
Abstract
Cystic fibrosis (CF) is caused by a mutation of the Cystic Fibrosis Transmembrane Conductance Regulator (CFTR) gene, which disrupts an ion channel involved in hydration maintenance via anion homeostasis. Nearly 5% of CF patients possess one or more copies of the G542X allele, which results in a stop codon at residue 542, preventing full-length CFTR protein synthesis. Identifying small-molecule modulators of mutant CFTR biosynthesis that affect the readthrough of this and other premature termination codons to synthesize a fully functional CFTR protein represents a novel target area of drug discovery. We describe the implementation and integration for large-scale screening of a homogeneous, 1536-well functional G542X-CFTR readthrough assay. The assay uses HEK 293 cells engineered to overexpress the G542X-CFTR mutant, whose functional activity is monitored with a membrane potential dye. Cells are co-incubated with a CFTR amplifier and CFTR corrector to maximize mRNA levels and trafficking of CFTR to the cell surface. Compounds that allow translational readthrough and synthesis of functional CFTR chloride channels are reflected by changes in membrane potential in response to cAMP stimulation with forskolin and CFTR channel potentiation with genistein. Assay statistics yielded Z′ values of 0.69 ± 0.06. As further evidence of its suitability for high-throughput screening, we completed automated screening of approximately 666,000 compounds, identifying 7761 initial hits. Following secondary and tertiary assays, we identified 188 confirmed hit compounds with low and submicromolar potencies. Thus, this approach takes advantage of a phenotypic screen with high-throughput scalability to identify new small-molecule G542X-CFTR readthrough modulators.
Introduction
Cystic fibrosis (CF) is an autosomal recessive disease caused by mutations in the cystic fibrosis transmembrane conductance regulator (CFTR) gene, which encodes an epithelial cell surface transmembrane protein responsible for hydration homeostasis via the transport of chloride and bicarbonate. 1 The significant reduction in CFTR protein at the plasma membrane resulting from CF-causative mutations leads to challenges in maintaining chloride ion homeostasis. Furthermore, beyond reducing levels of properly functioning, mature protein at the plasma membrane, this impairment of CFTR protein leads to a reduction of chloride transport, imbalance of extracellular fluid, and overall thickening of mucus. The buildup of mucus, particularly in the airway and lungs, leads to the pathological hallmark of the CF disease state, which is characterized by chronic bacterial infections, inflammation, and subsequent scarring of the lungs. However, a range of other pathologies can also manifest as part of the disease, such as intestinal obstruction syndromes, liver and pancreatic dysfunction, and male infertility. 1 In the absence of medical intervention, CF can lead to premature death.2,3
The mutations in CFTR that cause CF fall into five different classes based on their consequences for the encoded protein. Class II represents mutations that result in defective maturation and trafficking of CFTR to the cell surface. Class III and IV include mutations that result in reduced gating of the channel or conductance of the chloride ions through it, respectively. Class V mutations reduce overall CFTR levels by affecting the synthesis of the protein. Class I mutations prevent synthesis of CFTR mRNA or protein, and included within this class are premature termination codons, a subclass of mutations that is the ultimate therapeutic target of this study. Of the >2,000 mutations found to occur in CFTR, ~5% of CF patients possess at least 1 copy of the G542X mutation. This mutation encodes a stop codon at residue 542, resulting in incomplete protein synthesis.4–7
Currently, therapeutics that target the underlying cause of CF, the specific mutational defect in the CFTR protein responsible for its reduced function, are available only for certain mutations or classes of CFTR mutation. CFTR modulators and combinations of modulators are now approved drugs for a subset of CF genotypes and/or mutations. The first available, KALYDECO®, addresses the gating defect of Class III mutations, and its active ingredient is the potentiator ivacaftor (VX-770). 8 Subsequently, two therapies consisting of combinations of a corrector CFTR modulator with the ivacaftor potentiator were approved; ORKAMBI® for CF patients who are homozygous for the class II mutation resulting from the deletion of phenylalanine 508 (ΔF508-CFTR) and SYMDEKO®/SYMKEVI® also for ΔF508 homozygous, as well as ΔF508 compound heterozygous patients with a second allele of select residual function mutations. Most recently, a triple combination of two correctors with ivacaftor, TRIKAFTA, has been approved for patients who have one allele of ΔF508 and any other CF-causing mutation on the second allele.
Previously, we reported a validated, 384-well assay developed for identification of molecules that increase the functional readout of ΔF508-CFTR.
9
Although the ΔF508-CFTR assay used a halide-sensitive YFP quenching reporter, we chose an alternative, albeit similar, strategy for the detection of readthrough compounds of the G542X mutants, which result in a functioning CFTR channel
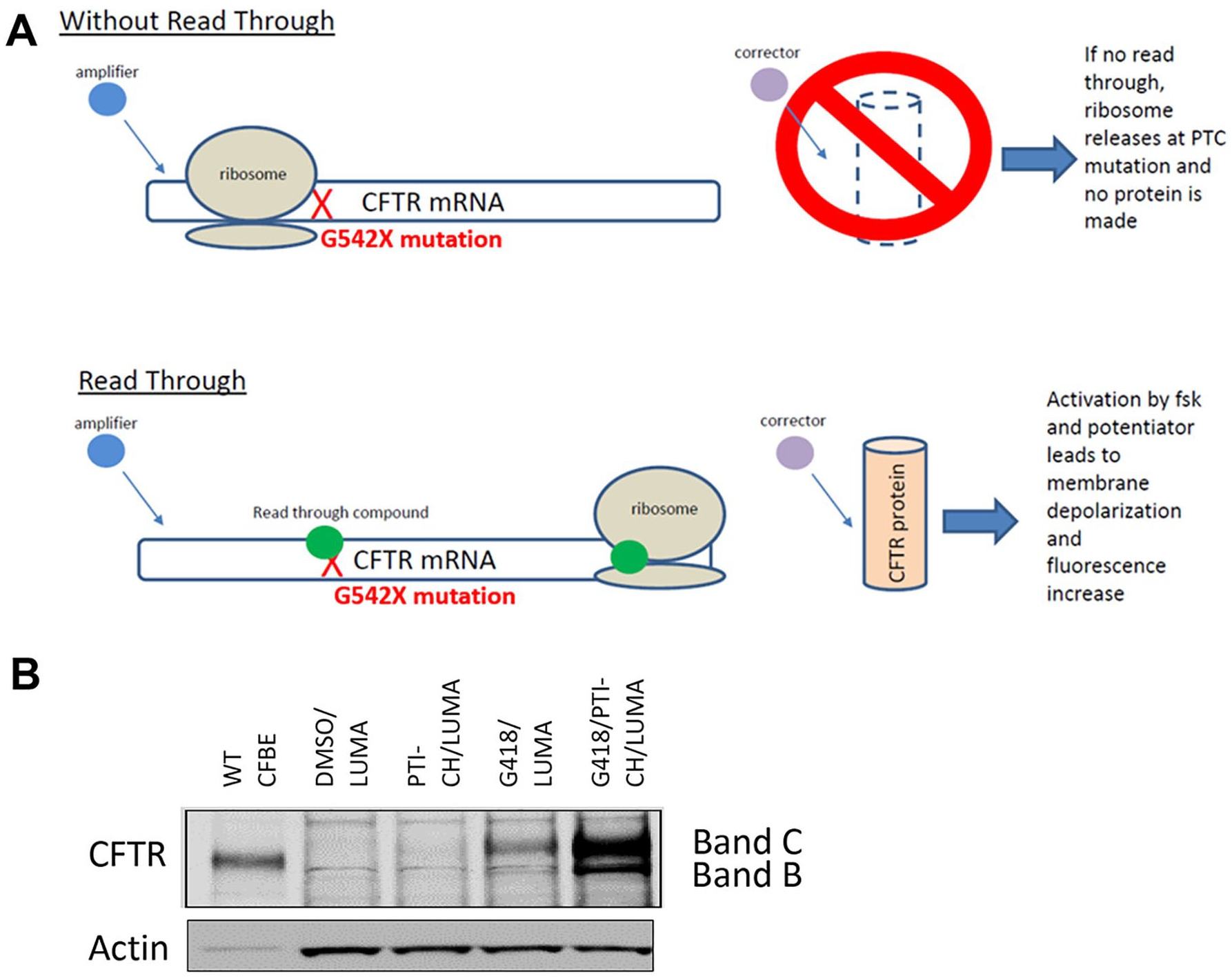
Schematic representation of the mechanism by which readthrough compounds restore channel function in G542X-CFTR–expressing cells. PTC refers to the premature termination codon. (
Here, we report the development and optimization of a G542X-CFTR functional readout assay that is automated, ultra-high throughput, and a 1536-well homogeneous no-wash assay. This should improve the chances of identifying potential drugs by dramatically improving throughput while maintaining assay quality and reducing overall costs. To validate this system, we completed a high-throughput screening (HTS) campaign, testing approximately 666,000 individual compounds for the ability to restore CFTR protein function. To determine the outcome of the reporter assay, a novel data analysis method was developed, and potential readthrough molecules were identified.
Materials and Methods
CFTR Small-Molecule Modulation for Functional Assay Development
We prioritized the development of a functional CFTR high-throughput assay over other more indirect reporters of readthrough to maximize the potential value of any identified translational readthrough hits. Because the efficiency of the raw hits may not be sufficient to generate detectable CFTR activity, our strategy was to include CFTR modulators of complementary mechanisms of action in the assay. To increase the levels of CFTR mRNA and its translation in the assay, we included the PTI-CH amplifier in the 48-h incubation. Similarly, the corrector VX-809 is included in the 48-h incubation to help improve the resultant CFTR protein’s folding and trafficking in the event that the translational readthrough lead to a misincorporated residue and thus a missense mutant protein. In addition, 2 acute-acting small molecules are used to maximize CFTR protein functional activity. The CFTR protein contains a domain that, when nonphosphorylated, prevents the channel’s activation. Phosphorylation of this regulatory domain by protein kinase A leads to an activated CFTR channel, a phosphorylation affected by the acute addition of forskolin. The second acute-acting small molecule is a CFTR potentiator, in this case genistein, that increases the opening time of the CFTR channel, allowing for greater chloride ion flow.
To establish an assay window for the translational readthrough of the G542X-CFTR described below, we used the aminoglycoside G418 as a positive control. Aminoglycosides promote translational readthrough of nonsense mutations with some local context dependence but largely independent of the mRNA that contains them. G418 has been shown to promote translational readthrough of G542X and other nonsense mutations in CFTR. 13 Although G418 on its own produces some functional CFTR, its synergy with amplifier, corrector, and potentiator provides a much better assay window. In addition, we anticipate novel translational readthrough compounds will also be complemented by these additional modulators and have included them in the assay and will continue to do so in the preclinical development of advanced leads as the project progresses.
Cell Lines and Cell Culture
HEK 293 cells were transiently transfected with pNLF1-C (CMV/Hygro) plasmid (Promega Corp., Madison, WI) containing the CFTR-G542X minigene (full-length cDNA of CFTR with three partial introns, 14, 15, and 16) inserted as described in Masvidal et al. 14 The plasmid contains CMV promoter and synthetic polyA signal. It carries hygromycin and ampicillin resistance markers. Cells were routinely cultured in five-tier flasks (Falcon, Thermo Fisher Scientific, Waltham, MA) at 37 °C and 95% relative humidity. Unless indicated otherwise, all cell culture reagents were sourced through Life Technologies (Carlsbad, CA). Cells were cultured in DMEM (Glutamax), 10% HI-FBS, and 1X Anti-Anti. The cells were passaged every 3 d at a seeding density of 15 million cells per flask. TrypLE was used to dislodge the cells during passaging.
Maxcyte Transient Transfection
All transfection reagents and equipment were obtained from Maxcyte Inc. (Gaithersburg, MD). HEK 293 cells were harvested at 70% confluency as previously described. 9 The cells were spun down to remove all TrypLE and media and resuspended with Maxcyte electroporation buffer at 1 × 108 cells/mL. DNA was added at 400 µg/mL and electroporated on the Maxcyte according to the manufacturer’s specifics. The cells were transferred to a 10 cm cell culture dish and incubated for 20 min at 37 °C and 95% relative humidity. The cells were then resuspended in growth media, counted, spun down, and frozen in cryovials using cell recovery freezing media (Life Technologies). The cells can then be used as a freezer-ready component in assays as one large stock of matched and validated cells, thus providing a freezer ready assay.
Minigene Stable Cell Line Generation
HEK 293 cells were plated in a six-well plate (CellBind, Corning, Corning, NY) at 350,000 cells per well. The following day, cells were transfected with 2 µg of CFTR-G542X minigene plasmid, described above, and with 10 µL Lipofectamine 2000 reagent, according to the manufacturer’s protocol (Life Technologies). Transfection media were removed from cells the next day and replaced with DMEM growth media supplemented with 10% bovine serum albumin and 1% penicillin/streptomycin for an additional 24 h. At this time, cells were subjected to selection media containing 800 µg/mL hygromycin for 7 d. Hygromycin-resistant cells were pooled from all wells and expanded for an additional 14 d. The HEK 293 CFTR-G542X stable cell line was shown to express G542X-CFTR protein by Western blots following incubation with a known aminoglycoside readthrough agent (G418) 15 (Fig. 1B). The cell line was shown to express functional CFTR following G418 incubation in an FMP membrane potential assay in comparison with the parental HEK 293 cells to establish G542X-CFTR responsiveness to readthrough modulators.
Compound Library
The Scripps Drug Discovery Library (SDDL) was profiled in the HTS campaign. This library currently consists of 666,120 unique compounds, representing a diversity of druglike scaffolds targeted to traditional and nontraditional drug-discovery biology. The SDDL has been curated from more than 20 commercial and academic sources and contains more than 40,000 compounds unique to Scripps. The SDDL compounds have been selected based on scaffold novelty, physical properties, and spatial connectivity. In its current iteration, the SDDL has several focused sublibraries for screening popular drug-discovery target classes (e.g., kinases/transferases, G-protein–coupled receptors, ion channels, nuclear receptors, hydrolases, transporters), with diverse chemistries (e.g., click chemistry, PAINS free, Fsp3 enriched, covalent inhibitors, and natural product collections) and desirable physical properties (rule-of-five, rule-of-three, polar surface area, etc.).16–21 All samples in the SDDL have been tested for purity and structural confirmation via liquid chromatography–mass spectrometry (LC-MS) and/or nuclear magnetic resonance to provide adequate quality assurance/quality control. In addition, after completion of the HTS campaign, all compounds that proceeded through the titration phase were subjected to LC-MS analysis using samples obtained directly from the same source plates as used in the assay. This allows for reconfirmation of hit sample purity and mass.
Results and Discussion
The 384-well format had several limitations that needed to be addressed to implement the assay to the 1536-well homogeneous high-throughput format (
Lipofectamine versus Maxcyte Transfection
At the time of initial optimization, there was not a stable cell line available (
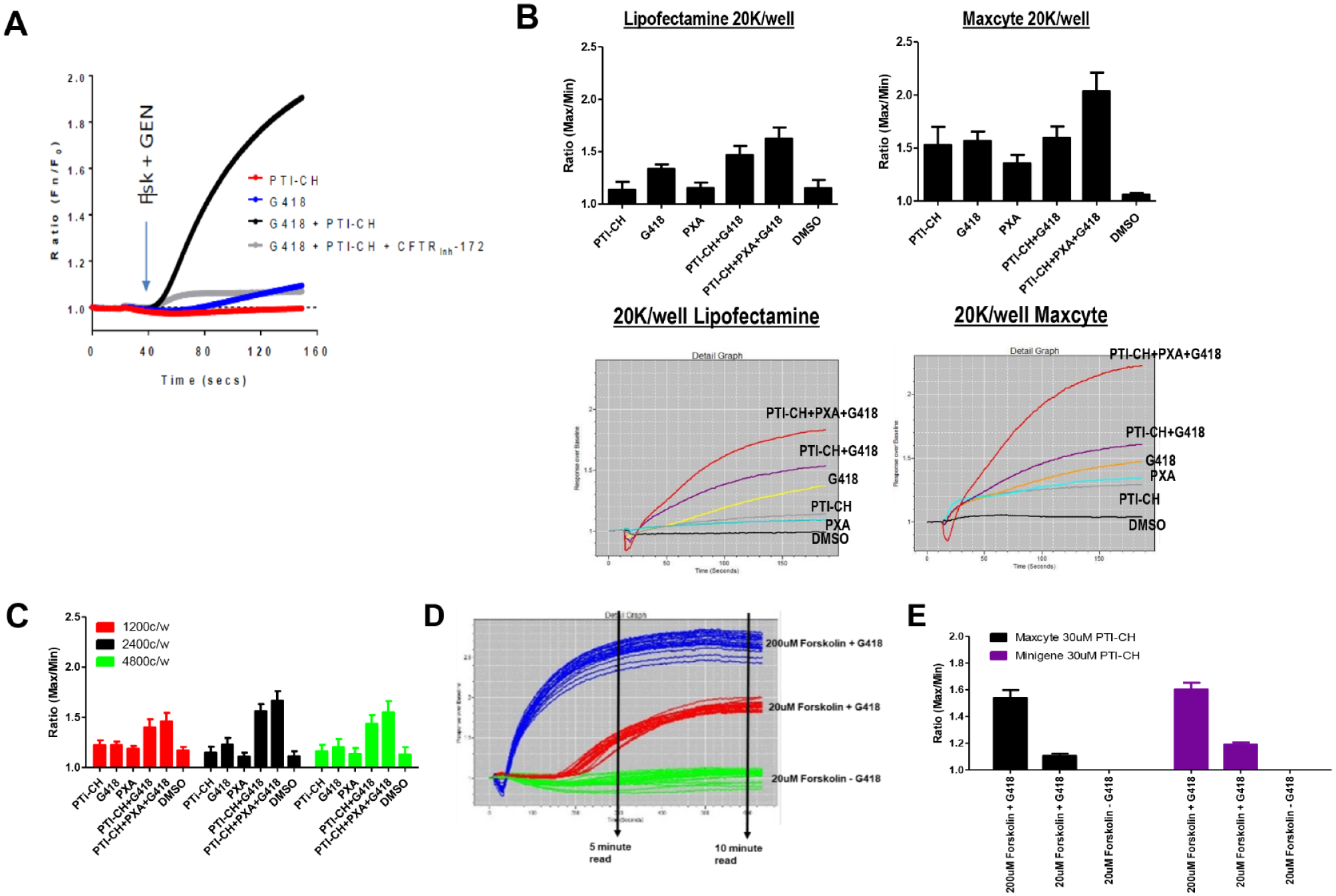
Initial recapitulation of the readthrough assay 384-well format of transient transfected G542X-CFTR cDNA and 1536-well optimization. (
Optimizations: Cell Titer
The initial Maxcyte transfected cells were tested in 1536-well format at 1200 cells per well (c/w), 2400 c/w, and 4800 c/w (
PTI-CH Titer and Forskolin/Genistein Stimulation Read Times
PTI-CH and forskolin stimulus can affect the overall response of the readthrough assay. PTI-CH is a small molecule of the amplifier class of CFTR modulators that increase the amount of CFTR protein.10,11 Forskolin is used to stimulate the CFTR channel through phosphorylation, enabling the channel to open and allow the release of chloride. Optimizing the concentration and timing of the response is needed to improve the signal window and Z′ of the assay (
Transient versus Stable Cell Line of Minigene
For the HTS screen, we chose to include the PTI-CH amplifier to increase the CFTR mRNA levels10,11 and provide more substrate for potential readthrough compounds. In addition, the amino acid that is inserted at position G542 by a readthrough compound may be different from glycine, thus creating a missense mutation at residue 542, as has been previously shown for G418-mediated readthrough.
13
We therefore also included a corrector, VX-809, to potentially improve the maturation of the missense CFTR resulting from the readthrough incorporation. During the optimization process, a stable cell line, transfected with the G542X minigene, was created to respond to higher concentrations of PTI-CH. These new cells were tested and compared with the Maxcyte transfected cells using a 30-min stimulation time with 30 µM PTI-CH (
Stable Cell Line LOPAC PILOT Data
To test the feasibility for an HTS screen, it is typically good practice to perform a pilot screen using the LOPAC 1280 library.
22
We did this using 200 µM forskolin as a nonspecific high control to assess the overall cell plating and reagent behavior. We also used 20 µM forskolin with 1 mM G418 as the 100% normalized control to determine the readthrough activity in the assay (“1 mM G418 Reference” in the equation below). The 20 µM forskolin with DMSO was used as the 0% normalization control and low control for cell plating and reagent behavior. An example of a column view scatterplot of activity taken from the DMSO plate can be found in
Figure 3A
. The LOPAC pilot was run in triplicate. The activity scatterplot and statistical data are found in
Figure 3B
. The correlation plot (
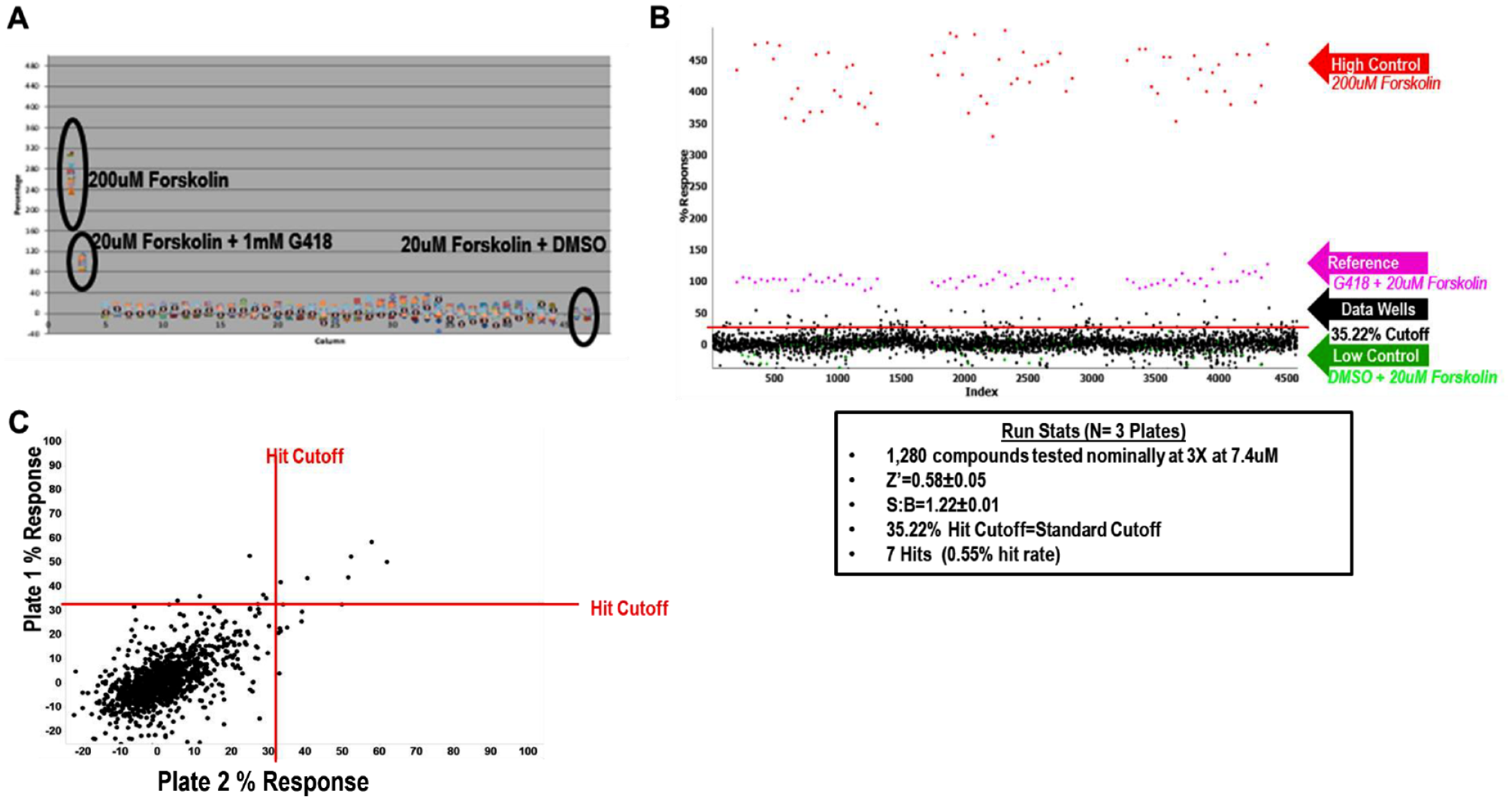
Final conditions for high-throughput screening (HTS) along with LOPAC pilot screen data. (
Cystic Fibrosis Transmembrane Conductance Regulator (CFTR) Readthrough 1536 High-Throughput Screening Campaign.
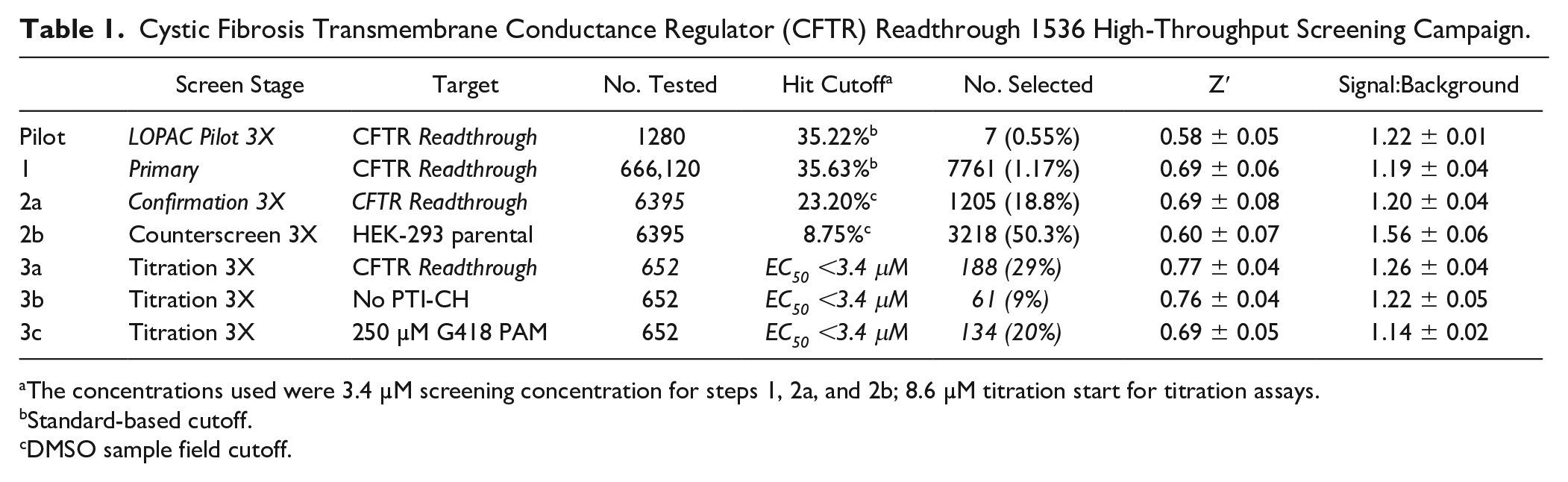
The concentrations used were 3.4 µM screening concentration for steps 1, 2a, and 2b; 8.6 µM titration start for titration assays.
Standard-based cutoff.
DMSO sample field cutoff.
Primary HTS
The first step of the HTS campaign was to screen the CFTR G542X readthrough assay against the SDDL. In this primary screen, 666,120 compounds were tested at a single concentration in singlicate, with a final nominal concentration of 3.4 µM for the library molecules. Raw assay data were imported into the Scripps database and subsequently analyzed using Symyx software (Santa Clara, CA). The activity of each compound was calculated on a per-plate basis using the following equation:
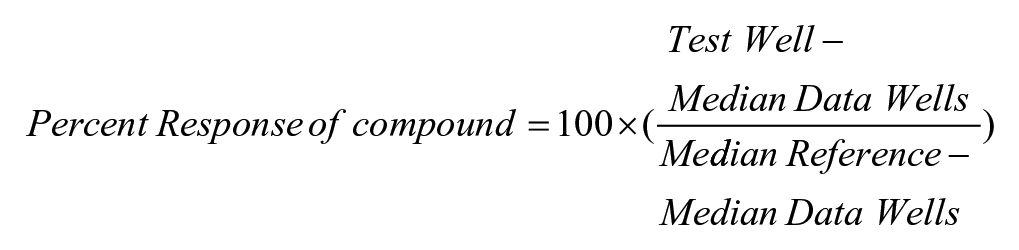
where the data wells contained cells, addition of test compound, 30 µM PTI-CH, and 1 µM VX-809 in media followed by stimulation with 20 µM forskolin and 10 µM genistein. The reference wells contained the same constituents as the low control described below, but we also added 1 mM G418. We also included controls that were used on each plate for n = 24 each, which were used to determine Z′ and S:B. “High control” represents wells containing cells with DMSO, 30 µΜ PTI-CH, and 1 µM VX-809 in media followed by stimulation with 200 µM forskolin and 10 µM genistein. “Low control” represents wells containing cells, DMSO, 30 µM PTI-CH, and 1 µM VX-809 in media followed by stimulation with 20 µM forskolin and 10 µM genistein. The Z′ was calculated from the high and low control, but the S:B for this assay was calculated using the reference versus low control wells. 23 Assay performance demonstrated an average Z′ of 0.69 ± 0.06 and an average S:B of 1.19 ± 0.04 (n = 536 plates). A summary of the results of the primary screening assays is shown in Figure 4A . A mathematical algorithm was used to determine the standard hit cutoff to identify active compounds. Two values were calculated: (1) the average percentage activity of compounds tested and (2) three times their standard deviation. The sum of these two values was used as a cutoff parameter; that is, any compound that exhibited a greater percentage activity than the cutoff parameter was declared active. Using this standard cutoff, the primary assay yielded 7761 active compounds (hits) with greater than 35.63% activity. A promiscuity index was also calculated using former HTS primary data; the expression “X out of Y” was interpreted as “this compound was a hit in X out of Y total primary screens.” The X value includes hits in the current screen. Compounds with high X values relative to Y values can be considered promiscuous (nonspecific activators, detection artifact, etc.). A total of 6400 compounds were selected for confirmation, of which 6395 compounds were available (see Table 1 ).
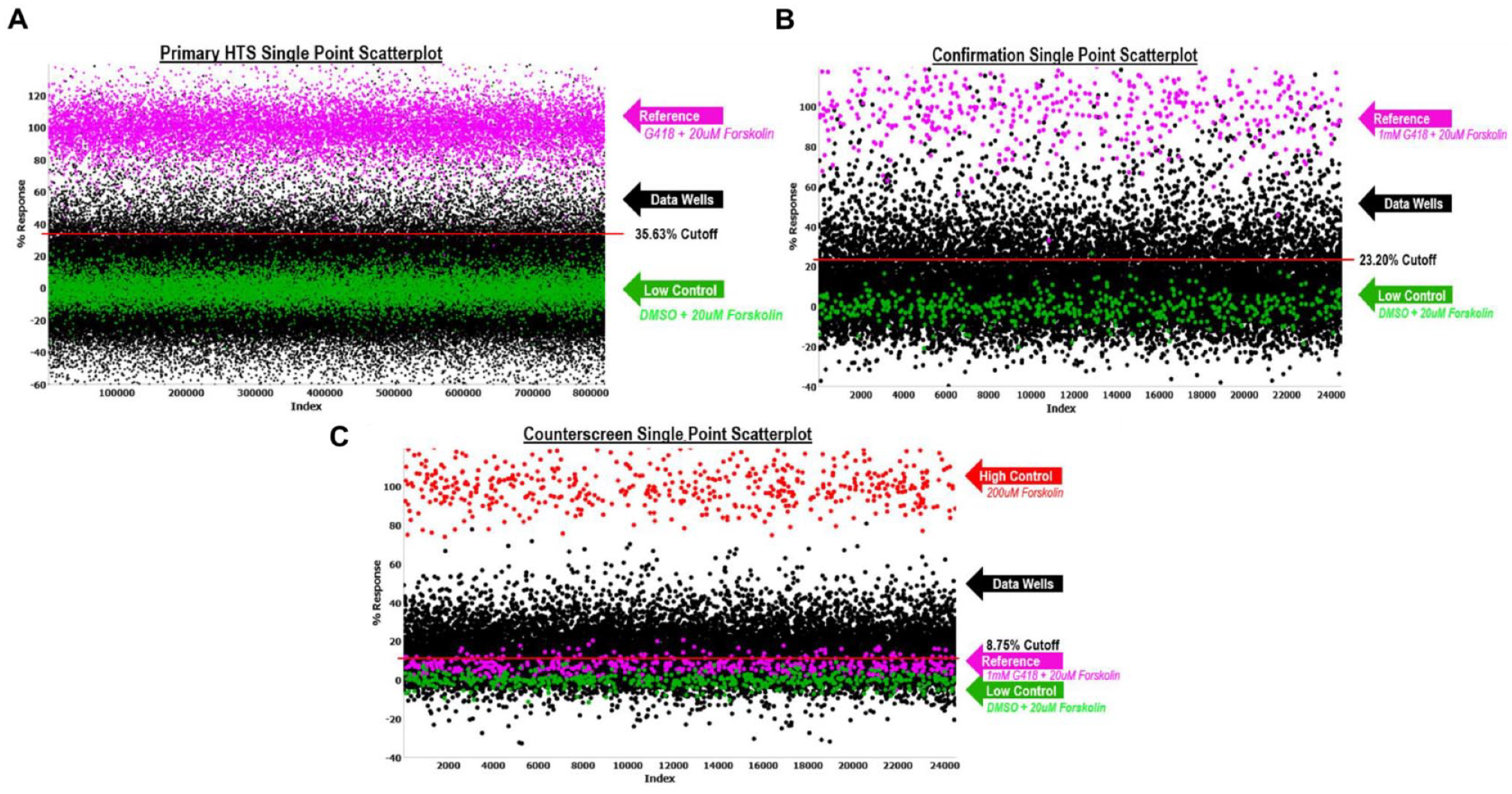
Primary and confirmation screens. The scatterplot of all wells tested during the primary screen. One hundred percent normalization is shown in pink. The high control is G418 + 200 µM forskolin (not shown in scatterplot – above 120% activity). The low control and 0% normalization are shown in green. All data plots are shown in black. The red line indicates the hit cutoff used for that screen. (
Confirmation/Counterscreen
After completion of cherry-picking, the secondary assays were run. The confirmation screen used the same reagents and detection system as the primary screening assay but tested each of the 6395 compounds at a single concentration (nominally 3.4 µM) in triplicate. The counterscreen assay employed the same assay conditions but used the parental HEK 293 cells that do not express G542X-CFTR. This was chosen to eliminate hit compounds showing CFTR-independent activity. The counterscreen also employed the same high control as the primary assay high control. The low control and sample field used DMSO or compounds with no stimulus or potentiator. The reference was not able to be used for the 100% control because the counterscreen cells, lacking G542X-CFTR, do not respond to G418. This counterscreen assay serves to identify sundry off-target hits as well as compounds that optically affect fluorescence measurements (colored compounds, etc.).
Similar to the primary screen, the CFTR G542X readthrough confirmation assay exhibited an average Z′ of 0.69 ± 0.08 and an S:B of 1.20 ± 0.04. Using the DMSO cutoff of 23.20% response (n = 5 plates), 1205 hits confirmed activity equal to 18.84% (
Titration Assays
The titration assays employed the same reagents, protocols, and detection systems as the primary assay but tested each of the compounds as 10-point dose-response titrations (threefold dilutions) in triplicate. In addition, two different formats were tested to provide a profile of the types of readthrough compounds that were identified in the primary screen and confirmed in the secondary screen.
The first of the three tertiary screens was a direct repeat of the secondary confirmation screen but with a 10-point dose response to evaluate potency in the original assay format. The second titration format, the no-amplifier assay, used the same cells and reagents as the primary and secondary screens but omitted the PTI-CH amplifier addition in the low control and sample field wells to identify whether compounds are able to provide a functional readthrough in the absence of the amplifier-mediated increase in CFTR mRNA. The third titration format, the G418 PAM, used the same cells and reagents as the primary and secondary screens, except the low control and sample fields also contained 250 µM G418 to evaluate whether the readthrough compounds were complemented by a submaximal G418 concentration or whether they were in fact antagonistic with this aminoglycoside-mediated readthrough activity. In this sense, we envision the assay being tested in a manner that is similar to other positive allosteric modulator assays (PAM) because it was optimized to select the maximal response and then adjusted to provide a submaximal concentration to allow hits to act through the same G418 mechanism.
The performance of the CFTR G542X readthrough tertiary screen titration assay was again statistically robust, with an average Z′ of 0.77 ± 0.04 and an S:B of 1.26 ± 0.04. The no-amplifier tertiary screen titration assay behaved similarly, with an average Z′ of 0.76 ± 0.04 and an S:B of 1.22 ± 0.05. Lastly, the G418 PAM tertiary screen titration assay performance was also suitable, with an average Z′ of 0.69 ± 0.05 and an S:B of 1.14 ± 0.02. For each test compound, the percentage activation was plotted against compound concentration. A four-parameter equation describing a sigmoidal dose-response curve was then fitted with adjustable baseline using Assay Explorer software (Symyx Technologies). The reported EC50 values were generated from fitted curves by solving for the X-intercept value at the 50% activity level of the Y-intercept value. The following rule was used to declare a compound as active or inactive: Compounds with an EC50 greater than 3.4 µM were considered inactive. Compounds with an EC50 equal to or less than 3.4 µM were considered active. Of those, 88 compounds demonstrated an EC50 <3.4 µM in the CFTR G542X readthrough assay and an EC50 >3.4 µM in the no-amplifier and G418 PAM assay formats. All titration assay results are tabulated in
Table 1
. The Venn overlap of the three assays is shown in
Figure 5A
. Clustering was done on the 188 active compounds to show the structural similarities of the various hits (
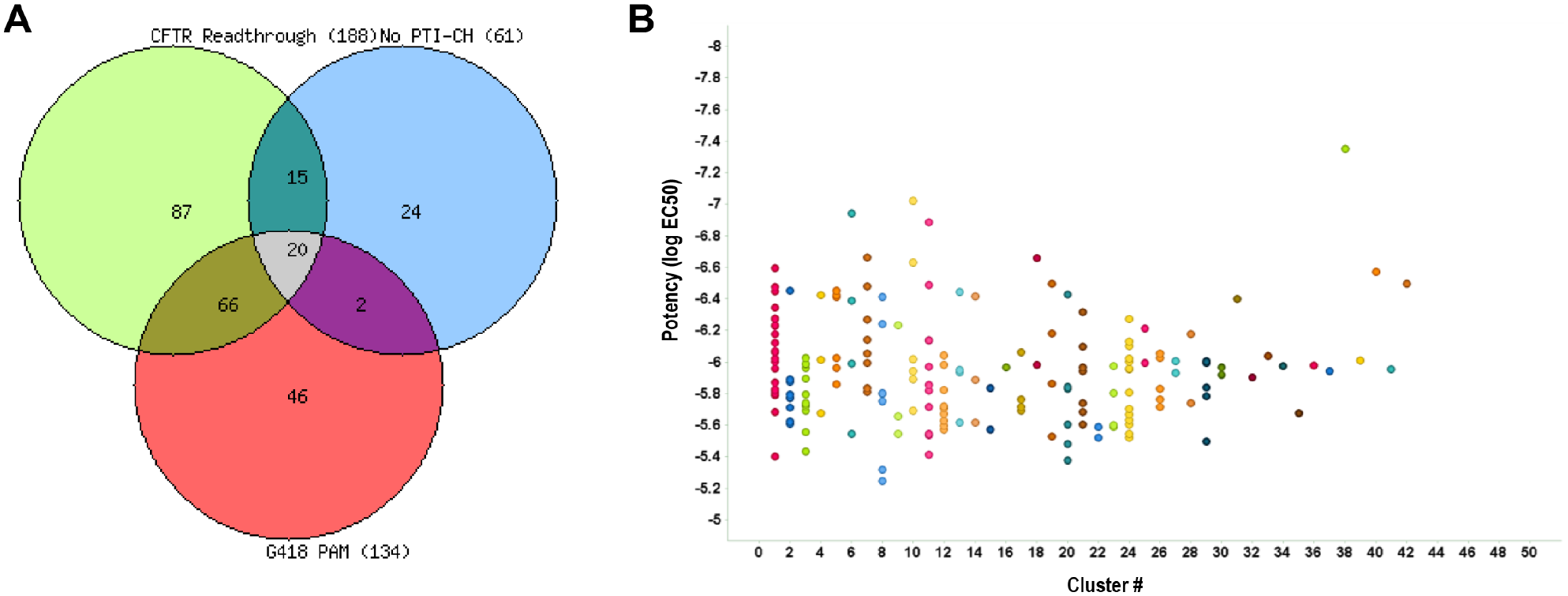
Outcome of the titration assays. (
In summary, the phenotypic HTS assay described here provides a framework for identifying compounds that can promote the readthrough of premature termination codons. Because such an activity may not be efficient enough on its own to produce detectable levels of functional readthrough protein, complementary modulators are co-incubated in the assay and the screen to maximize mRNA levels and folding and trafficking of the target protein to synergize with an active hit of limited initial activity. Compounds that allow translational readthrough and synthesis of functional protein are detected using a dye sensitive to changes in membrane potential, making the assay adaptable for other channels or transporters that affect that cellular phenotype. To our knowledge, this is the first study to report the methods and outcomes of a large-scale HTS target CFTR readthrough of the G542X mutant that resulted in 188 compounds with low and submicromolar potencies. These compounds are now being advanced to primary cell testing to evaluate their ability to provide translational readthrough in a more physiologically relevant context.
Monogenic diseases caused by nonsense mutations represent a particular challenge for therapeutic development. As in the case for G542X-CFTR, the encoded protein, if made, is truncated, resulting in an incomplete polypeptide that lacks essential domains for function and may be unable to fold sufficiently to avoid rapid degradation. Upstream of translation into protein, the quality control pathway of nonsense-mediated decay may recognize the nonsense mutation-containing transcript and direct the mRNA to be degraded, thus reducing the levels available to make even the truncated protein product. Translational readthrough, if achieved, circumvents both of these synthesis defects and, if efficient enough, could restore function to levels approaching the nonmutated disease gene. However, as seen in our preclinical cellular system, the combination of a translational readthrough agent such as G418 with modulators with complementary mechanisms enhances the functional restoration.
A compound optimization campaign may result in the development of a stand-alone translational readthrough drug candidate. However, the therapeutic development of CFTR modulators targeting F508del-CFTR has thus far shown that a single molecule may not be able to provide sufficient functional rescue to CFTR for therapeutic benefit. Therefore, in pursuit of a therapeutic for patients who have at least one nonsense allele and who do not have a second CF-causing allele for which there is an approved therapeutic, it will be important to explore the possibility of developing a combination of modulators as that therapeutic. Such a strategy includes performing hit-to-lead and lead optimization in the presence of clinical-stage CFTR modulators to identify and ensure their complementarity. The efficacy of promising leads for CFTR nonsense mutations beyond G542X will also be explored in the hope that translational readthrough compounds identified in this study may have efficacy that extends beyond their benefit for the G542X-CFTR allele.
Supplemental Material
Supplement – Supplemental material for Identification of Compounds That Promote Readthrough of Premature Termination Codons in the CFTR
Supplemental material, Supplement for Identification of Compounds That Promote Readthrough of Premature Termination Codons in the CFTR by Emery Smith, Danijela Dukovski, Justin Shumate, Louis Scampavia, John P. Miller and Timothy P. Spicer in SLAS Discovery
Footnotes
Acknowledgements
Supplemental material is available online with this article.
Declaration of Conflicting Interests
The authors declared no potential conflicts of interest with respect to the research, authorship, and/or publication of this article.
Funding
The authors received no financial support for the research, authorship, and/or publication of this article.
References
Supplementary Material
Please find the following supplemental material available below.
For Open Access articles published under a Creative Commons License, all supplemental material carries the same license as the article it is associated with.
For non-Open Access articles published, all supplemental material carries a non-exclusive license, and permission requests for re-use of supplemental material or any part of supplemental material shall be sent directly to the copyright owner as specified in the copyright notice associated with the article.