
Abstract
The so-called undruggable space is an exciting area of potential growth for drug development. Undruggable proteins are defined as those unable to be targeted via conventional small molecule drugs. New modalities are being developed to potentially target these proteins. Targeted protein degradation (TPD) is one such new modality, which over the last two decades has moved from academia to industry. TPD makes use of the endogenous degradation machinery present in all cells, in which E3 ubiquitin ligases mark proteins for degradation via ubiquitin attachment. This session explored the challenges and perspectives of using protein degraders as novel therapeutic agents. The session began with a general introduction to the modality, followed by considerations in evaluating their on- and off-target toxicities including data from an IQ Consortium working group survey. Unique absorption, distribution, metabolism, and excretion (ADME) properties of degrader molecules were presented in relation to their effect on drug development and nonclinical safety assessment. The role of transgenic models in evaluating hemotoxicity associated with cereblon-based therapies was then discussed. A case study to derisk dose-limiting thrombocytopenia was also presented. Finally, a regulatory perspective on the challenges of having toxicity associated with protein degraders was presented.
Introduction
This article represents a combined synopsis of 6 presentations given at session 5 titled “protein degraders” at the 43rd Annual symposium of the Society of Toxicologic Pathology in Baltimore, Maryland, in June 2024. The theme of the symposium was titled “Innovative Therapeutics: Biology, Toxicologic Pathology, and Regulatory Perspectives.” Drs Clare Hoover from AstraZeneca and Kiran Palyada from Pfizer Inc co-chaired the session. Six subject matter experts provided a comprehensive overview of protein degraders, on- and off-target toxicities, absorption, distribution, metabolism, and excretion (ADME) properties of protein degraders, and a regulatory overview of this exciting modality.
History, Mechanism of Action, and Classes of Protein Degraders
Dr Renee Hukkanen, Pathology Director at Amgen, gave the introductory presentation to set the stage for the session. Her talk was titled “Protein Degraders for the Pathologist: A primer on Hijacking the proteasome.”
Targeted protein degraders (TPDs) are molecules that induce the destruction of a protein of interest (POI) by harnessing an endogenous protein degradation system, such as the ubiquitin proteasome system (UPS) or the lysosome. Because TPDs are not restricted by the need for a binding pocket, they are able to target proteins which are “undruggable” using traditional modalities. TPDs are one category of molecules within a developing field of drugs acting through induced proximity (i.e., bringing together POIs with effector agents, such as the UPS).1,29 Both TPDs and the larger field of induced proximity drugs are growing areas of research in which toxicologic pathologists play an increasing role.
Proximity-induced degradation has been explored for nearly 25 years since the description of the first synthetic TPD in 2001.1,24 The first TPD to enter clinical trials was ARV-110, a heterobifunctional cereblon (CRBN)-based degrader of the androgen receptor. ARV-110 was developed by Arvinas and entered a Phase ½ clinical study in 2019. Currently, there are more than 45 degrader molecules at clinical stage, targeting multiple ligases and various proteins involved in cancer, inflammation, and neurodegeneration. 24 The majority of TPDs in clinical trials are heterobifunctional small molecules (hSM) or molecular glues targeting CRBN or VHL ligases.
To understand the mechanism of action for TPDs, key terminology for two common categories is discussed: hSM, also known as proteolysis targeting chimeras (PROTACs), and molecular glues. Both hSMs and molecular glues utilize ligases to target POI for degradation; hSMs can alternatively utilize chaperones, members of the Heat Shock Protein 60/70/90 family (HSP), to ubiquitinate targeted proteins for degradation. 29
hSMs
The hSMs are larger TPDs that have two unique (hetero) sides connected by a linker (Figure 1). Each side is capable of binding to a separate target (bifunctional): the POI and a ubiquitination system. The linker connecting each side of the molecule may be of variable length and compositional rigidity, conferring stability and degradation efficiency to the hSM. There are more than 100 known linkers that use a variety of chemical approaches to overcome molecular size and polarity. 1 The target-binding domain, which may also be known as the warhead, lies on one side of the linker. The target-binding domain is specific to the POI, of which more than 400 are under investigation. 23 The POI may be extracellular, membrane-bound, or intracellular. The combination of the target-binding domain and the POI is known as a “neo”substrate because the POI is serving as a novel substrate for ubiquitination, in the context of the hSM. On the opposite side of the linker lies the effector domain, capable of engaging the ubiquitination system. In the case of the UPS, this would be the ligase (or HSP) binding domain. There are over 600 known ligases which function in normal cellular processes including metabolism, proliferation, apoptosis, DNA damage, or repair. 22 The distribution of each ligase is both tissue- and species-specific, with CRBN and VHL being the most commonly used by hSM. Ligases form through assembly of multiple catalytic subcomponents (E1, E2, and E3, for example) integrated into an active form, capable of transferring ubiquitin. The E3 component contains a substrate-binding domain specific for a targeting element of a protein that identifies it for degradation. The combination of the ligase, hSM, and POI is called the ternary complex. Upon formation of the ternary complex, the POI is ubiquitinated by the ligase, targeting it for degradation by the 26s proteosome.
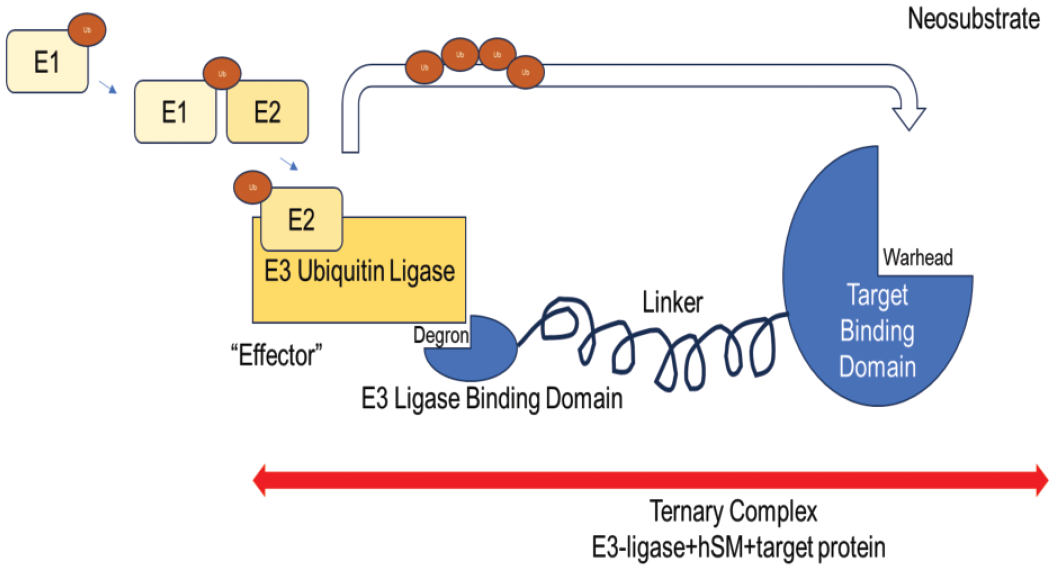
Heterobifunctional small molecules are targeted protein degraders capable of bringing an E3 ligase and protein of interest into close proximity, resulting in ternary complex formation and ubiquitination of the neosubstrate.
Molecular Glues
Molecular glues are smaller TPDs with far more favorable physiochemical properties (Figure 2). A molecular glue can directly join a POI to an effector (e.g., ligase). When a molecular glue binds a ligase, it causes conformational changes that promote binding of the POI. This neointeraction between the POI and the ligase forms a ternary complex, resulting in ubiquitination of the POI. 21
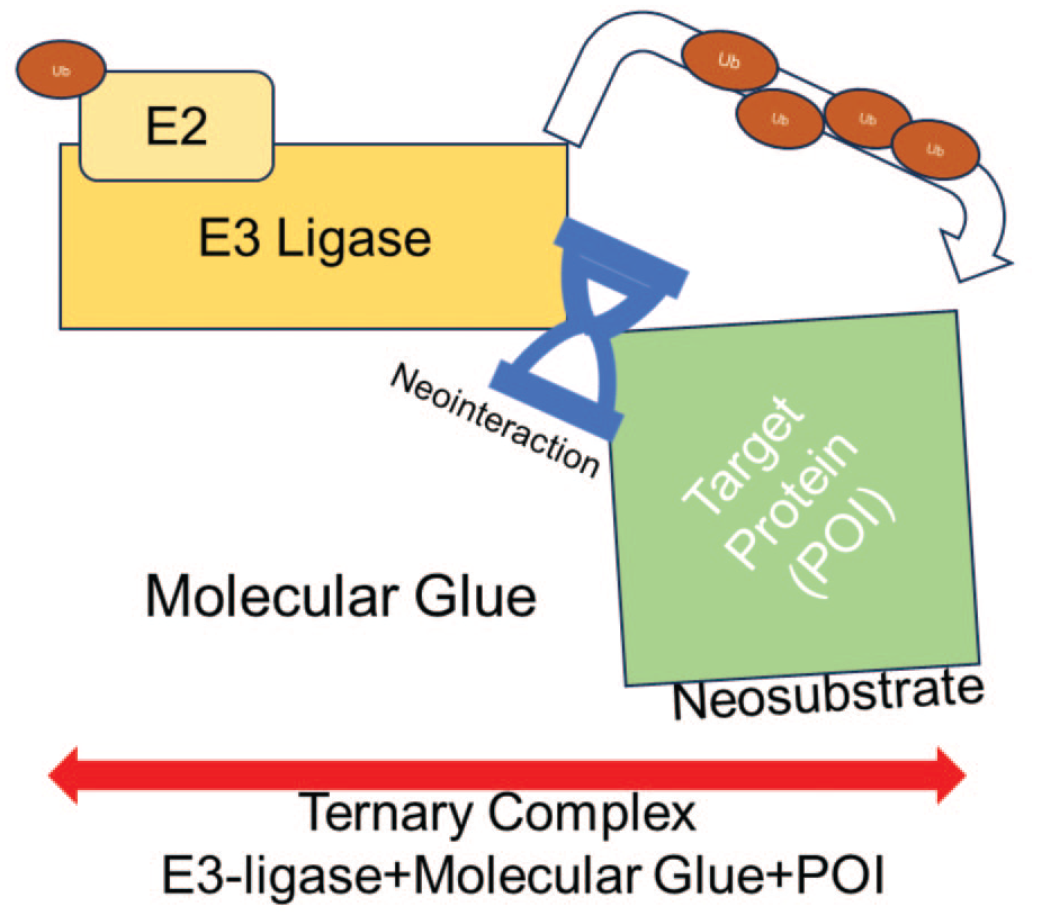
Molecular glues are targeted protein degraders capable of inducing a neointeraction between an E3 ligase and a neosubstrate, resulting in ternary complex formation and ubiquitination.
Proteosome
Regardless of how a POI is ubiquitinated, the next step in degradation is digestion via the 26S proteosome (Figure 3). The proteosome is a cytosolic proteolytic complex that recognizes proteins intended for degradation via a ubiquitin “tag.” The normal cellular function of the proteosome is to remove abnormal proteins, maintaining protein homeostasis and influencing the regulation of many cellular processes which depend on degradation (e.g., circadian rhythms). 10 TPDs are said to “hijack” the proteosome because the POI becomes a neosubstrate for proteolysis via proximity-induced ubiquitination.
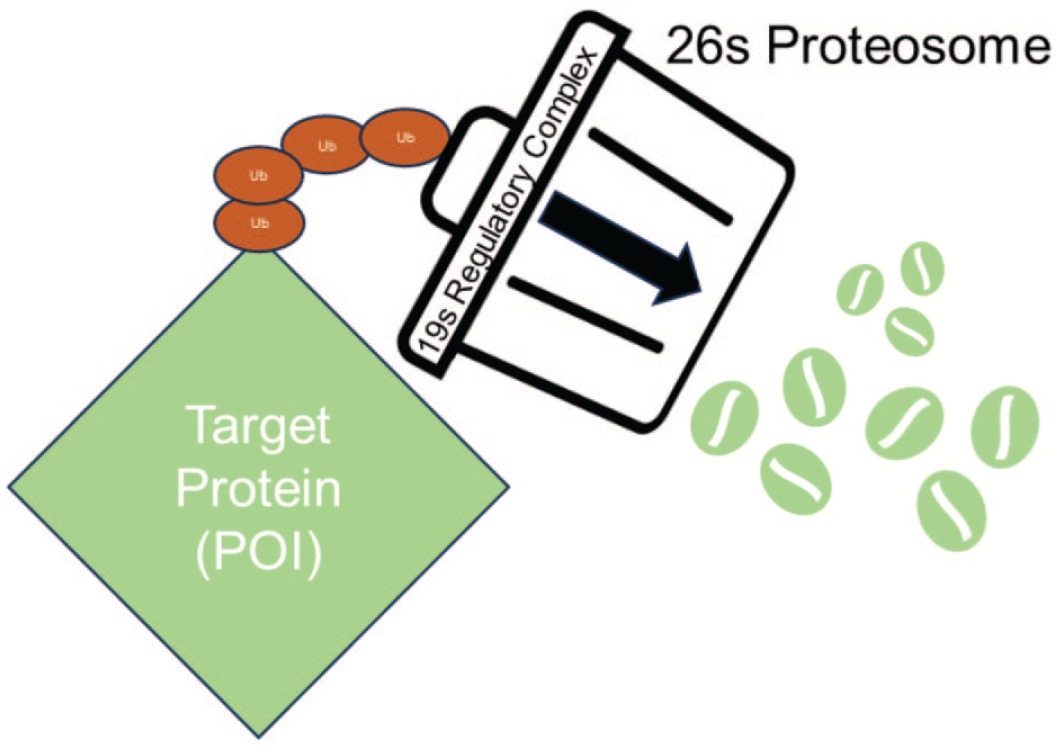
The 26S proteosome is a hollow, barrel-shaped complex of proteases which digest cytosolic (or nuclear) proteins. The 19S regulatory complex recognizes and removes ubiquitin tags and begins the process of protein unfolding and degradation as the protein moves through the proteosome.
The Next Generation of Proteosome-Targeting Degraders
Beyond hSM and molecular glues, there is an array of chimeric molecules using the proteosome to degrade POI. Chimeras have been developed with ligase-targeting degrons and warheads targeting RNA-binding sites or DNA nucleotides, such as transcription factors. Combining antibodies with TPDs provides further tissue specificity of degradation. Examples include proteolysis-targeting antibodies (targeting membrane-bound or cell surface proteins) and Antibody-drug conjugates (ADC) where the payload is a TPD.
Targeting the Lysosome
The concepts discussed above apply beyond TPDs which utilize the proteosome. Lysosomal-targeting degraders take advantage of cellular machinery including the endosome, phagocytosis, or autophagy (macro, micro, or chaperone-mediated) to degrade POI. TPDs using the lysosome for degradation include bifunctional molecules that bind an antibody or oligoglycopeptide transmembrane proteins to a POI (extracellular or transmembrane). Bispecific antibodies can also recruit membrane-bound ligases to degrade cell surface POI. Another approach is attaching a DNA aptamer to a POI and a cell surface lysosome-shuttling receptor (IGFIIR). As with chimeras targeting the proteosome, HSP can be utilized to target the lysosome. Chaperone-mediated autophagy targets members of the HSP to bring a POI into contact with an autophagolysosome. The potential to develop chimeric molecules is virtually unlimited, including examples such as the use of cell membrane–penetrating proteins, nanobodies, and tags linked to organelles. The reader is directed to review articles summarizing new technologies under development. 29
Proximity-Induced Protein Modifications: Beyond Degradation
Protein destruction via ligase or lysosomal activity is only one means of protein modification. Using post-translational methylation, phosphorylation, or allosteric changes, it is possible to modulate a protein’s function via activation, inactivation, or re-localization within the cell. Through all of these target outcomes, induced proximity molecules expand the druggable genome by leveraging the cell’s natural machinery.
Nonclinical Safety Considerations
Alongside the significant benefits afforded by TPDs, proximity-induced degraders pose unique drug development challenges and opportunities for nonclinical safety evaluation. The proteosome plays a critical function in cellular homeostasis; therefore, an excess of proteins labeled for destruction could potentially overwhelm the proteosome. Accumulation of normal cellular waste due to excessive intended (on-target) degradation is a theoretical liability of TPDs.
Off-Target Degradation
The specificity of TPDs must be exquisite to prevent ubiquitination and degradation of unintended proteins. Off-target degradation can occur with molecular glues capable of creating “neo”interactions between a ligase and multiple intended and unintended POIs. The most notable example of off-target degradation occurs with molecular glues targeting CRBN, specifically, thalidomide and structural/functional analogues (lenalidomide and pomalidomide), known as Immunomodulatory Drugs (IMiDs).5,9 The binding of IMiDs to CRBN and intended targets of IKAROS and AIOLOS, for example, results in efficacious treatments, including multiple myeloma and leprosy. Unfortunately, the binding of thalidomide to CRBN also results in unintended degradation of multiple proteins, including SALL4, a member of the spalt-like family of developmental transcription factors (Figure 4). SALL4 is responsible for gene expression and epigenetic modification of pluripotent cells. When it is absent during embryo-fetal development (EFD; specifically days 20-30 post fertilization), the lateral plate mesoderm does not differentiate, resulting in limb malformations known as phocomelia. Because of species-specific sequence variations, SALL4 is not degraded by IMiDs in rats, mice, or hamsters, but is degraded in rabbits and nonhuman primates (NHPs). Species-specific off-target degradation presents a teratogenicity liability at the platform level for CRBN-based TPDs. 18 Approaches to derisk CRBN-based TPDs include in vitro assays across species, SALL4 degradation screens, and performing in vivo embryo-fetal assays earlier in development process.
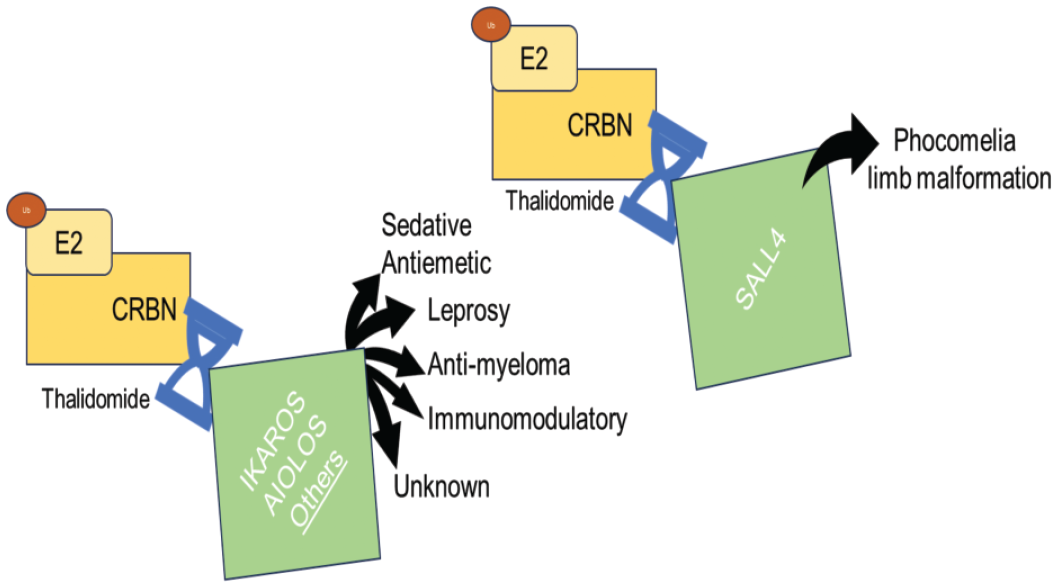
The neointeraction between thalidomide and cereblon E3 ligase promotes degradation of multiple proteins, including intended teraputic targets (IKAROS and AIOLOS) and unintended proteins (including SALL4 and p63).
Species Selection
Unlike the 1:1 ratio of traditional small molecules to their POI, TPDs rely on the interaction of three components: the ligase, the TPD, and the POI. Therefore, we need to understand patterns of expression, homology, and binding affinity across tissues and species for both the ligase and POI. Because the presence/activity of the ligase and POI may vary by species and tissue, the choice of nonclinical species for safety evaluation depends on the biological activity of the ternary complex. The biological effect of TPDs can be measured by various methods, such as Western blot, mass spectrometry, or functional assays, to assess the degree and duration of protein degradation. One common way of visualizing degradation across the cellular proteome is known as a volcano plot.
Pharmacology and Kinetics
The pharmacology of TPDs is event-driven, rather than occupancy-driven, based on a catalytic mode of action. Because one molecule can degrade multiple copies of the POI, efficacy can be achieved at low doses for an extended period of time. The duration of activity continues after metabolism of the TPD, to include the time required for cellular resynthesis of the POI. The POI and the degradation system are both required for efficacy; therefore, TPDs can be selectively active within specific tissues, cells, or disease states.
Unlike the linear kinetics of a traditional small molecule, the kinetics of TPDs are based on the concentration of four different molecules in equilibrium: free TPD, the ternary complex (ligase+TPD+POI), the TPD bound to the ligase alone, and the TPD bound to the POI alone. 15 At low concentrations or at a supersaturated state, single binding forms (ligase+TPD and/or TPD+POI) can predominate, with peak efficacy occurring at a mid-concentration range where the ternary complex (ligase+TPD+POI) predominates. A bell-shaped dose-response curve or “hook effect” may be observed in in vitro activity studies. Modifying the affinity of the warhead domain for the POI can reduce the hook effect. Importantly, the hook effect has not been observed in vivo.
Absorption, Distribution, Metabolism, and Excretion
The physiochemical properties of TPDs pose significant challenges for drug development. The ternary complex is a large (1.0 kDa on average for hSM) molecule with surface polarity, which does not conform to a set of in silico guidelines predicting high oral absorption, known as “the (Lipinksi’s) Rule-of-5.” 23 Solubility of most TPDs is low and oral bioavailability is limited. Phospholipidosis is associated with cationic and lipophilic compounds and may be a liability for some TPDs. Approaches to overcome physiochemical challenges are an investigative focus of the field. Examples of creative solutions include linker modifications to reduce molecular size and allow for dosing of molecular components separately (CLIPTAC), and nanoparticle delivery systems. 2
ADME and Nonclinical Safety Assessment of Protein Degraders
Dr Katie Stamp, a senior Director of Toxicology at Bristol Myers Squibb, gave a presentation titled “Nonclinical safety assessment of targeted protein degraders: An industry perspective.” Dr Stamp set the stage by summarizing the surveys from the International Consortium for Innovation and Quality in Pharmaceutical Development (IQ Consortium). Dr Laurie Volak, Director of DMPK at Rapport Therapeutics, presented on the DMPK issues when working with heterobifunctional protein degraders. The summary from Drs Stamp and Volak is given below.
Considering the theoretical ADME and safety challenges posed for TPDs (described above and in the work by Moreau et al 20 ), the International Consortium for Innovation and Quality in Pharmaceutical Development (IQ Consortium) Protein Degrader Working Group (WG) published the output of two online surveys that benchmarked current preclinical practices for TPDs in 2022.11,26 There were 18 respondents in total, none of which were working exclusively or primarily in degraders. Most companies had been working in the field for 5 years and were relatively early in drug development.
Of the perceived risks (based on those outlined in the work by Moreau et al 20 ), respondents ranked off-target protein degradation leading to toxicity and issues related to physical properties such as solubility and permeability as the greatest concerns. The hook effect was ranked of lowest concern, perhaps reflecting the fact it has been very rarely observed in vivo.
Considering in vitro safety screening, generally, the concepts are similar to typical small molecules with modifications including assessment of target protein degradation, extended assay duration, and the inclusion of additional assays to detect degradation of off-target proteins. Physical properties of heterobifunctional degraders such as low solubility, high protein binding, and nonspecific binding to plastics were factors limiting to in vitro safety assessments. Human cells are the most used species for in vitro off-target testing.
For in vivo safety assessment, concepts were again generally similar to standard small molecules with some modifications, such as assessment of on- and off-target protein degradation and extended duration of recovery. For selection of a toxicologically relevant species for this modality, the most important factors were assessment of target degradation in vitro that should be confirmed in vivo, and target and E3 ligase expression; the use of proteomics across cell types/species to determine relevance for both on- and off-targets should be considered. Importantly, when considering the relevant toxicology species, no companies default to the NHP as a toxicology species, even when CRBN is the ligase.
Modifications to developmental and reproductive toxicology (DART) assessment were more likely with CRBN ligase than others, which reflects the concern with potential teratogenicity associated with CRBN binders. A majority of respondents considered modifications to EFD assessment including species selection and/or earlier in vitro/in vivo studies and this was particularly prevalent for those targeting non-life-threatening indications.
There was limited regulatory/clinical experience among respondents, but to date, it appears that clinical plans and starting dose selection are similar to standard small molecules with the same therapeutic area.
The data captured generally focused on heterobifunctional CRBN-mediated TPDs and there are limited data at this stage to support the preclinical-clinical translation to aid in determination of best practice for the safety assessment of this modality. However, based on the data available, summarily, the nonclinical safety assessment for heterobifunctional degraders is similar to typical small molecules with some modifications in techniques used, assay conditions/study endpoints, and timing of assessments to address differences in mode of action of the class (Figure 5). Selection of a relevant toxicology species requires consideration for both the target protein and the E3 ligase and requires assessment of protein degradation. Off-target protein degradation is a primary concern, and when the ligase is CRBN, DART assessments are often modified including species selection, and earlier in vitro/in vivo studies.
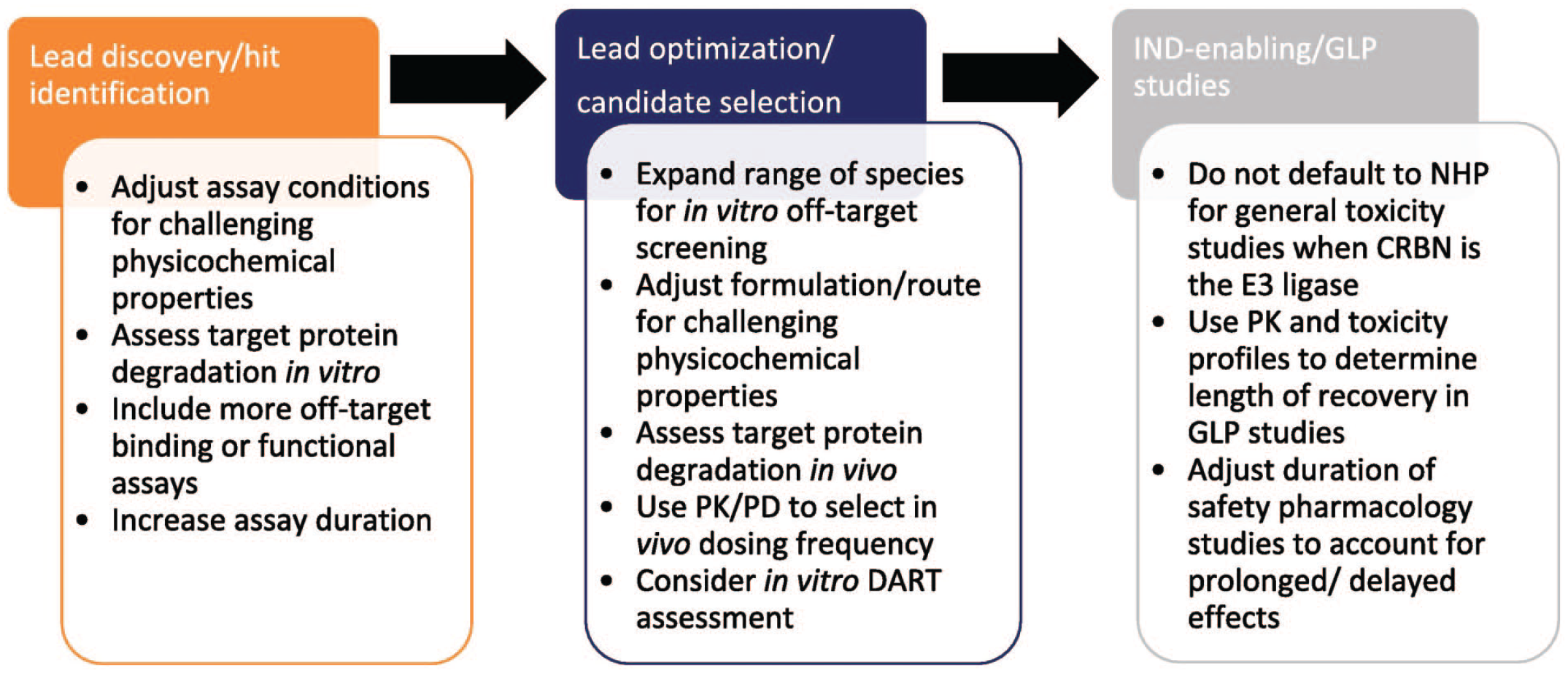
Considerations for the safety assessment of TPDs at different stages of development. CRBN indicates cereblon; DART, developmental and reproductive toxicology; GLP, good laboratory practice; IND, investigational new drug; NHP, nonhuman primate; PD, pharamacodynamics; PK, pharamcokinetics. (Reprinted with permission from Hemkens et al 11 )
Species selection and teratogenicity are persistent topics that warrant more efforts to determine best practices, particularly when the E3 ligase is CRBN. Conversely, the hook effect and proteosomal dysfunction appeared to be less impactful in safety assessment than originally hypothesized since effects have very rarely been observed in vivo.
The ADME concepts are similar to typical small molecules with some modifications related to the typical beyond rule-of-five properties of heterobifunctional degraders. These include additional assays such as proteomics and degradation assessments (with minimal protein in media) as part of the in vitro pharmacology; addition of chromatographic Log D, experimentally determined exposed polar surface area and MDCK permeability with protein modifiers as part of the in vitro ADME studies; microsomal metabolite identification (unless permeability allows use of hepatocytes), transporter substrate assessment with protein modifiers, and modified plasma protein-binding assessments as part of the in vivo/Tier 2 ADME; and finally, for the human dose projection, consideration of pharmacokinetics in the fed and fasted states.
An Overview of On- and Off-Target Toxicity of TPDs
Dr Richard Peterson, a Research Fellow at AbbVie, presented an overview of on- and off-target toxicity of TPDs. The toxicity seen with TPDs can be associated with on-target (degradation of target in normal tissues) and off-target (degradation of nontargeted proteins) mechanisms.
On-Target Toxicity
On-target toxicity with TPD in nondiseased tissues is not uncommon, especially in tissues where there is broad expression of the targeted E3-ligase (Table 1) (e.g., CRBN). On-target toxicity can be due to catalytic effects of degraders and/or prolonged target protein degradation.
The expression of the most common E3 ligases (i.e., CRBN and VHL) is generally consistent across NHP, dog, and rat.

- = no expression, + = low expression level, ++ = moderate expression level, +++ = high expression level.
The catalytic nature of heterobifunctional degraders can lead to on-target toxicity. After target protein degradation, the degrader molecule is released allowing it to bind to another target protein resulting in a catalytic process (i.e., sub-stoichiometrically induced ubiquitination). Degrader “recycling” results in a disconnect between pharmacodynamics and pharmacokinetics of the small molecule degrader. Benefits of the catalytic property of degraders include potentially increased efficacy, while downsides include safety risks due to prolonged protein degradation of on-target and potential off-target proteins and inability to accurately model the PK of degraders.
Depending on the target protein and function, prolonged degradation could lead to toxicity at the cell/organ level. The degrader mechanism of action/pharmacology is similar to that of an irreversible antagonist, where the pharmacodynamic response is prolonged over what would be anticipated from pharmacokinetic data (i.e., PK/PD disconnect).
The productive ternary complex formation occurs at the optimal degrader concentrations leading to degradation of the target protein. However, at higher degrader concentrations binary complexes predominate preventing the target ubiquitination and degradation leading to the “hook” effect (Figure 6). This could lead to increased neosubstrate degradation (i.e., decreased specificity) or potentially cause the degrader to act as an agonist/antagonist with associated toxicity. The onset and kinetics of the “hook” effect observed in in vitro experiments is likely different from what is observed in cell-based degradation assays and in vivo studies. The hook effect is likely more of a concern for efficacy than safety and has not been reported in vivo.
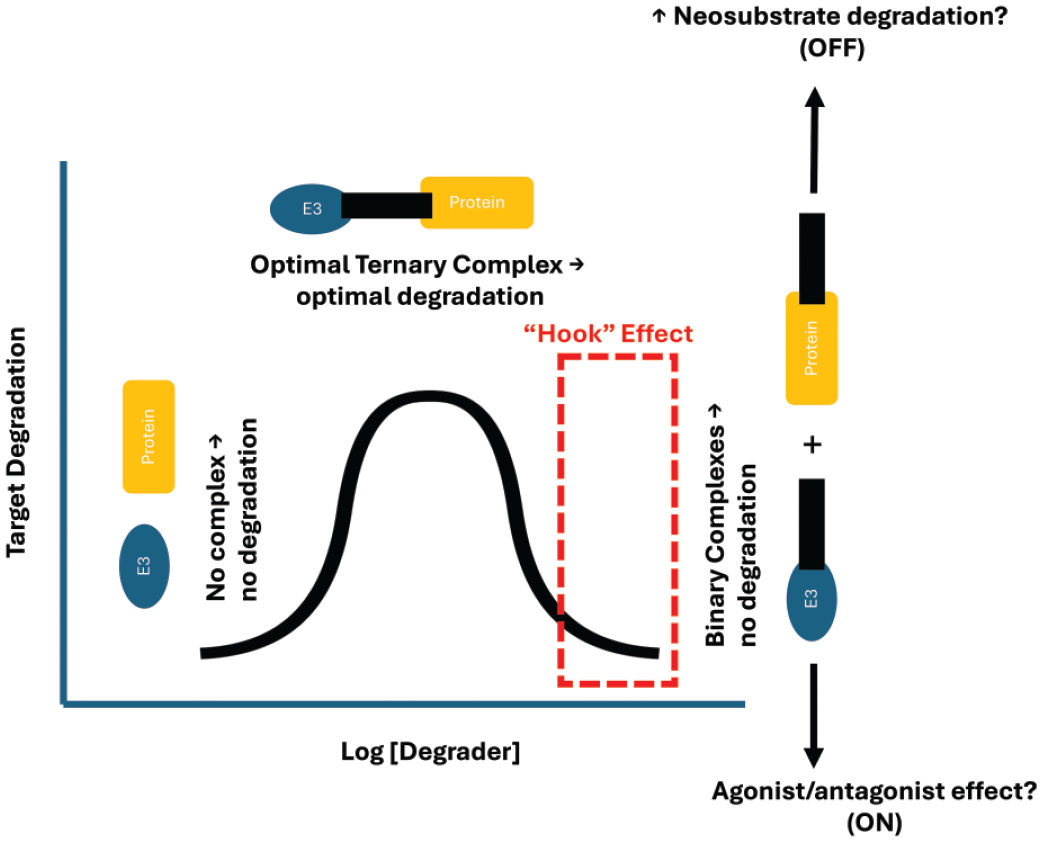
Representation of hook effect with protein degraders. Optimal degrader concentration leads to the formation of ternary complex leading to maximal degradation of the target. Higher concentration of degraders leads to the formation of excessive binary complexes with decreased target degradation and increased neosubstrate degradation. (Adapted from Moreau et al 20 )
Off-Target Toxicity
Off-target toxicity with TPDs can be associated with (1) selectivity, (2) proteasome involvement in MHC class 1 antigen presentation—possible autoimmune effects, and (3) physicochemical properties of the target-binding ligand (TBL), E3-ligase-binding ligand (LBL), and/or linker.
Degraders are not completely selective and can degrade other proteins. Degraders are small molecules prone to typical off-target binding/metabolism that should be assessed (e.g., Cerep/bioprofiling). (1) Neo-substrates: certain molecules (e.g., IMiDs [immunomodulatory drugs in the thalidomide series]) bind to E3 ligases (e.g., CRBN) and recruit substrates (e.g., “neosubstrates”; zinc finger transcription factors such as IKZF1 and IKZF3 [Ikaros/Aiolos family members: lymphocyte biology], and SALL4 [i.e., developmental transcription factor]) for ubiquitination and proteasomal degradation. (2) “Bystander degradation”: a protein that is not bound to the degrader, but in the general vicinity can induce neo protein-protein interactions and can be ubiquitinated and then degraded via the proteosome. (3) Binary engagement (vs ternary) of the TBL of the degrader to other proteins leading to proteosomal degradation. Examples of off-target degradation with potential safety concerns include selectivity-based (e.g., SMARCA-4 vs SMARCA-2; IRAK-4 vs IRAK-2) and neosubstrate degradation of SALL4/PLZF/p63 with degraders that bind to CRBN (i.e., teratogenicity concerns, etc.). Bioinformatic tools (in silico approaches) and proteomic tools such as TMT quantitation (11-plex Isobaric Tandem Mass Tag Labeling with analytical mass spectroscopy) and mass spectroscopy–based data-independent acquisition (DIA) analysis methods are valuable in identifying potential off-target proteins.
Proteasome involvement in MHC class 1 antigen presentation can lead to possible autoimmune effects. MHC class I peptides are derived from proteolytic degradation via the proteasome. Experimental BET-PROTAC treatment induces the presentation of specific MHC-I peptides from target bromodomain (BRD) proteins, as well as peptides from other proteins that could be mapped to known pathways associated with BET inhibition and degradation. This effect can be beneficial for targets involving adaptive immune response; however, this effect also has theoretical potential to result in immune reactions against the target and related proteins. To date, there has been no internal evidence of degrader-induced autoimmune effects.
Heterobifunctional degraders typically are in the beyond rule-of-five (bro5) chemical space (TBL, LBL, and linker structure) with cationic-amphiphilic physicochemical characteristics and are associated with increased incidence of phospholipidosis and pharmacokinetics-related tissue accumulation leading to toxicity (Table 2 and Figure 7).
Incidence of phospholipidosis with protein degraders across different species.

Phospholipidosis was observed in the foamy macrophages in the lung, liver, and lymphoid tissues.
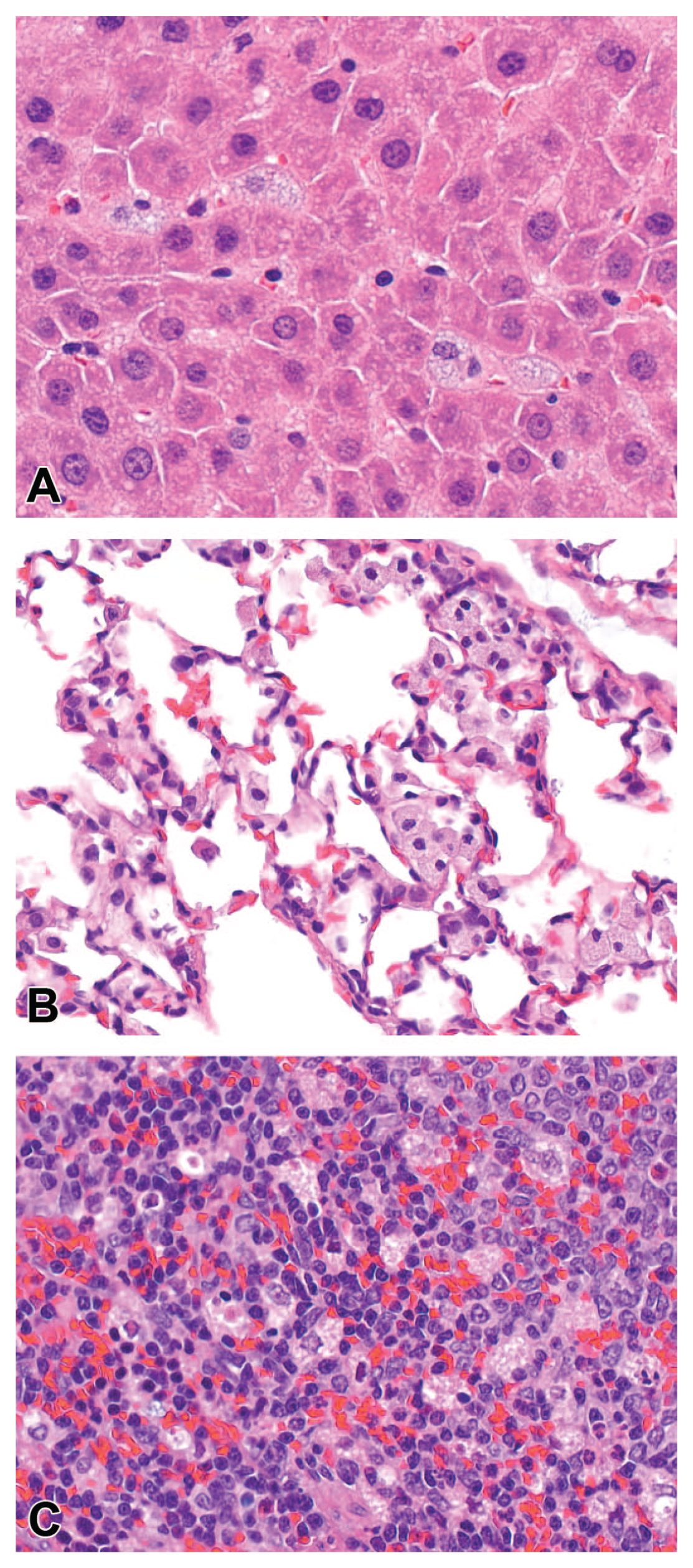
Phospholipidosis was observed in the liver (A), lung (B), and spleen (C) with protein degraders.
CRBN-Associated Teratogenicity
CRBN neosubstrate degradation is associated with toxicity including teratogenicity. This is evident with IMiDs that bind CRBN (e.g., thalidomide) and degrade proteins (neosubstrates such as SALL4 that is important to EFD). Internally and externally reported CRBN LBLs do not degrade SALL4 or other key neosubstrates involved in EFD and thus have reduced risk of teratogenicity compared with IMiDs. This is more of a regulatory/safety concern for non-oncology indications (e.g., Immunology and Neuroscience). Highly conservative regulatory expectations from health authorities may impact future labeling (i.e., black box warning/REMS program). Due to species differences in CRBN and SALL4 aminoacid sequences, rabbit and NHP are more relevant than rodent or dog in EFD studies.
In vitro assays to profile protein degraders for derisking potential CRBN-related teratogenicity include SALL4 degradation high-throughput screening assay (HTS), hiPSC assay for SALL4 degradation and markers for lateral plate mesoderm differentiation, and Toxys ReproTracker.
An in vivo EFD study in rabbits can be performed to derisk CRBN-related teratogenicity and will assess target and SALL4 degradation in the rabbit, confirm maternal and fetal exposures, tolerability, and protein degradation, identify maternal toxicity (e.g., increased embryo resorptions and post-implantation loss), and any effects on fetal external, visceral, or skeletal development (i.e., teratogenicity). This study can be performed earlier in development, prior to investigational new drug (IND) submission if useful, to identify potential EFD issues early in the rabbit, although NHP EFD studies will likely have to be performed at a later timepoint in the program in this speaker’s opinion.
Peripheral Neuropathy
Clinical peripheral neuropathy (PN) is seen with IMiDs (i.e., thalidomide> pomalidomide/lenalidomide) and could be considered a potential platform toxicity. Pharmaceutical companies have described PN clinical and/or microscopic findings anecdotally (primarily dorsal root ganglion (DRG)-focused) in rodent, dog, and cynomolgus monkeys with subsets of CRBN-recruiting degraders (Table 3). These findings appear to be dependent on CRBN expression. There is high expression of CRBN in spinal cord and DRG. Anecdotal reports suggest that CRBN-KO protects from PN. To date, there is no evidence of PN with in-house heterobifunctional degraders targeting CRBN.
Incidence of peripheral neuropathy (CRBN-associated) with different compounds in the rat and dog.

The approach to investigating peripheral nerve toxicity slightly differs between non-GLP investigative studies and GLP studies with CRBN-targeting degraders. For non-GLP toxicity studies in rodents, sciatic nerve and lumbar spinal column with sacrum (vertebrae = L3-L7, cord = L4-S3) is collected. In non-rodents, sciatic nerve and representative levels of the spinal column (cervical [intumescence; vertebrae/cord = C5-T2], thoracic [T3-13], and lumbar [intumescence; vertebrae = L4-S3]) are collected. In non-GLP studies, DRGs are dissected at trimming and histopathology is performed on 2-4 DRGs from lumbar intumescence in both rodents and non-rodents. In GLP studies or non-GLP studies with clinical signs (all species), sciatic nerve, DRGs, and cervical, thoracic, and lumbar spinal cord are evaluated. For general details on the best practices for sampling and processing of the peripheral nervous system, the reader is referred to the STP position paper from Bolon et al. 3
QTc Prolongation
QTc prolongation (e.g., Kymera’s IRAK4 degrader and Foghorn’s FHD-609, a BRD9 CRBN degrader) has resulted in partial clinical holds without evidence of hERG degradation or CRBN/target involvement.
Developing Safer Protein Degraders
To develop safer degraders with better safety profiles, it is recommended to focus on the E3 ligases and linkers. It is crucial to select E3 ligases that have limited expression pattern that includes organs that are critical in a particular disease/indication (i.e., take advantage of differential expression patterns to improve efficacy and mitigate toxicity). It is important to keep in mind the tissue expression differences and species expression differences for CRBN, VHL, or other novel E3 Ligases. It is also useful to optimize the E3 ligase–binding domain. Linker structure optimization (length and attachment sites) can be associated with safer and more efficacious degraders. Longer linkers allow for efficient ternary complex formation and optimization of linker attachment sites can improve degrader selectivity leading to decreased neosubstrates/off-target effects. The physicochemical property optimization of linkers has shown some success in modulating safety issues.
CRBN-Targeting PROTAC Safety Considerations
Dr Clare Hoover, a Director of Pathology at AstraZeneca, gave a presentation on preclinical safety assessment of CRBN-targeting PROTAC therapeutics.
CRBN is the most frequently reported E3 ligase target in clinical stage PROTACs and typically incorporates an IMiD or IMiD-derivative as the E3 ligase warhead chemistry design. 1 Targeting CRBN provides a chemistry design advantage with known structural information on binding sites, biophysically validated E3 ligase affinity for target protein structures, and can be targeted with small molecular weight chemistry. 27 Unfortunately, targeting CRBN can introduce off-target safety risks due to broad tissue expression and CRBN-mediated off-target protein degradation.
IMiD drugs, such as thalidomide and its derivatives lenalidomide and pomalidomide, provide clinical benefit in B-cell malignancies and autoimmune diseases through modulation of CRBN substrate binding. 16 Unintended consequences of IMiD CRBN modulation were clinical toxicities of teratogenicity, hematotoxicity (neutropenia and thrombocytopenia), and PN. 16 Recently, the mechanism behind these toxicities has been shown to be IMiD recruitment of key zinc finger transcription factors to CRBN for ubiquitination and subsequent degradation, specifically SALL4 and IKAROS (IZKF) family proteins.17,18 When considering PROTAC safety, it is important to understand if an IMiD-based chemistry design targeting CRBN initiates bystander protein degradation of these key transcription factors. Reports of PROTACs with pomalidomide or lenalidomide as the E3 ligase warhead have demonstrated off-target degradation of IZKF proteins, highlighting the possibility of CRBN-mediated off-target degradation.4,31
CRBN off-target protein degradation can be evaluated through in vitro screens for degradation of neosubstrates, including SALL4 and IKAROS, monitoring of the global proteome, and in vivo assessment in a relevant animal model. 8 One consideration for preclinical in vivo species selection is that a CRBN polymorphism at amino acid V391I renders common safety species, including pig, mouse, rat, dog, hamsters, and ferrets, resistant to IMiD toxicities. 8 In contrast non-human primates and rabbits have a conserved CRBN sequence with humans and develop thalidomide teratogenicity in toxicity studies. 25 One possible alternative for preclinical in vivo studies is the use of a CrbnI391V transgenic mouse model that recapitulates teratogenicity and hematotoxicity following IMiD treatment. 8 Mitigation strategies to avoid or minimize effects of off-target CRBN-mediated protein degradation include modifying the chemical PROTAC linker design to alter the binding affinity between CRBN and neosubstrates or employing a more selective IMiD warhead in the chemistry design to avoid an off-target degradation profile.
Derisking Dose-Limiting Thrombocytopenia With Protein Degrader Technology
Dr Allison Vitsky, a Research Fellow in Global Drug Safety Research and Development at Pfizer, was a clinical pathologist on a number of in vivo studies involving protein degraders at her organization. She presented a case example of using protein degrader technology to derisk dose-limiting thrombocytopenia.
As previously described, PROTACs may improve therapeutic index through precision targeting of a POI when compared with small molecule modalities, achieved in part through target selection with subsequent confirmation of binding and degradation. In addition, common E3 ubiquitin ligases recruited by PROTACs may deliver varying outcomes, as enhanced cell and/or tissue selectivity may occur if the ligase being recruited is differentially expressed across tissues or cell types. Dysregulation of the paralog chromatin regulators/histone acetyltransferases, CREB-binding protein (CBP) and/or EP300, is implicated in a variety of cancers, making them attractive oncology targets; 12 however, their synthetic lethal relationship renders systemic inhibition difficult, and thrombocytopenia has emerged as one of the dose-limiting toxicities of traditional small molecule inhibitors of the BRD of CBP/EP300.13,19 In an effort to derisk this platelet effect, a series of rat studies was performed to compare heterobifunctional degraders of CBP and/or EP300 with a BRD inhibitor. Compounds evaluated included dual CBP/EP300 degraders using either VHL or CRBN E3 ligases, as well as a selective EP300 degrader using a VHL E3 ligase. Animals administered the BRD inhibitor or either dual degrader had notably lower platelet counts that were thought to be attributable to an impact on megakaryocyte maturation in the bone marrow. Conversely, the selective EP300 degrader showed little apparent effect on megakaryocytes, with nominal resultant lowering of platelet counts. 7 These observations were similar to those that have been reported by other investigators evaluating selective EP300 degraders.6,30 Although selectivity for EP300 appeared to be critical to mitigating the impact on megakaryocytes and platelets in this series of investigations, VHL-linked PROTACs have also been reported to diminish thrombocytopenia caused by traditional inhibitor compounds due to lower expression of VHL on platelets.14,28
Regulatory Considerations
Dr Stephanie Leuenroth-Quinn, an Associate Director for Pharmacology and Toxicology at FDA, gave a TPD presentation from a regulatory perspective and provided data-mining results of toxicity profiles across products that target the E3 ligase, CRBN.
Assessing nonclinical toxicity of TPD therapies can be complicated from a regulatory perspective. The source of toxicity may be due to one or more factors including the degradation (vs inhibition) of the intended target protein leading to a prolonged PD activity and loss of all functionality of the protein, the potential for off-target or bystander degradation effects, and/or the unintended effects of targeting an E3 ligase. In addition, heterobifunctional degraders also have the potential for the “hook effect,” which could impact interpretation of a test article–related dose response and setting a NOAEL (No Observable Adverse Effect Level). Consideration of the assays and methodologies to assess and interpret the pharmacological and toxicological profile of the TPD is important to establish a meaningful nonclinical assessment to ensure clinical safety for both oncology and non-oncology indications. Currently, the FDA follows the guidance documents ICH M3(R2) “Nonclinical Safety Studies for the Conduct of Human Clinical Trials and Marketing Authorization for Pharmaceuticals” and ICH S9 “Nonclinical Evaluation for Anticancer Pharmaceuticals” for the regulatory recommendations for TPD products, as they are considered small molecules. Although TPDs have unique considerations for toxicity interpretation in a nonclinical study, they also share many similarities with traditional small molecules, such as the potential for off-target or secondary pharmacologic effects, differences in nonclinical species specificity, and systemic exposure profiles (Figure 8). Regardless, data obtained from submitted nonclinical TPD pharmacology and toxicology studies are used to ultimately establish safety margins to the most sensitive nonclinical species to ensure patient safety and to help inform patient monitoring in the clinic.
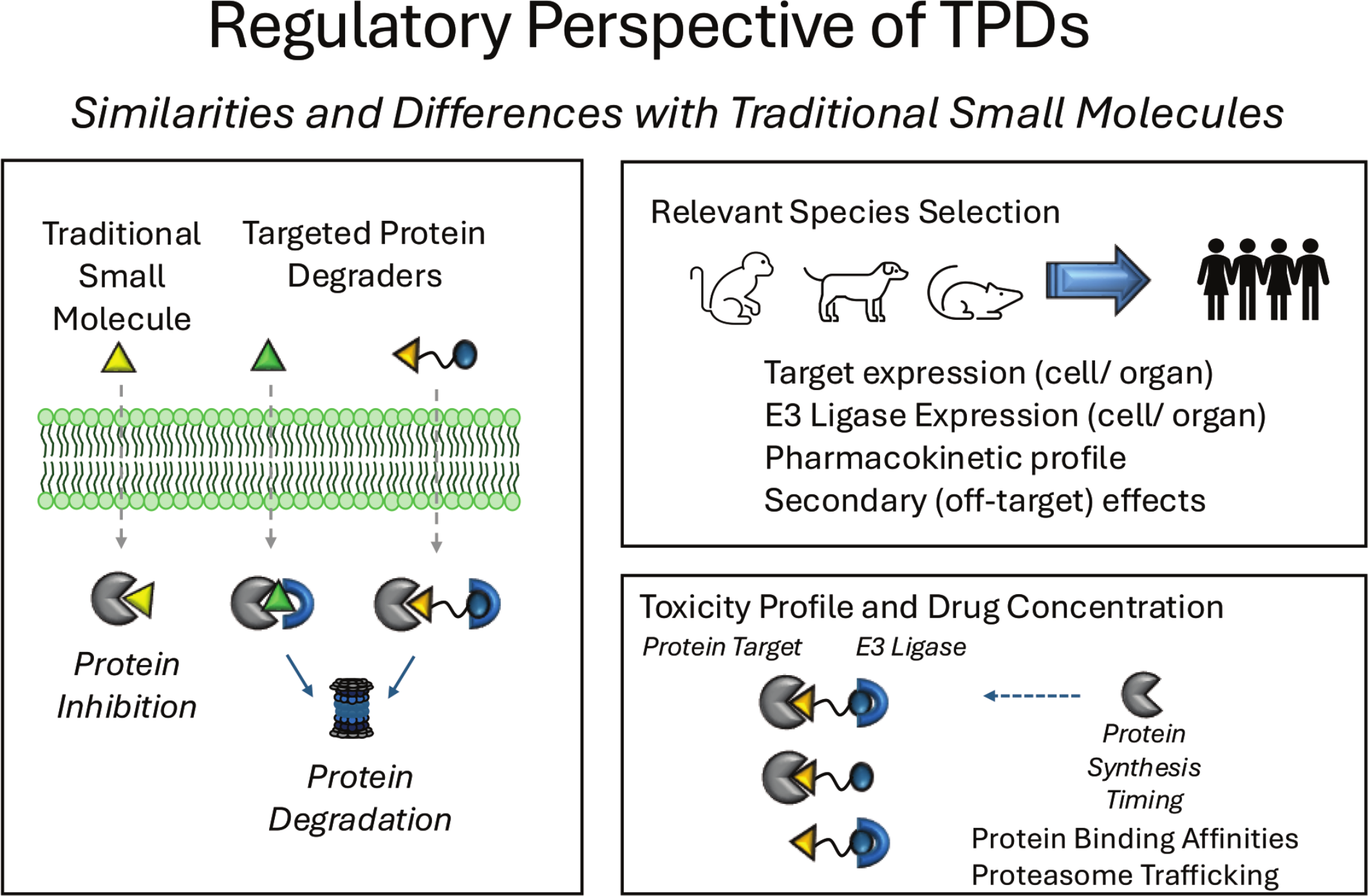
Regulatory perspectives of TPDs. While there are many similarities between assessing toxicities of traditional small molecules and TPDs, such as considerations for relevant species selection and the potential for off-target effects, important differences exist. Some of these differences include assessing potential toxicities due to degradation versus inhibition of a target protein and the time for resynthesis, the possibility of the hook effect, and impacts of targeting the protein of interest as well as an E3 ligase.
As the scientific community continues to gain experience with TPD therapeutics, shared toxicity profiles across species, E3 ligases, or common proteins of interest may become evident. A data-mining initiative was performed comparing different TPD products (heterobifunctional degraders and molecular glue degraders) for target organ toxicities identified by histopathology analyses. The CRBN E3 ligase was a common target across all applications analyzed with varying proteins of interest or intended neosubstrates. To establish criteria for analysis, ten different heterobifunctional degraders were first analyzed for any evidence of the hook effect from histopathology and hematology data. As there were only a few equivocal results where an incidence or severity of a finding appeared to be greater at low- or mid- versus high-dose, a decision was made to conduct the overall analysis between control and high-dose groups only. Histopathology data were collected from a total of 12 applications, where eight were for heterobifunctional degraders and four were for molecular glue degraders (Figure 9). This dataset included a total of 24 one-month general toxicity studies in rodent and non-rodent species, where male and female groups were combined together by dose, to conduct the analysis. Several conclusions were made from this limited dataset which include the following: (1) The immune system (spleen, thymus, lymph nodes) was a common target of toxicity for heterobifunctional degraders in both the rodents and non-rodents. These histopathology findings were frequently associated with changes in cellular hematology parameters, but more commonly observed in rodent species. Immune system toxicity was also detected in the non-rodents for molecular glue degraders. (2) The gastrointestinal (GI) tract was a common site for toxicity for non-rodents for all TPDs assessed. (3) The heart and kidney were common target organs of toxicity across all applications (heterobifunctional degraders and molecular glue degraders) in both the rodents and non-rodents. (4) Increased toxicity did not necessarily correlate with increased systemic exposure when comparing between species for a single product. (5) For molecular glue degraders, the toxicity profile in the non-rodents tended to be more severe than for the rodents, although systemic exposures in the rodents were higher in all four applications analyzed. (6) When comparing the toxicity profiles of products that share both the same E3 ligase and POI target, although there are similarities based on the intended pharmacologic effect, there was noteworthy variability in target organs of toxicity and severity of findings across products and species.
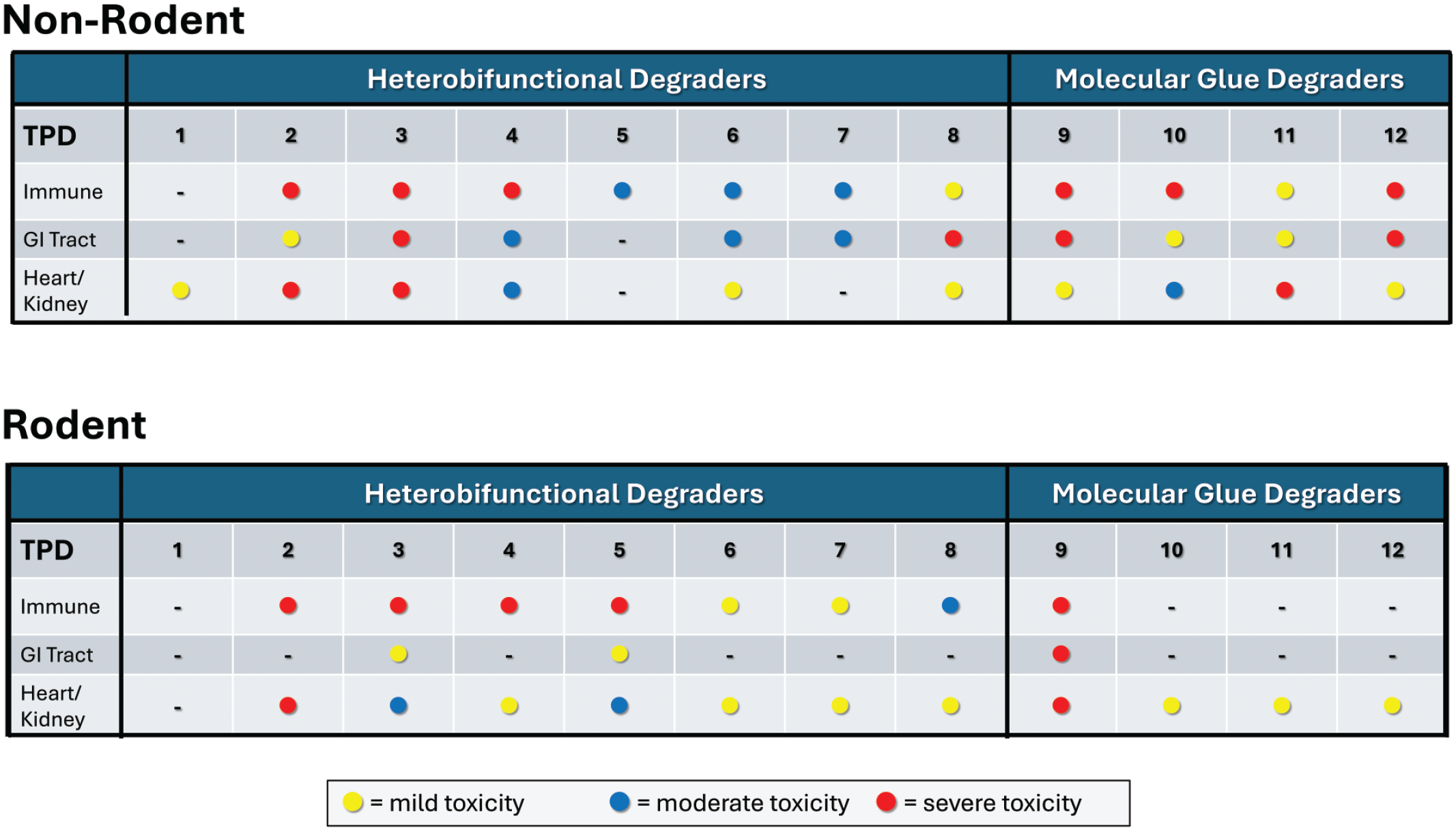
Assessment of target organs of toxicity by histopathology findings. Twelve different TPD products, all targeting the E3 ligase cereblon, were compared for select target organs of toxicity based exclusively upon histopathology analysis. Results were separated out between rodent and non-rodent species as well as heterobifunctional degraders and molecular glue degraders.
Based upon the completed data-mining analysis, several points to consider were presented to assess and interpret TPD nonclinical toxicity profiles, as follows. While in vitro assessments that evaluate the degradation of the intended target protein in species used in toxicity studies should be submitted for Agency review, additional protein degradation assays or proteomics assessments could be conducted to interpret or derisk suspected off-target degradation-induced toxicity. In addition, to better understand human relevance of target organs of toxicities identified in nonclinical species, including supportive information on target protein and E3 ligase expression in humans by organ/tissue (e.g., public literature, databases, assays) would be helpful. To fully assess the potential for the hook effect in nonclinical in vivo studies, there are several points to consider when conducting and interpreting a study such as (1) histopathology examination of all dose groups (i.e., low- and mid-dose), regardless of high-dose findings; (2) examination of hematology parameters as a possible indication of the hook effect; (3) preliminary assessment of wide-ranging dose levels, to fully assess the potential for the hook effect prior to final dose selection for a pivotal toxicology study. As a final study design consideration, when assessing reversibility of toxicity, the time needed for target protein resynthesis should be factored into the recovery duration period.
Footnotes
Acknowledgements
KP would like to thank Dr Monica Rodrigo who was the co-chair for the session in the initial stages of organizing the symposium for her time and expertise.
Author Contributions
I am not sure what to add here. Please use the standard verbiage used for all other sessions for the symposium.
Declaration of Conflicting Interests
The author(s) declared no potential conflicts of interest with respect to the research, authorship, and/or publication of this article.
Funding
The author(s) received no financial support for the research, authorship, and/or publication of this article.