
Abstract
We previously developed a panel of one-step real-time quantitative reverse transcription PCR (one-step qRT-PCR; hereafter referred to as qRT-PCR) assays to assess compound efficacy. However, these high-cost, conventional qRT-PCR manual assays are not amenable to high-throughput screen (HTS) analysis in a time-sensitive and complex drug discovery process. Here, we report the establishment of an automated gene expression platform using in-house lysis conditions that allows the study of various cell lines, including primary T cells. This process innovation provides the opportunity to perform genotypic profiling in both immunology and oncology therapeutic areas with quantitative studies as part of routine drug discovery program support. This newly instituted platform also enables a panel screening strategy to efficiently connect HTS, lead identification, and lead optimization in parallel.
Introduction
Drug discovery is a challenging process that requires iterative assessment of compound pharmacological activity during candidate optimization. Lack of efficacy and unanticipated adverse events are two main causes of drug attrition that can result, in part, from poor in vitro biological assessment of compound activities.
In the past decade, monitoring the functional consequences of compound activity through studying gene expression profiles has become a key component in both drug discovery and development efforts to understand complex biological responses in normal and pathological processes. The impact of gene expression profiling has been observed across all phases of the drug discovery process: molecular stratification of disease states, potential drug target selection, patient selection, and target engagement in clinical studies.1,2
Traditional PCR methods have proved to provide useful tools for the development of DNA- and RNA-based applications in drug discovery and development. However, cost and throughput challenges have limited the use of PCR as a transcript screening method for high-throughput screen (HTS) applications. A novel approach for quantitative reverse transcriptase and PCR using real-time detection (qRT-PCR) has been developed. 3 The one-step qRT-PCR method enables the reverse transcription of mRNA and cDNA synthesis in a single well with a shared buffer using a reverse transcriptase along with a DNA polymerase.4,5 This latest development has provided opportunities to minimize the sample handling, decrease bench time, and reduce assay variability. Those benefits have made the one-step qRT-PCR an especially strong choice for quantitating gene expression in HTS applications.
In order to improve our ability to better understand desired versus undesired pharmacological profiles of compounds during lead optimization, we have developed a high-throughput platform for the assessment of gene expression signatures in key primary in vitro cellular models. Furthermore, we have leveraged automation to develop a capability to simultaneously screen multiple high-throughput platforms6,7 in a parallel fashion in order to significantly improve the speed of hit identification, evaluation, and optimization as part of delivering novel drug candidates into clinical development. 8
While introducing the one-step qRT-PCR technology into our platform we found that high cost and low throughput were significant challenges due to the use of expensive commercial reagents and a manual assay operation mode. Here we report our effort to leverage automation and process innovation for the establishment of a high-throughput automated one-step qRT-PCR platform.
Materials and Methods
Reagents
Human lymphotoxin beta R/TNFRSF3 antibody was purchased from R&D Systems (Minneapolis, MN). All Bristol-Myers Squibb (BMS) test compounds were made internally at BMS. BIRC3, CTAG1A, MAGE-A4, CCL3, RPL32, GAPDH Taqman probes, and PCR plates were obtained from Applied Biosystems (Waltham, MA). qRT-PCR cell (assay) plates were obtained from VWR (Bridgeport, NJ). Taqman Fast virus one-step master mix was provided by Thermo Fisher Scientific (Waltham, MA). Ultrapure distilled water (DNAse and RNAse Free) and RNAsecure were from Invitrogen (Carlsbad, CA). ImmunoCult Human CD3/CD28 T Cell Activator was purchased from STEMCELL Technologies (Kent, WA). CD3+ T cells were obtained from Biological Specialty Corporation (Colmar, PA).
Cell Cultures
HCT116 and U2OS cell lines were purchased from the American Type Culture Collection (Manassas, VA). U2OS (ATCC, cat. HTB-96) was cultured in Dulbecco’s modified Eagle’s medium (DMEM; Invitrogen, cat. 12430-047) supplemented with 10% fetal bovine serum (FBS; Seradigm, cat. 1500-500) and 1% pen/strep (Invitrogen, cat. 15140-122). HCT116 (ATCC, cat. CCL-247) was grown in RPMI 1640 (Invitrogen, cat. 11875-085) plus 10% FBS (Invitrogen, cat. 16140-047), 10 mM HEPES (Invitrogen, cat. 15630-080), and 1% anti/anti (Invitrogen, cat. 15240-096). CD3+ T cells were maintained in RPMI 1640 (Invitrogen, cat. 11875-085) supplemented with 5% FBS (Invitrogen, cat. 16140-047).
For the one-step BIRC3/U2OS qRT-PCR assay, 3000 U2OS cells were seeded onto the 384-well assay plate (Greiner Bio-One, cat. 781090-3B) in 40 µL of assay medium with DMEM (Gibco, cat. 12430-047) plus 10% FBS (Seradigm, cat. 1500-500).
The cell plate was then incubated overnight at 37 °C, 5% CO2 environment. For the one-step CTAG1A/MAGE-A4/HCT116 qRT-PCR assay, HCT116 cells were plated in a 384-well assay plate (Greiner Bio-One, cat. 781090-3B) at a density of 1000 cells/well in 40 µL of assay medium with RPMI 1640 (Gibco, cat. 11875), 10% FBS (Gibco, cat. 0173), 1% pen/strep, and 1% HEPES. The cell plate was then incubated overnight at 37 °C, 5% CO2 environment.
Test Compound Preparation
For the one-step qRT-PCR, test compounds were serially diluted in DMSO with fivefold dilutions for seven concentration points ranging from 1.28 × 10–6 to 2 × 10–2 M.
One-Step qRT-PCR Manual Assays
One-Step BIRC3/U2OS qRT-PCR Assay
Cell plates with U2OS cells were treated with 100 nL of serially diluted compounds using an Echo 550 liquid handler (Labcyte, Sunnyvale, CA). The U2OS cells were incubated with test compounds for 1 h in a humidified 37 °C, 5% CO2 incubator. Then 10 µL of human lymphotoxin beta R/TNFRSF3 (R&D Systems, cat. AF629) antibody was added to each well in columns 1 and 3–24 to a final concentration of 333 ng/mL. Ten microliters of the media was added onto wells in column 2 and the stimulation was 5 h at 37 °C, 5% CO2 incubator. After anti-human lymphotoxin/beta R induction, cells were washed two times using Dulbecco’s phosphate-buffered saline (DPBS) at room temperature; then cells were lysed using 10 µL of lysis buffer (0.3% Triton [Bio-Rad, cat. 1610407] with 1× RNAsecure [Invitrogen, cat. AM7005] in DPBS [Invitrogen, cat. 14190-144]) at room temperature for 20 min. Partial lysate was transferred to a 384-well PCR plate, and then mRNA was reverse transcribed and cDNA was amplified with Taq polymerase using TaqMan Fast virus one-step master mix (Thermo, cat. 4444432), target probe BIRC3 (Applied Biosystems, cat. Hs00985031_g1/4351370), and housekeeper probe GAPDH (Applied Biosystems, cat. Hs02786624_g1/4448490). The PCR volume was 6.7 µL in a 384-well PCR plate (Applied Biosystems, cat. 4483285) consisting of 0.7 µL of lysate, 1.7 µL of the 4× master mix, 3.7 µL of RNase free water, and 0.3 µL of each probe. Amplification, detection, and analysis were performed by a QuantStudio 12K Flex Real-Time PCR System (Applied Biosystems, cat. 4471050). The mRNA reverse transcription step involved an incubation at 50 °C for 5 min. The PCR cycling conditions included an initial denaturation of 95 °C for 20 s, followed by 40 cycles of 95 °C for 3 s and 60 °C for 30 s.
One-Step CTAG1A/MAGE-A4/HCT116 qRT-PCR Assay
Cell plates with HCT116 cells were treated with 100 nL of serially diluted compounds using an Echo 550 liquid handler for 72 h at 37 °C, 5% CO2 incubator. After 72 h of incubation, cells were washed two times using DPBS at room temperature; then cells were lysed at room temperature for 20 min using 0.3% Triton (Bio-Rad, cat. 1610407) in DPBS (Invitrogen, cat. 14190-144) with 1× RNAsecure (Invitrogen, cat. AM7005). Partial lysate was transferred to a 384-well PCR plate, mRNA was reverse transcribed, and cDNA was amplified with Taq polymerase using the TaqMan Fast virus one-step master mix (Thermo, cat. 4444432), target probe CTAG1A (Applied Biosystem, cat. Hs01126052_m1/4351370), MAGE-A4 (Applied Biosystems, cat. Mm00522322_s1/4351370), and housekeeper probe GAPDH (Applied Biosystems, cat. Hs02786624_g1/4448490). The PCR consisted of 0.7 µL of lysate, 1.7 µL of the 4× master mix, 3.7 µL of RNase free water, and 0.3 µL of each probe. The PCR program conditions were 1 cycle at 50 °C for 5 min and 1 cycle at 95 °C for 20 s, followed by 40 cycles of thermal cycling at 95 °C for 3 s and 60 °C for 30 s.
One-Step CCL3/CD3+ T-Cell qRT-PCR Assay
CD3+ T-cell (Biological Specialty Corp., cat. 215-01-10.M6521) cells were thawed in RPMI 1640 medium (Invitrogen, cat. 11875-085) with 5% FBS (Invitrogen, cat. 16140-047). A total of 40,000 cells/well in 20 µL of RPMI 1640 media with 5% FBS were seeded in a 384-well assay plate, and the cells were incubated at 37 °C for 1 h. After 1 h of settling, 100 nL of compounds or DMSO was added in the presence or absence of 100 nL of stimulator. The assay plate was incubated for 16 h in a 37 °C, 5% CO2 incubator, and then cells were lysed at room temperature for 20 min using 10 µL of Roche lysis buffer (Roche, cat. 05943523001) according to the manufacturer’s recommendation. TaqMan Fast virus one-step master mix (Thermo, cat. 4444432) with target probe CCL3 (Applied Biosystem. Hs00234142_m1/4351370) and housekeeper probe RPL32 (Applied Biosystems, cat. Hs00851655_g1/4448490) was prepared. Lysate (0.7 µL) was transferred to a 384-well PCR plate. Then, 1.7 µL of the 4× master mix, 3.7 µL of RNase free water, and 0.3 µL of each probe were added into the 384-well PCR plate to a total volume of 6.7 µL for PCR. The PCR cycling reaction was carried out at 50 °C for 5 min for the reverse transcription; the cDNA amplification was directly followed by 40 cycles at 95 °C for 3 s and 60 °C for 30 s.
Data Analysis
For the assay data correlation, GraphPad Prism (version 7.03; GraphPad, La Jolla, CA) was used to generate the r2 values, p values, and 95% confidence intervals in a regression model. For the assay performance evaluation, all shown p values were calculated in GraphPad Prism (version 7.03) using a logistic regression model with the significance threshold set to p < 0.05.
For the one-step qRT-PCR data analysis, the 2[–ΔΔC(T)] method 9 for determining relative quantification of changes in gene expression relative to a reference sample was used. We defined Ct as the fractional PCR cycle number at which the reporter fluorescence is greater than the threshold. The Ct values were obtained from real-time PCR instrumentation Quant Studio 12K Flex (Applied Biosystems) and retrieved to the BMS data engine through the BMS in-house data processor. 10 Precalculation for 2[–ΔΔC(T)] was set up in BMS proprietary software, Toolset, and dose–response curves of compounds and IC50 values were quantified using the four-parameter logistic equation built into BMS proprietary software, Curvemaster.
Results and Discussion
Automation System Design and Execution
We have established a robotic platform Workstation 7 (WS7) containing liquid handlers and instruments capable of reagent dispensing, plate washing, and aspiration. The WS7 main deck ( Fig. 1A ) is based on a Thermo Scientific Fanuc F7 robot system (Waltham, MA) ( Fig. 1B ), controlled by Thermo Scientific Momentum software for robot scheduling and execution. The integrated automated WS7 system has additional components, including a Thermo Scientific CRS automated carousel and two Thermo Scientific Cytomat incubators set at 4 and 37 °C gassed with 5% CO2. The additional components comprise a Velocity11 PlateLoc sealer (Agilent Technologies, Santa Clara, CA), three Velocity11 Vspin centrifuges (Agilent Technologies), two Thermo Scientific CRS regrip turntables, two sets of EL406 (BioTek, Winooski, VT) washer and dispenser stations, and three Thermo Scientific Multi-drops for reagent dispensing. In the WS7 system, a Tempest liquid handler (Formulatrix, Bedford, MA), a Bio-shaker (Q Instruments, Jena, Germany), and CyBio Felix liquid handler (Analytik-Jena, Jena, Germany) ( Fig. 1C ) are also incorporated.

Automated WS7 system and Thermo Scientific QuantStudio Robotic Station at BMS. (
All components of the WS7 system are fully integrated and provide a walk-away operation that delivers an assay completion plate. The gene expression profiling ready plate is then transferred for the qRT-PCR performed on the Thermo Scientific Spinnaker Microplate Robot controlled with QuantStudio (Applied Biosystems, Foster City, CA) automation platform ( Fig. 1D ).
The manual one-step qRT-PCR assays were described in the Materials and Methods section. The automated one-step qRT-PCR assays inherited the same assay conditions, including assay volumes, cell density, compound concentrations, incubation times, and handling of the sample.
Automated One-Step BIRC3/U2OS qRT-PCR Assay Workflow for Compound Profiling
Traditionally, the BIRC3/U2OS qRT-PCR assay was performed manually and involved labor-intensive processing. During the automated assay, cell plates were manually loaded to a 37 °C SteriStore incubator (HighRes Biosolutions, Beverly, MA) in the automated PrintStation, which is an automated ACell platform (HighRes Biosolutions) controlled by the proprietary scheduling software package Cellario. Following a 24 h incubation, the assay was begun with the compound stamp using the ATS Gen 5 (EDC Biosystems, Fremont, CA) in PrintStation. The assay plates were then transferred to a 37 °C Cytomat incubator on the WS7 platform for completion of the assay.
The integrated WS7 system provides a wash/aspiration capability that enables an automated biocomplexity platform. The fully automated BIRC3/U2OS qRT-PCR assay method was established as a mirror image of the manual assay. The automation workflow was depicted in Figure 2 for profiling compound inhibitory activity of BIRC3 expression in U2OS cells.
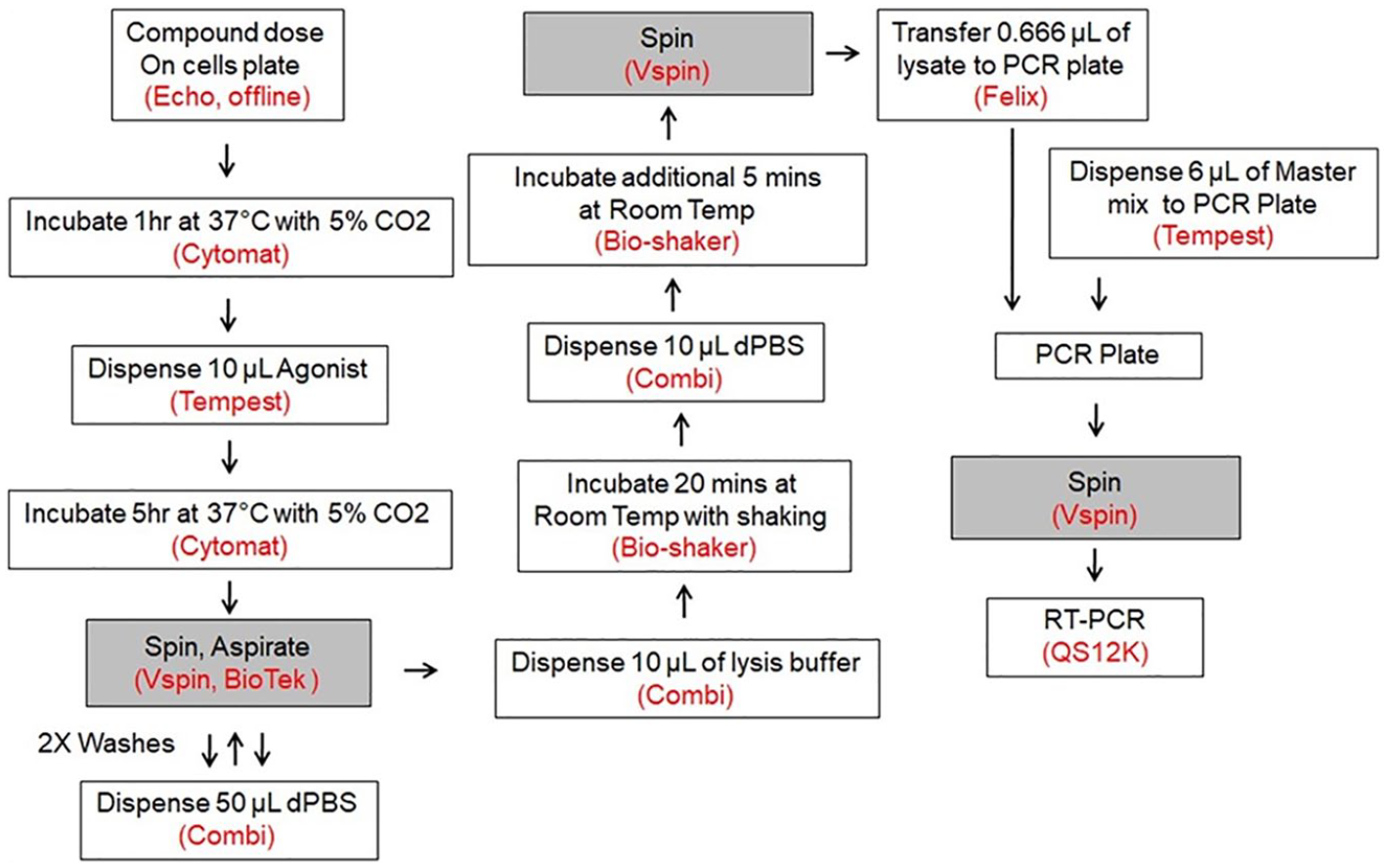
Flowchart of the robotic BIRC3 one-step qRT-PCR assay protocol. After the compound was dosed offline, the assay plate was placed on the WS7 platform. Ten microliters of agonist was added onto the assay plate post the 1 h incubation. Cells were washed and lysed at the end of 5 h of induction; lysate and qPCR master mix were then transferred into the PCR plate. The PCR plate was transferred to the one-step qRT-PCR robot station, and the RT-PCR was performed on the QuantStudio 12K.
Validation of the In-House Lysis Conditions
The process for the manual BIRC3/U2OS qRT-PCR assay used a commercially available (Roche) lysis buffer that proved to be cost-prohibitive for use in high-throughput applications. To reduce cost, we looked to develop an optimized in-house one-step lysis buffer. Several approaches were explored, including Triton and 1× RNAsecure in PBS.11,12 Triton titration and Roche lysis buffer were tested in parallel in the qRT-PCR assay to measure GAPDH amplification in the U2OS cells; the GAPDH CT means, standard deviations, and coefficients of variation (CVs) were compared ( Suppl. Table S1 ). The optimized in-house lysis buffer consists of 0.3% Triton and 1× RNAsecure (Invitrogen, cat. AM7005) in PBS. To evaluate the in-house lysis condition, reference control compounds and compounds 1 and 2 were studied using either the in-house lysis reagent or the commercially available lysis reagent ( Fig. 3A,B ). Compounds 1 and 2 showed comparable activity using the in-house lysis conditions (IC50 = 94.3 and 13.9 nM, respectively) compared with the commercial reagent conditions (IC50 = 109.0 and 16.6 nM, respectively). Additionally, assay statistics including the means for the assay total (T) and background (B), the standard deviations (SDs) for the assay total (T) and background (B), and the CVs for the assay total (T) and background (B) were presented. Comparing those statistical parameters showed no significant difference between the two lysis conditions (p < 0.05). Also, the Z′ value and assay window (S/B) were not significantly different (p < 0.05) when comparing the two cell lysis methods in the assay protocol ( Fig. 3C ).
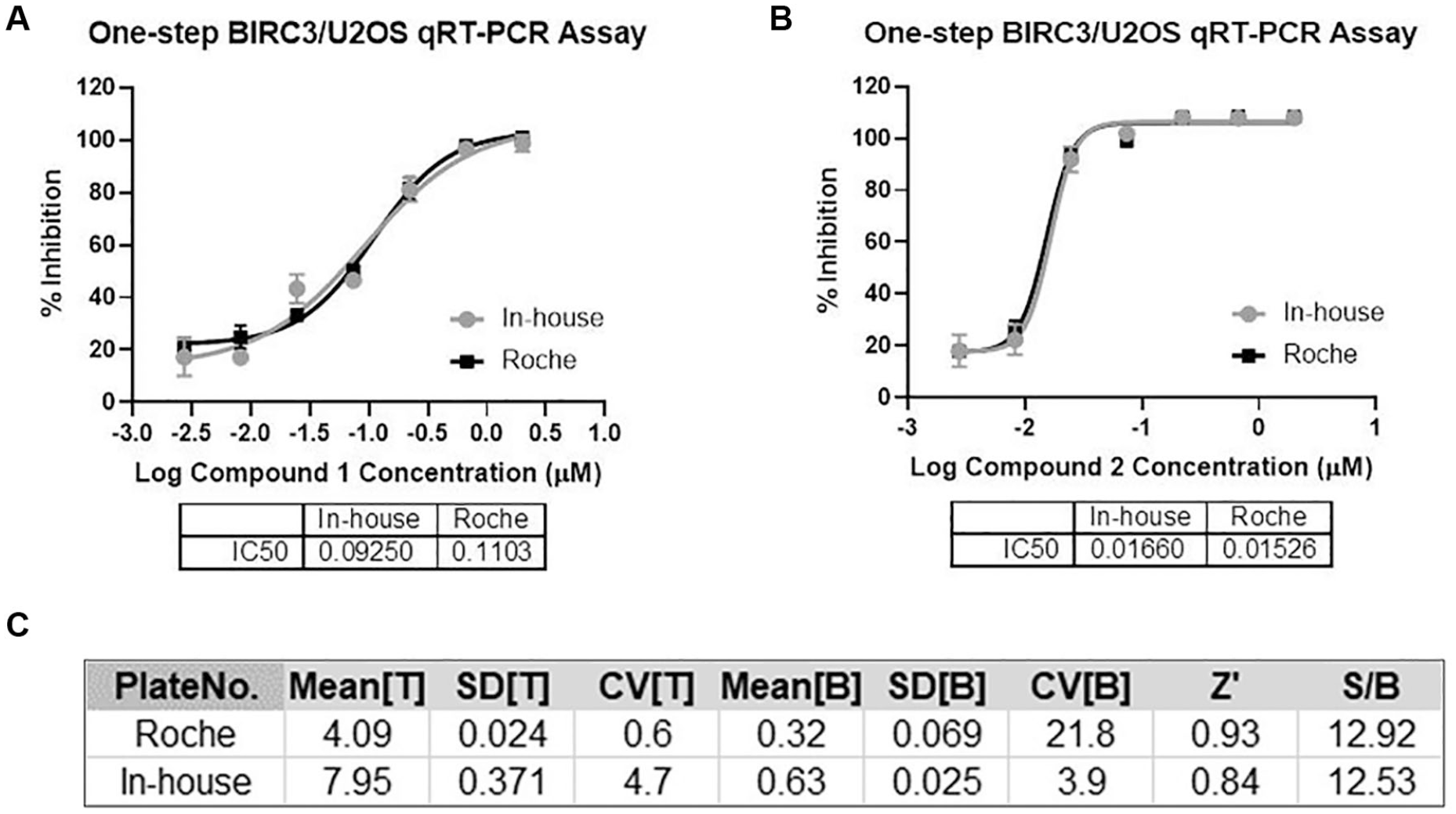
Validation of the lysis condition for one-step qRT-PCR assay. Comparison of the Roche lysis buffer and in-house lysis conditions for reference control compounds 1 (
Coupled with automation capability, the lysis buffer developed in-house dramatically improved the broad applicability of the one-step qRT-PCR platform in a cost-effective manner.
Validation of the Automated One-Step qRT-PCR Assay
Automated one-step qRT-PCR platform validation was carried out using the in-house lysis conditions via the BIRC3/U2OS, CTAG1A/HCT116, and MAGE-A4/HCT116 one-step qRT-PCR assays.
Baculoviral inhibitors of apoptosis protein (IAP) repeat containing 3 (BIRC3; also known as AIP1, API2, MIHC, cIAP2, HAIP1, HIAP1, IAP-1, MALT2, RNF49, and c-IAP2) encodes a member of the IAP family of proteins that inhibits apoptosis by binding to tumor necrosis factor receptor-associated factors TRAF1 and TRAF2. 13
Multiple lines of evidence have shown TRAF1 to have a role in various cancers, autoimmunity, rheumatoid-arthritis associated sepsis, and cardiovascular disease. 14 This study has found that TRAF2 promotes the invasiveness of pancreatic cancer cells. 15
BIRC3 is a multifunctional protein that regulates apoptosis and modulates inflammatory signaling/cell proliferation, as well as cell invasion and metastasis. 16 Studies have shown that BIRC3 disruption is implicated in fludarabine-refractory chronic lymphocytic leukemia. 17 BIRC3 upregulation has also been observed in gastrointestinal stromal tumors. 18 In addition, cellular inhibitors of apoptosis proteins (cIAPs) have been reported to be key regulators of nucleotide-binding and oligomerization domain-containing (NOD) signaling in support of innate immunity. 19 Based on the important role of BIRC3 in inflammatory and apoptotic signaling, we have utilized it as a key biomarker in our gene expression panel for drug discovery programs.
For the automated BIRC3/U2OS one-step qRT-PCR assay validation, we compared IC50 values obtained from both the manual and automated assays for 94 compounds that covered a wide range of potencies. Comparison of relative potencies in the two assay formats produced an average correlation coefficient of 0.91 (p < 0.0001) ( Fig. 4A ). Comparing the statistical parameters for each protocol revealed no significant difference between the two assay methods (p < 0.05) ( Fig. 4B ). These results clearly demonstrate that converting the assay format from manual mode to automation mode provides consistent pharmacological data for compounds with comparable assay quality.
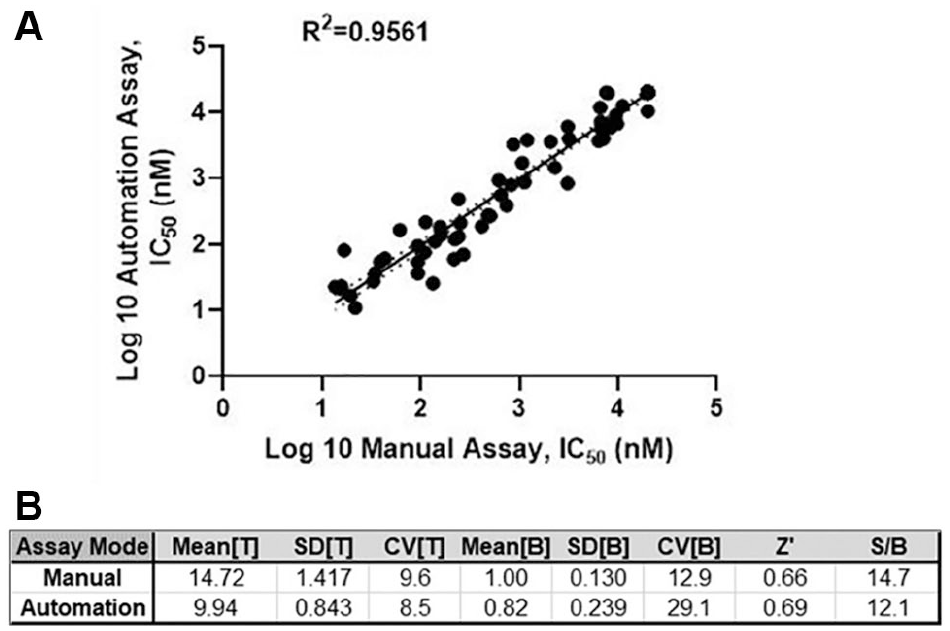
Automated one-step qRT-PCR assay validation. (
Trans-lesion synthesis (TLS) is an important DNA damage tolerance mechanism that permits continuing DNA synthesis in cells harboring damaged genomes. 20 Melanoma antigen A4 (MAGE-A4), a tumor cell-specific protein, contributes to TLS pathway activation, DNA damage tolerance, and genome maintenance in cancer cells. 21 MAGE-A4 has been reported in diverse human malignancies, including uterine malignancies, ovarian neoplasms, hepatocellular carcinomas, mucosal melanomas of the head and neck, esophageal adenocarcinomas, and colorectal cancer. 22
Cancer-testis antigen 1A (CTAG1A; also known as NY-ESO-1) is a tumor cell antigen found in various types of cancers. CTAG1A expression was restricted to normal testis and ovary, which is activated in a wide range of tumors, provoking both antibody and cell-mediated immune responses in cancer patients. 23
Both MAGE-A4 and CTAG1A are members of cancer-testis (CT) antigens; their expression has been found solely in the testis among the normal adult tissues. In malignancy, this gene regulation is disrupted, resulting in CT antigen aberrant expression in various types of malignant tumors. This expression pattern has made both MAGE-A4 and NY-ESO-1 targets for cancer immunotherapy.22,24
We have included CTAG1A and MAGE-A4 as part of our automated, high-throughput gene expression profiling panel. In order to validate assays for these biomarkers in our panel, six compounds exhibiting a range of potencies were evaluated, IC50 values were obtained and compared for the automated CTAG1A ( Suppl. Table S2 ) and MAGE-A4 ( Suppl. Table S3 ) qRT-PCR assays using manual assays in the HCT116 cell background. SDs of concentration–response experiments comparing IC50 values from three independent tests are presented. The automated one-step qRT-PCR assay was within a threefold difference of values obtained from manual assays. Pharmacology data demonstrated good alignment comparing these assay formats.
With the aid of automated technology and innovative science, 20 steps of assay procedures were fully integrated into the qRT-PCR platform to provide a walk-away operation that eliminates much of the tedium of manual lab work. As a result, a total of 6–8 h can be saved for assay scientists for the BIRC3/U2OS qRT-PCR assay.
Evaluation of the Automated One-Step qRT-PCR Assay Performance
Assays employed in HTS and lead optimization in drug discovery research should be rigorously validated for both biological and pharmacological relevance, as well as for robustness and consistency of assay performance. In an HTS environment, assay performance is an important measurement for assay risk evaluation and mitigation strategy for potential assay variability. Statistical reproducibility is a measurement for robustness and reliability for an ideal HTS assay format. 25 To determine the assay reproducibility, 96 compounds with a wide range of potencies have been tested through the WS7 BIRC3/U2OS one-step qRT-PCR assay; IC50 data for 94 compounds (2 compounds displayed visual precipitation and their data were excluded for the comparison) were compared with experiment 1 automation assay. Comparison of the two independent tests yielded an excellent correlation in terms of observed potencies (r2 = 0.99, p < 0.0001) for automation experiment 2 versus experiment 1, indicating an excellent automation assay reproducibility ( Fig. 5A ). Similarly, other statistical parameters were in line, comparing experiment 2 versus experiment 1 automation assays (p < 0.05) ( Fig. 5B ).
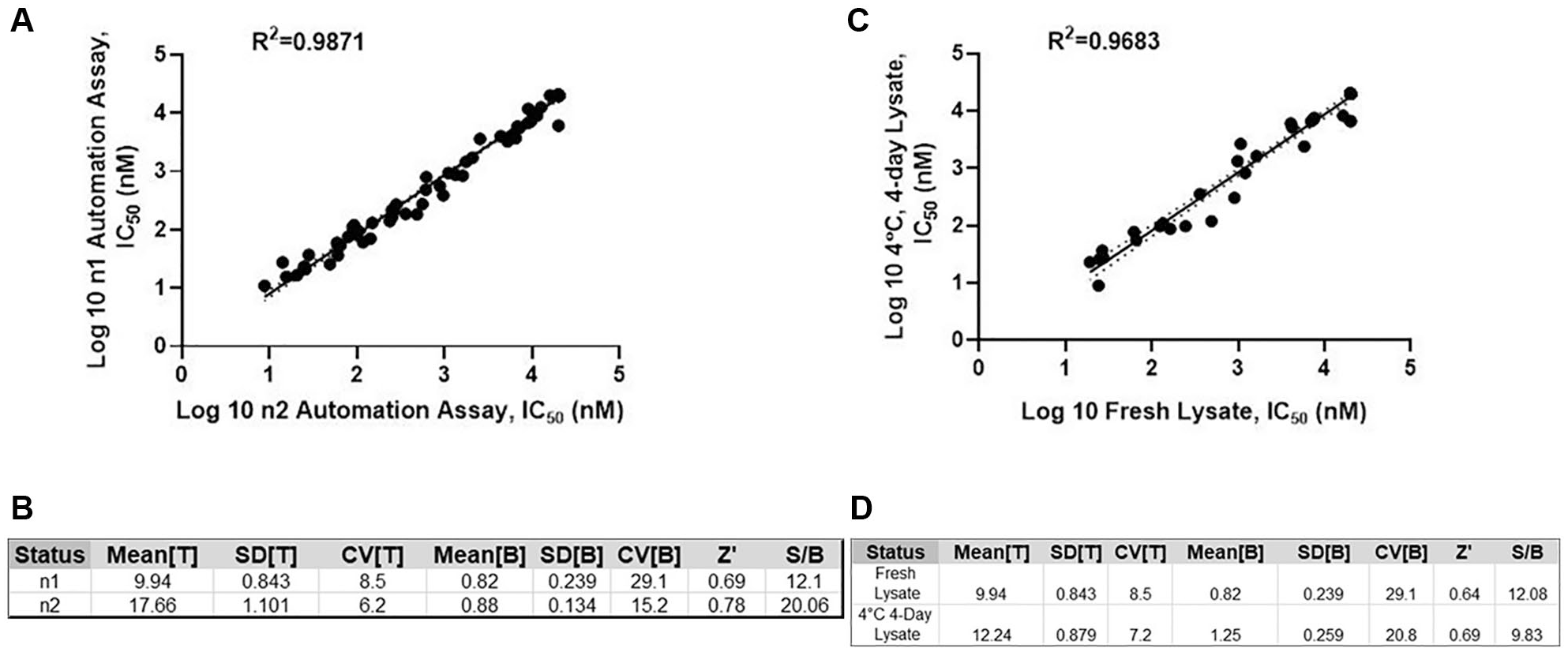
Automated one-step qRT-PCR assay performance. (
Reagent stability can have important effects on assay fidelity, and it is critical particularly for our panel assay design and operation in an HTS environment. 26 In addition, reagent and sample durability plays a vital role in the quality assurance and quality control of an assay. This is especially important for the one-step qRT-PCR profiling since each of the PCRs takes 1 h per plate, necessitating storage of sample lysates and/or PCR mixes until they can be processed and run in the qRT-PCR and detection. To develop our gene profiling strategy for our HTS workflow, the stability of samples and reagents under storage and assay conditions have been determined. In our initial approach, stability was validated through lysate storage stability tests at both 4 °C and room temperature for the WS7 BIRC3/U2OS one-step qRT-PCR assay. We also tested the premix stability for the lysate and qPCR master mixtures at both 4 °C and room temperature. We observed that both the lysate and premix lost activity within 1 h at room temperature compared with freshly made lysate and qPCR mix (data not shown), which is most likely due to RNA degradation at room temperature.
In order to evaluate the storage tolerance of the cell lysates at 4 °C, 48 compounds were tested through the BIRC3/U2OS one-step qRT-PCR assay using the freshly made lysate compared with the lysate stored at various time points for up to 4 days. IC50 values were plotted using the linear regression analysis in GraphPad and yielded an r2 of 0.97 (p < 0.0001), demonstrating good stability of the lysate for the 4-day storage ( Fig. 5C ). The assay performance parameters displayed a p value of <0.05, which indicated good alignment between both preparations of lysates in the assay ( Fig. 5D ). Lysate storage longevity has not been evaluated beyond a 4-day storage at 4 °C.
To assess the qPCR premix stability at 4 °C, 12 compounds were evaluated for their inhibition of the human lymphotoxin beta R/TNFRSF3 antibody-induced U2OS BIRC3 one-step qRT-PCR assay. IC50 data of 12 test compounds were examined; the IC50 values of 12 compounds demonstrated good correlation when comparing the freshly made mix with the premix at 4 °C storage for 6 h. Pharmacology data represented the mean ± SD (n = 3) ( Suppl. Table S4a ). Assay performance comparison exhibited compatibility indicating good stability and functionality for up to 6 h of storage at 4 °C (p < 0.05) ( Suppl. Table S4b ).
The stability results shed light for us on the strategic planning for gene expression profiling. Also, these results have enabled the Momentum software scheduling with flexibility for either one compound plate with multiple genes, several compound plates with one gene, or several compound plates with multiple genes within the limits we determined for reagent stability. The lysate stability findings also provide flexibility in order to retest compounds in the event that there is a failure during the gene expression profiling operation run.
CCL3/CD3+ T-Cell One-Step Automated qRT-PCR Assay for Screening Effort
We have found increased T-cell activity and antitumor efficacy in target knockout (KO) mice compared with wild-type (WT) littermates in one of our oncology programs, and elevated CCL3 expression in the target KO correlated with an antitumor phenotype.
A representative deck of 40,414 compounds was assessed at 10 µM through the primary HiBit target screen. After triaging for cytotoxicity and selectivity follow-up characterization, a collection of 373 compounds from the primary screen were evaluated through the CCL3/CD3+ T-cell one-step qRT-PCR assay. Eighty-two hits were identified that exhibited IC50 values of <1 µM; the screening results are summarized in Figure 6A . Compound 27 was identified from the CCL3/CD3+ T-cell one-step qRT-PCR screen with an IC50 of 63 nM. Compound 27 displayed an IC50 of 80 nM from the HiBit primary target screening shown in Figure 6B . A graphical representation of a CCL3/CD3+ T-cell qRT-PCR screening plate is illustrated in Figure 6C . All 83 hits have been confirmed in follow-up tertiary screening, and new compound syntheses around the structure cores of these hits are ongoing to identify the leading compounds.
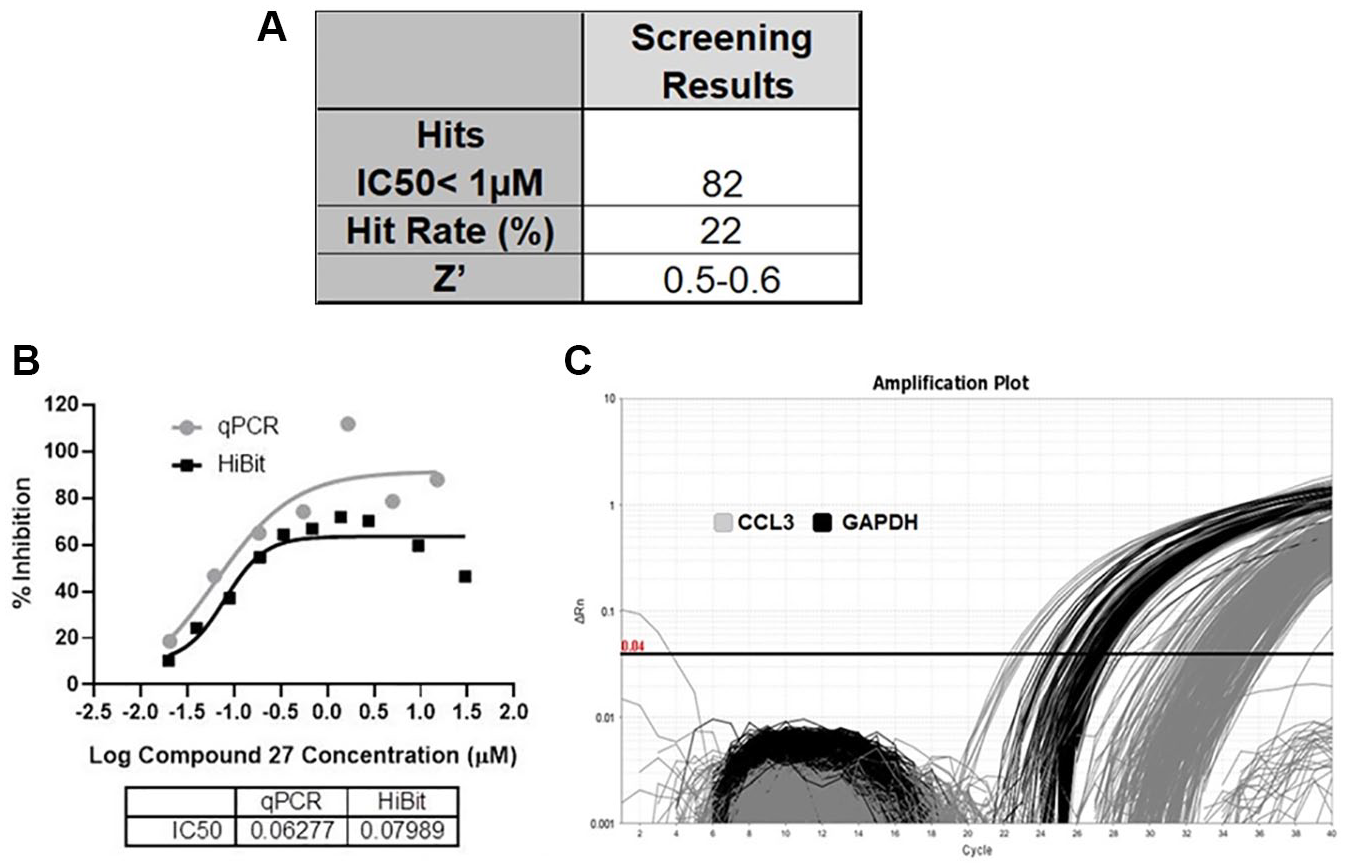
CCL3/CD3+ T-cell one-step qRT-PCR screening result from a representative deck. (
By leveraging automation, we have launched an automated CCL3/CD3+ T-cell one-step qRT-PCR assay to enable rapid parallel screening of hits from a primary HiBit target screen. Our data have provided in vitro evidence that CCL3 is a biomarker gene for the target of interest.
By technology advancement and process innovation, we have developed, validated, and launched BIRC3/U2OS qRT-PCR and CTAG1A/MAGE-A4/HCT116 one-step qRT-PCR assays as an automation platform using in-house optimized lysis conditions. Currently, we are in the process of refining the in-house lysis conditions for other types of primary cell qPCR assays. Additional parallel HTSs with compound libraries using the single-point concentration format are also ongoing.
Our newly validated automated one-step qRT-PCR platform is a streamlined process with an increased capacity that reduces the hands-on time and repetitive tasks for the assay operator. The automation system not only allows us to effectively use automation white space for genotypic profiling, but also enables us to fulfill a parallel screening strategy. Also, the scientific competent in-house lysis conditions add a cost-efficient, powerful fuel to our technology advancement. Altogether, the automated one-step qRT-PCR platform potentially would accelerate the time frames for compound discovery and optimization and enable more efficient searches in the small-molecule drug space.
Drug discovery is a fast-paced, continually evolving field. In recent years, gene editing, CRISPR, and gene therapy have become hot spots for both drug discovery and development in the areas of drug target validation, drug candidate characterization, improvement of patient stratification, therapy selection, and disease monitoring. 27
In addition, emerging single-cell technologies have become increasingly important in biological analysis to provide a more detailed picture of complex biology and unmask the heterogeneity that is present in tissues. 28
The newly established automated gene expression capability will enable the expanding landscape of BMS HTS research to engage in gene editing, CRISPR, and single-cell technologies to facilitate drug discovery for HTS, lead evaluation, and optimization. These new capabilities will help to identify new drug targets using new technologies and perspectives.
Supplemental Material
Supplemental_Material_for_Leveragig_Automation_Toward_by_Chen__et_al_0529_2020 – Supplemental material for Leveraging Automation toward Development of a High-Throughput Gene Expression Profiling Platform
Supplemental material, Supplemental_Material_for_Leveragig_Automation_Toward_by_Chen__et_al_0529_2020 for Leveraging Automation toward Development of a High-Throughput Gene Expression Profiling Platform by Jing Chen, Alan Futran, Austin Crithary, Sha Li, Alex Wolicki, Kylie Fogarty, Joe Baldick, Peter Chase, Brian J. Arey and Jonathan Lippy in SLAS Discovery
Footnotes
Acknowledgements
The authors thank Jordan Blum and Mark Fereshteh for their critical contribution as team members in this effort at BMS. We are thankful for the valuable feedback of Ying-Kai Wang for this manuscript. We are appreciative of Christian D. Ferrante, Brittany Guerriero, and John Marrazo for the creation of compound plates for experimentation. Also, we recognize the efforts of Humberto D. Ribeiro, Monica Kwok, and Bruce Braender for the cell culture support. We are sincerely grateful to Litao Zhang for her support, and we thank the BMS Immunology and Oncology Discovery teams for compound supplies for this study.
Supplemental material is available online with this article.
Declaration of Conflicting Interests
The authors declared no potential conflicts of interest with respect to the research, authorship, and/or publication of this article.
Funding
The authors received no financial support for the research, authorship, and/or publication of this article.
References
Supplementary Material
Please find the following supplemental material available below.
For Open Access articles published under a Creative Commons License, all supplemental material carries the same license as the article it is associated with.
For non-Open Access articles published, all supplemental material carries a non-exclusive license, and permission requests for re-use of supplemental material or any part of supplemental material shall be sent directly to the copyright owner as specified in the copyright notice associated with the article.