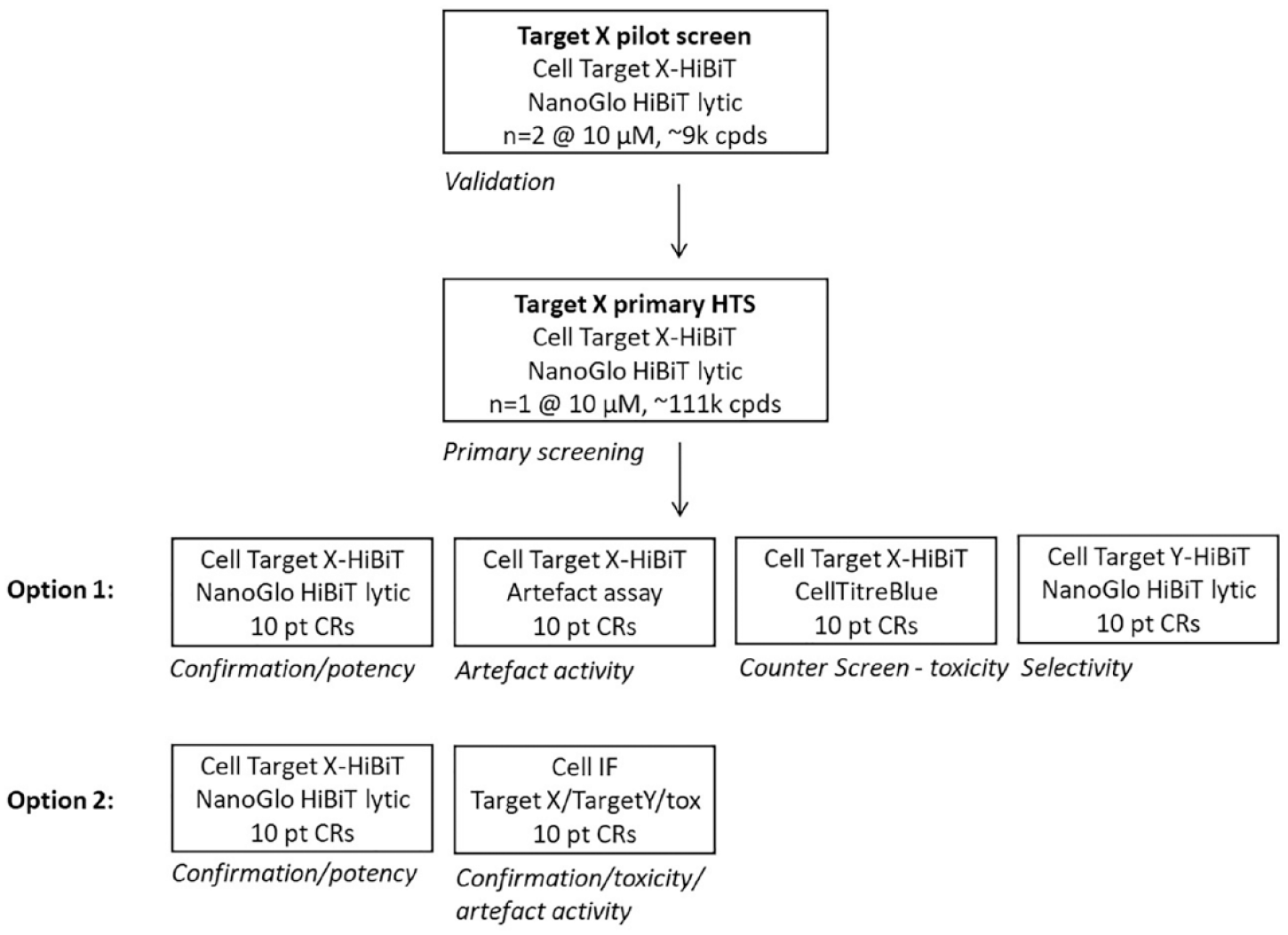
Potential screening cascade options when starting hit finding with a phenotypic HiBiT primary assay. Options 1 and 2 can be adapted based on selectivity requirements, availability of specific antibodies, and cell lines.
During triage of the primary screen, we found that ~85% of confirmed primary hits (>35% activity) were either toxic or active in the artifact assay, indicating that these assays provide a rapid and highly efficient strategy for the assessment of hits directly after the primary HTS. Comparing the outputs from the diversity library versus the focused set from the HTS output, we observed a ~20-fold enrichment with hit rates of 0.1% for the diversity set versus 2% for the identified binders, following the toxicity and artifact filters. These data suggest that a significant number of hits from biochemical screening approaches might indeed elicit degradation of their target in cells.
After this first, relatively simple triaging stage, the much more challenging part of mechanistic characterization has to follow. At this stage, it first needs to be assessed whether the selected compounds lower target levels in cells by inducing protein degradation or by modulating protein expression. To deconvolute these mechanisms, rescue experiments with different inhibitors probing the proteasomal or lysosomal cellular pathways for degradation can be set up. A reversal of the effect of a small molecule on the protein level of the target in these experiments could confirm protein degradation rather than effects on any other cellular pathway. Concomitantly, such experiments can be used to deconvolute the mechanism of degradation, provided that sufficiently specific inhibitors are available and toxicity at the tested time point is not limiting. Alternatively, focused CRISPR libraries could be used in an attempt to identify essential components in the degradation pathway induced by the small molecule by measuring reversal of the compound-induced effect. Mayor-Ruiz et al. successfully used this approach to deconvolute their small-scale screen. 20 However, the experimental procedure as well as the extensive data analysis might render it challenging for routine application in a screening cascade. Once it has been established that a compound is indeed inducing degradation of the target protein in cells, the question of whether this is via direct engagement with the target or an indirect mechanism arises.
Biophysical methods such as differential scanning fluorimetry (DSF), surface plasmon resonance (SPR), and other lower-throughput biophysical methods, including isothermal titration calorimetry (ITC) and SEC-MALS, may potentially provide the critically important information that is needed to confirm direct target engagement. These methods are likely to be challenging for the majority of degraders. For example, SERD-like degraders might destabilize their target upon binding, making direct measurement of ligand binding challenging. Target destabilization is additionally likely to make CETSA data difficult to interpret, and the known molecular glues do not even bind to their target in isolation with an affinity that could be measured by DSF or SPR. Also, the usual limitations for these methods arise: as they typically require the use of truncated isolated protein domains, they may not contain all of the recognition interaction structure of the full-length target found in cells. Similar challenges arise for targets that are part of large multicomponent complexes that are often impossible to reconstitute outside of the cell. For these reasons, positive confirmation of binding will provide additional annotation, but observation of a lack of binding cannot be used to exclude hits.
Apart from direct target engagement, the other big challenge is to determine selectivity and potential off-target effects of a putative SMODE. Probably the only way to fully elucidate off-target effects is a proteomics approach to measure the change in levels of mature proteins upon degrader treatment. 39 It will be critical to efficiently prioritize chemistry through a suite of different assays to select a small set of compounds from successful series for this type of analysis. It remains to be proven whether cell-based assays with phenotypic readouts are enough to drive chemistry efforts to establish an initial structure–activity relationship (SAR). This would be the most inclusive way to fully leverage the unbiased nature of the initial HiBiT degradation assay and would allow the identifications of novel, so far not described mechanisms of SMODEs.
Future Outlook
Preliminary data from our PoC work described in this publication suggest that an unbiased cell-based screening approach can successfully identify putative degraders from a diversity screening collection. We show how simple counter-screen strategies can be used to efficiently triage for toxic compounds as well as technology hitters and are currently working on further characterizing hits in terms of kinetics and mechanisms of degradation. Deconvoluting the output of such a cell-based phenotypic screen is with no doubt challenging, but tools like CRISPR screening or specific inhibitors might help to develop a pragmatic way to quickly prioritize the most promising chemistry. The strengths of this approach are that it is unbiased and inclusive of all possible MoAs of degraders ( Table 1 ) and generally applicable to all target classes. Even more, for targets where a biochemical approach to finding ligands as a starting point for a bifunctional degrader is not feasible, this approach might be the only option to identify a degrader. This might be the case for a number of relevant targets for which it is not possible to generate purified protein in a physiologically relevant form because of size limitations or them being part of large multiprotein complexes. An additional application of the HiBiT assay could be as an MoA annotation to profile outputs from biochemical and biophysical screens. Again, our preliminary data together with previous examples in the literature10,38 suggest that degradation in a cellular context is an MoA often found in biochemical screens. In either case, it will be exciting to see if degrader molecules with improved physiochemical properties are identified as starting points against more difficult targets in the portfolio and the degrader MoA becomes a routine complementary hit-finding strategy in hit identification.
Footnotes
Declaration of Conflicting Interests
The authors declared the following potential conflicts of interest with respect to the research, authorship, and/or publication of this article: All authors are employed by AstraZeneca, and their research and authorship of this article was completed within the scope of their employment with AstraZeneca.
Funding
The authors received no financial support for the research, authorship, and/or publication of this article.