
Abstract
Objective
Osteoarthritis (OA) is a multifactorial disorder, in which genetic factors are strongly associated with its development. However, the pathogenesis of OA is still unclear, and recently it has been observed that epigenetic modifications are also involved in the pathogenesis of OA. This study aims to study the potential role of m6A-related genes in the occurrence and development of OA.
Design
We downloaded the OA expression profile data (GSE55235) from the Gene Expression Omnibus database. First, function enrichment analysis of 17 representative m6A methylation regulatory factors was performed using the DAVID database and Metascape online tool. Then, we analyzed the expression of 17 m6A methylation regulatory factors in OA and the correlation between regulatory factors using Perl software. Finally, receiver operating characteristic (ROC) curve analysis and the area under the ROC curve were used to evaluate the diagnostic effectiveness of m6A-related genes for discriminating patients with OA and healthy.
Results
We first identified that 12 of the 17 genes were differentially expressed in OA. ALKBH1, EIF3, IGF2BP3, WTAP, and YTHDC1 were associated with early diagnosis and prognosis of OA.
Conclusions
m6A RNA methylation regulator factors are key players in the progression of OA and have potential role in the stratification of prognosis and the formulation of treatment strategies.
Introduction
Osteoarthritis (OA) is the most common form of arthritis; the main characteristics are matrix degradation and collagen synthesis imbalance, cartilage cell metabolism disorder, inflammation activation, and cartilage aging.1,2 According to statistics, the prevalence rate of OA in people over 60 years old can reach 60% to 65%, and with the aging of the population and the increase in obesity, its prevalence is increasing, which seriously threatens human health and quality of life.3,4 There are many factors that affect the occurrence of OA, and its etiology and pathogenesis are unclear.
OA is a complex disorder, in which environmental and genetic factors are strongly related to its development. Genetic heterogeneity does not seem to explain all the characteristics of OA. Therefore, it is important to study the epigenetic factors and mechanisms related to OA progression and treatment response. N6-methyladenosine (m6A) is a universal modification of RNA molecules in eukaryotes, which plays key roles in regulating fundamental biological processes such as gene expression control and regulation of mRNA stability and homeostasis.5,6 Recent studies suggested that m6A is involved in various aspects of RNA metabolism including pre-mRNA splicing, 3′-end processing, translation regulation, and mRNA decay processing. 7 Similar to DNA methylation, m6A modification is reversible and catalyzed by corresponding enzymes, namely, “Adenosine methyltransferases (m6A writers), RNA-binding proteins (m6A readers) and Demethylases (m6A erasers).” The m6A RNA methylation regulator regulates gene expression in epigenetics, thereby playing role in regulating biological processes. Emerging evidence reported m6A modification is involved in the progression of various human diseases, such as rheumatoid arthritis (RA), 8 cancer, 9 obesity, 10 and atherosclerosis (AS). 11 However, little is known about the role of m6A in OA, and the expression and prognostic value of m6A RNA methylation regulator factors have not been reported.
In the present study, we applied bioinformatics methods to analyze the expression patterns of m6A methylation regulators in OA. Furthermore, the relationship between differential m6A genes and the prognosis and early diagnosis of OA was analyzed. We found that the expressions of m6A genes play key roles in the process of OA and firstly identified five genes as potential biomarkers.
Methods
Data sets
The expression profile data sets from the OA data sets (GSE55235) were collected from the Gene Expression Omnibus (GEO) databases, based on the inclusion and exclusion criteria of the GEO. Inclusion criteria include data sets involving human synovial tissue and expression profiling by array. R software was used in pre-treating and transforming gene.
Selection of m6A RNA Methylation Regulator Factors
A list of 17 m6A RNA methylation regulator factors were sorted from the published literature (shown in Table 1),12-14 including m6A writers (METTL3, RBM15, WTAP, and ZC3H13), m6A readers (EIF3, HNRNPA2B1, HNRNPC, IGF2BP2, IGF2BP3, YTHDC1, YTHDC2, YTHDF1, YTHDF2, and YTHDF3), and m6A erasers (FTO, ALKBH1, and ALKBH4). Then, we systematically compared their expression in OA.
m6A-Related Genes.

Bioinformatic Analysis
The OA data sets were converted into expression estimates. Then the background correction, quartile data normalization and probe summarization were performed by the Robust Multi-array average algorithm in the R(v3.6.0) affy package.
The differential expression of m6A-related genes was analyzed using limma method. Two-sided Wilcoxon rank-sum test was used to evaluate the gene expression difference between two groups. Heat maps of differential expression for m6A-related genes were generated using heatmap package, and Corrplot package was used to analyze the correlation between m6A-related genes themselves in R. Functional enrichment was performed using the Sangerbox tools (http://www.sangerbox.com/tool).
To determine the prognostic value of m6A RNA methylation regulators, we performed receiver operating characteristic (ROC) analysis of the expression of m6A RNA methylation regulators in GEO database, and calculated the area under the curve (AUC), as well as the corresponding sensitivity, specificity. P <0.05 is considered statistically significant, and all statistics were performed with SPSS 22.0 software.
Result
Functional Enrichment Analysis
We analyzed 17 m6A-related genes by protein-protein interaction (PPI) analysis using Metascape and STRING online database, and the results are shown in Suppl. Figure S1, which suggests that there is a strong relationship between m6A-related genes. Then GO enrichment analysis showed that they are mainly involved in the regulation of mRNA metabolic process, nuclear speck, and mRNA binding, which contain biological process (BP), cellular component (CC), and molecular function (MF), respectively (Table 2, Fig. 1).
The Top 10 GO Terms of m6A.
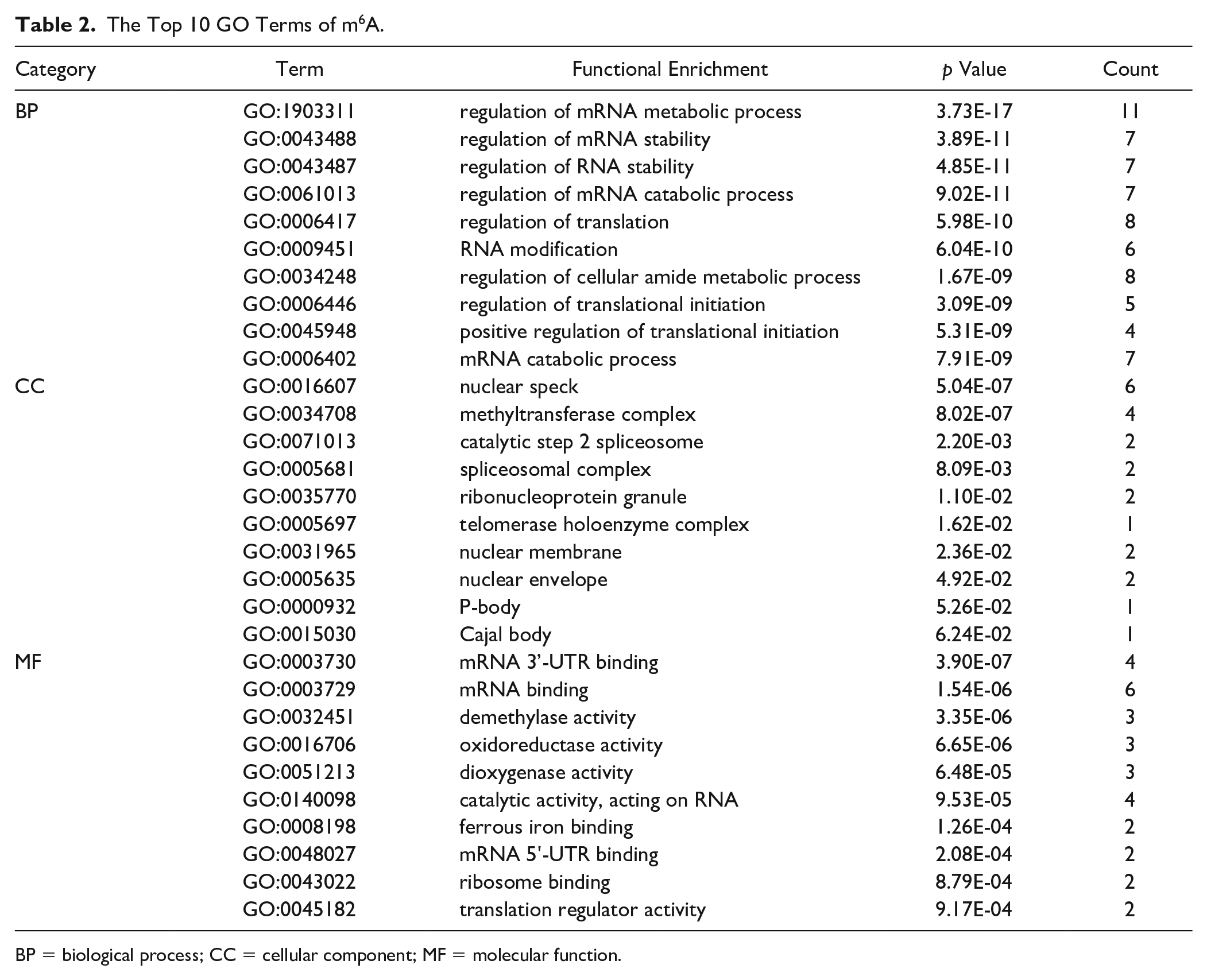
BP = biological process; CC = cellular component; MF = molecular function.
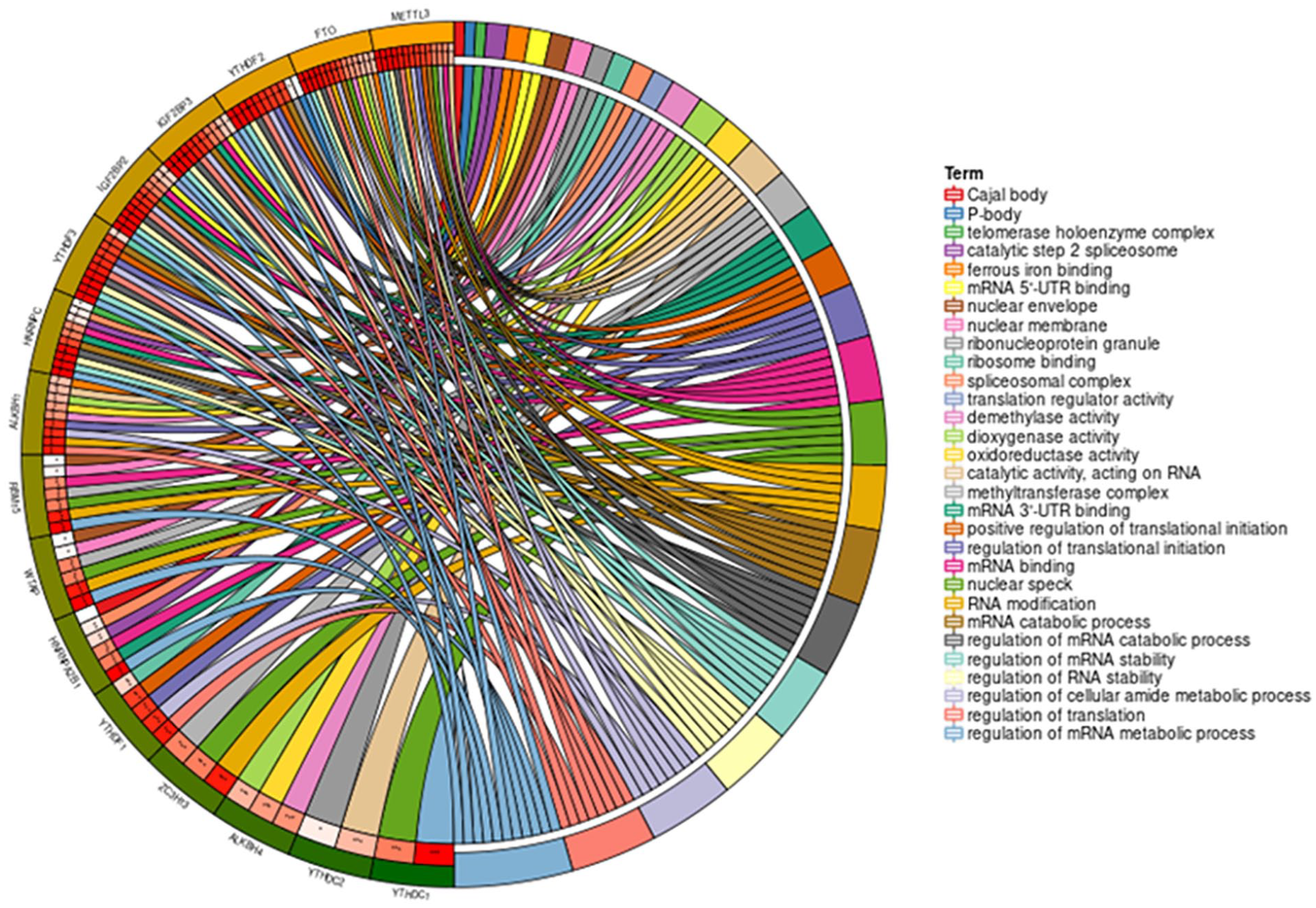
The top 10 GO terms of m6A.
The Expression of m6A RNA Methylation Regulator Factors between Normal and OA Patients
The result of the expression of m6A-related genes between the normal and OA patients is shown in the heat map (Fig. 2), which identified that 12 of the 17 genes were differentially expressed between normal and OA patients (Table 3). Then, quantitative analysis showed that in patients, the expression levels of ALKBH1 (P < 0.0001), EIF3 (P = 0.0005), FTO (P = 0.0337), HNRNPA2B1 (P = 0.0118), IGF2BP3 (P = 0.0001), RBM15 (P = 0.0093), WTAP1 (P < 0.0001), YTHDC1 (P < 0.0001), and YTHDF1 (P = 0.0195) are downregulated, while METTL3(P = 0.0167) and IGF2BP2 (P = 0.0418) are upregulated (Fig. 3). Furthermore, to better understand the interactions of 17 m6A RNA methylation regulators in OA, we further analyzed the correlations among these genes (Suppl. Fig. S2). Both many positive correlations correlation were observed, such as HNRNPA2B1 and EIF3, EIF3 and WTAP, YTHDC1 and IGF2BP3, and so on.
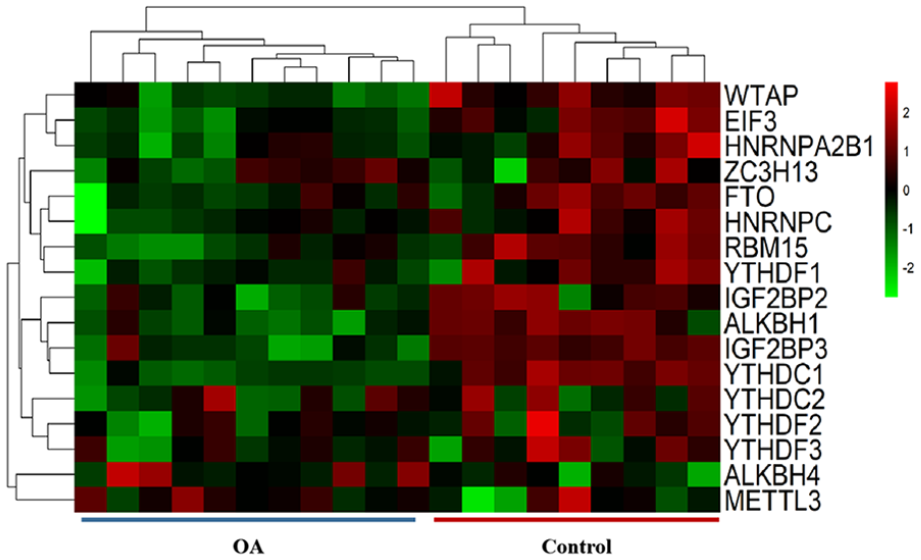
Heat maps of m6A-related genes. The column of the heat map represents samples and the row represents m6A-related genes. Red represents upregulated genes and green represents downregulated genes. OA = osteoarthritis.
The Relative Expression of m6A-Related Genes.
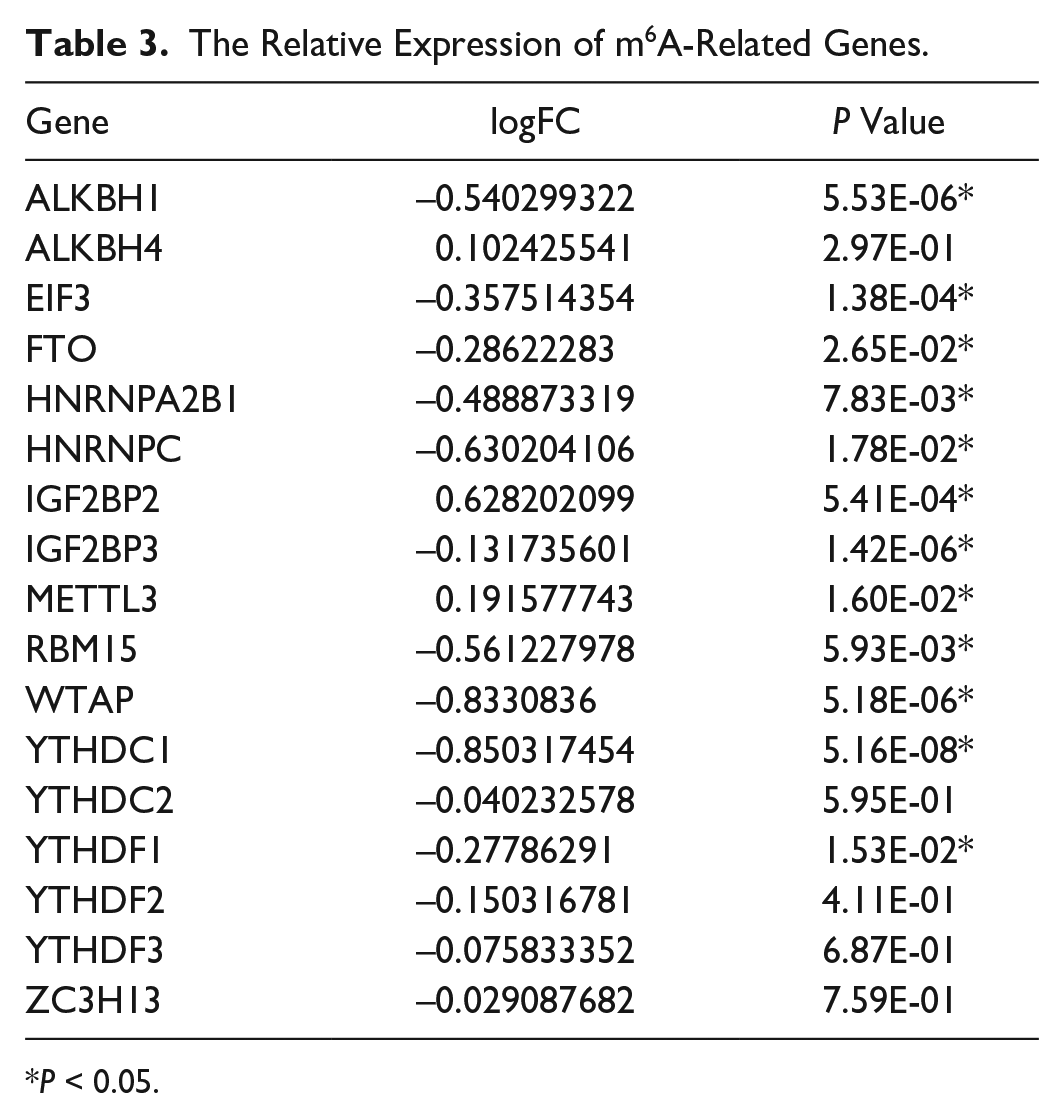
P < 0.05.
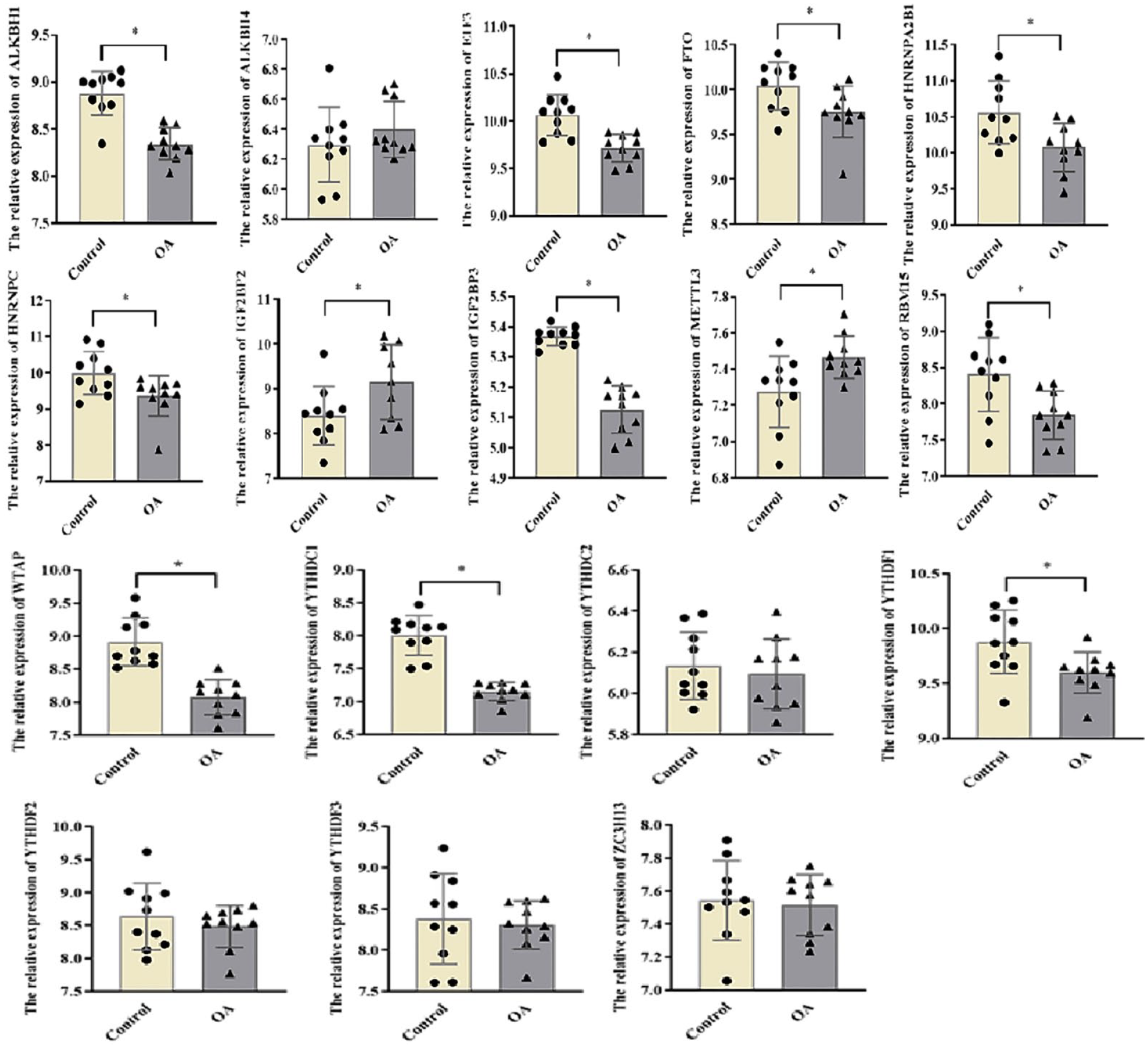
The expression of m6A-related genes. OA = osteoarthritis.
Diagnosis Value of m6A RNA Methylation Regulators in OA
To explore the relationship between m6A-related genes and early diagnosis of OA, we used ROC and the area under the curve to analyze and evaluate the diagnostic effectiveness of genes for discriminating patients with OA and normal samples. The results presented that ALKBH1, EIF3, IGF2BP3, WTAP, and YTHDC1 genes were associated with OA, which could serve as valuable biomarkers. Among them, the AUC was 0.96 (95% confidence interval [CI], 0.8747-1.045, P < 0.0001) for ALKBH1, 0.9100 (95% CI, 0.7854-1.035, P = 0.0005) for EIF3, 1 (95% CI, 1.000-1.000, P < 0.0001) for IGF2BP3, 1 (95% CI, 1.000-1.000, P < 0.0001) for WTAP, and 1 (95% CI, 1.000-1.000, P < 0.0001) for YTHDC1, respectively, suggesting that five differential genes have diagnostic value in OA (Fig. 4).
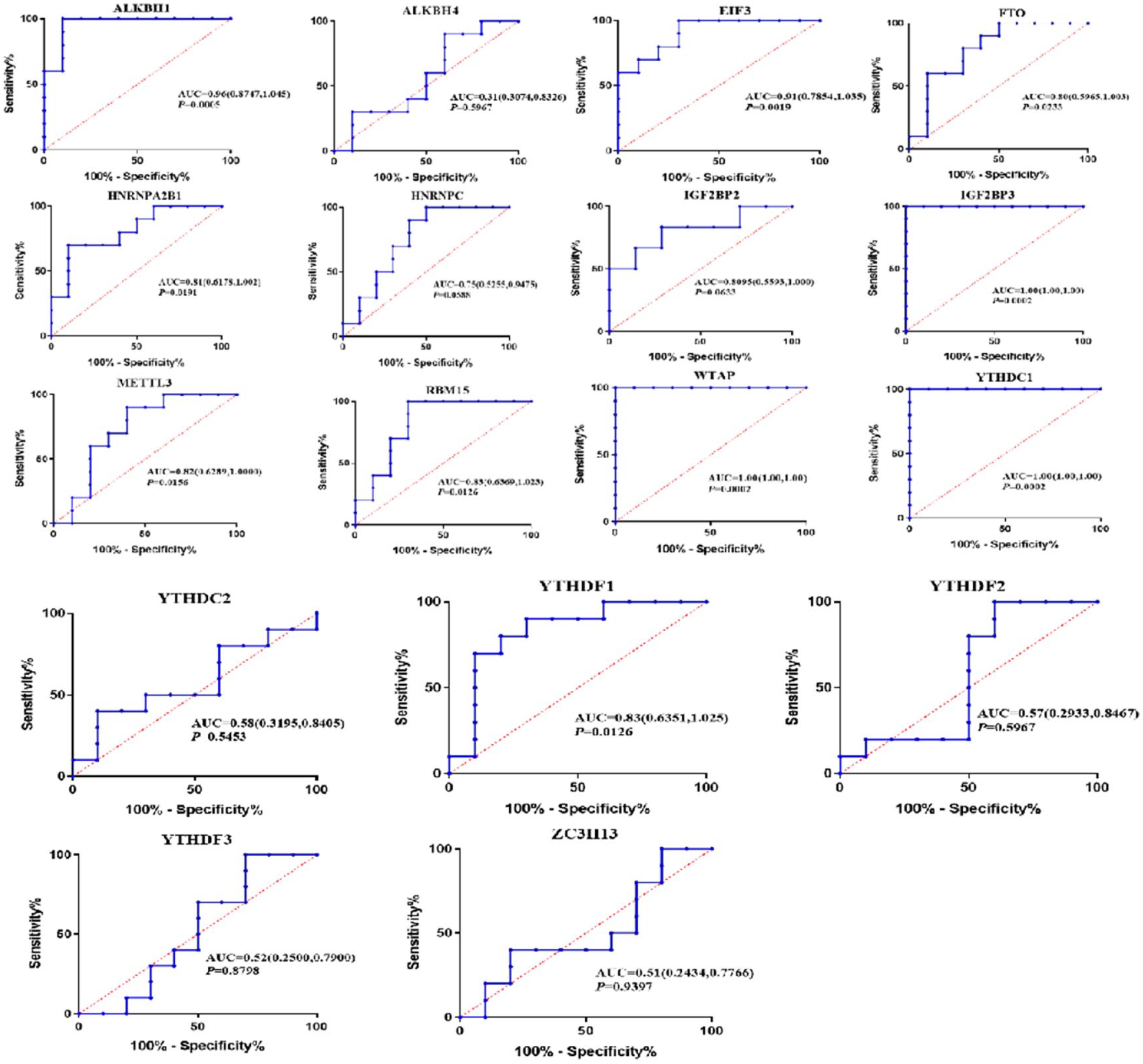
ROC curves of m6A RNA methylation regulators for OA diagnosis. ROC = receiver operating characteristic; OA = osteoarthritis.
Discussion
OA is a chronic degenerative osteoarthropathy, many studies have explored the pathogenesis of OA, but the mechanisms of different epigenetic alterations are not fully understood, so its etiology remains unclear. m6A modification is a type of epigenetic modification, which is necessary for the biogenesis and function of RNA and is involved in the occurrence and development of many diseases. In this study, we used the gene expression profile data set in the GEO database to conduct bioinformatics analysis to identify the m6A differential genes involved in the pathogenesis of OA. A total of 17 genes were analyzed, including 2 upregulated and 10 downregulated genes. In the prognostic analysis, it was found that the gene expression levels of 5 genes are closely related to the prognosis and early diagnosis of OA. This should provide a comprehensive environment for OA, which helps to develop its diagnosis and treatment.
Functional enrichment analysis revealed that there is a strong relationship between m6A-related genes. GO enrichment analysis revealed a number of biological processes and pathways related to the occurrence and development of OA, such as mRNA metabolic process regulation, nuclear speck and mRNA binding, all of which are also closely related to its function. We revealed that m6A RNA methylation regulators are involved in many biological processes and signal transduction pathways, indicating their important role in the initiation and development of OA, which is consistent with previous research that m6A modification is closely related to bone and joint diseases such as RA. 15
We identified that 12 of the 17 genes were differentially expressed in OA, including 2 upregulated genes and 10 downregulated genes. Methyltransferase 3 (METTL3) is the key enzyme for m6A methylation modification. 16 Studies have reported that the methylation mediated by METTL3 is tissue and cell specific.16,17 METTL3 was highly expressed in osteoarthritis and can promote experimental osteoarthritis by regulating the inflammatory response and apoptosis of chondrocytes, and can attenuate lipopolysaccharide-induced inflammation in macrophages through NF-κB,18,19 which was consistent with our results. The major function of insulin-like growth factor 2 mRNA-binding protein 2 (IGF2BP2) is to regulate cell metabolism; IGF2BP2 was highly expressed in pancreatic cancer, and upregulation of IGF2BP2 was associated with poor prognosis of pancreatic cancer patients. 20 FTO (fat mass and obesity-related protein) is a well-known eraser enzyme that is involved in mediating methylation reversal and is related to inflammation. Relevant studies have shown that decreased expression of FTO in peripheral blood is a risk factor for RA, and the expression of FTO may be used as an indicator of activity and inflammation.8,21 Heterogeneous nuclear ribonucleoprotein C (HNRNPC), a member of the nuclear protein family, has evidence that HNRNPC participates in the p53 regulatory network by interacting with long non-coding RNA (lncRNA). 22 Furthermore, we analyzed the correlations among these genes, and many positive correlations correlation were observed, such as HNRNPA2B1 and EIF3, EIF3 and WTAP, YTHDC1 and IGF2BP3, and so on. Studies found that m6A-related genes were highly correlated in pancreatic cancer, cervical squamous cell carcinoma, and endometrial adenocarcinoma,23,24 which is consistent with this study.
Furthermore, we analyzed the factors related to the early diagnosis and prognosis of OA. ROC analysis showed that the area under the ROC curve of ALKBH1, EIF3, IGF2BP3, WTAP, and YTHDC1 genes was more than 0.9, which were associated with the prognosis of OA. ALKBH1, a member of ALKB family, is a 2-ketoglutarate and Fe2+ dependent hydroxylase. ALKBH1 is indispensable for the osteogenic differentiation of human mesenchymal stem cells (MSCs) and indicates that DNA N6-mA modifications are a new mechanism for the epigenetic regulation of stem cell differentiation. 25 Other studies have shown that ALKBH1 interacts with the core transcriptional pluripotency network of embryonic stem cells and participates in the regulation of pluripotency and differentiation. 26
Eukaryotic translation initiation factor 3 (EIF3) is a multi-subunit complex, which plays a key role in translation initiation. Recent studies have shown that EIF3b silencing can inhibit the growth of osteosarcoma cells and induce apoptosis. 27 IEIF3b can inhibit cell proliferation and migration and promotes apoptosis by downregulating Wnt signaling pathway in acute myeloid leukemia. 28 Studies have shown that the osteosarcoma cells with DNA methyltransferase inhibitors or histone deacetylase inhibitors treated could upregulate the expression of insulin-like growth factor2 mRNA-binding protein3 (IGF2BP3). In addition, IGF2BP3 activity can be influenced by mTORC, which is major downstream effectors of phosphoinositide 3 kinase (PI3K) and mitogen-activated protein kinase (MAPK) pathway. 29 YTHDC1, a N6-methyladenosine-binding protein, which is adjacent to nuclear speckles, regulated mRNA splicing by recruiting splicing factors to the targeted mRNA. 30
Wilms tumor 1-associated protein (WTAP) is a putative splicing regulator. Study showed that Smad2/3 interacted with METTL3-METTL14-WTAP, and Smad2/3 could act as a hub for coordinating proteins that play roles in mRNA modification, apoptosis, DNA repair, and transcriptional regulation. 31
In conclusion, this study first demonstrates the critical role of ALKBH1, EIF3, IGF2BP3, WTAP, and YTHDC1 in OA which provides novel insights into recognizing the pathogenesis of OA and a promising biomarker for OA. However, little is known about the function and molecular mechanism of these key m6A methylation genes in OA. Therefore, functional experiments are needed to further explore its potential role and mechanism.
Supplemental Material
sj-docx-1-car-10.1177_19476035221137722 – Supplemental material for Expression of m6A Methylation Regulator in Osteoarthritis and Its Prognostic Markers
Supplemental material, sj-docx-1-car-10.1177_19476035221137722 for Expression of m6A Methylation Regulator in Osteoarthritis and Its Prognostic Markers by Di Zhang, DanDan Zhang, XiaoLi Yang, Qiang Li, RongQiang Zhang and YongMin Xiong in CARTILAGE
Footnotes
Acknowledgments and Funding
We acknowledge the funding support from the National Natural Science Foundation of China (82073494, 81773372, 81573104). The author(s) disclosed receipt of the following financial support for the research, authorship and/or publication of this article: This study was supported by the National Natural Science Foundation of China (82073494, 81773372, 81573104).
Declaration of Conflicting Interests
The author(s) declared no potential conflicts of interest with respect to the research, authorship, and/or publication of this article.
Ethical Approval
GEO belongs to public databases, and the patients involved in the database have obtained ethical approval. User can download relevant data for free for research and public relevant articles. Our study is based on open source data, so there are no ethical and other conflict.
References
Supplementary Material
Please find the following supplemental material available below.
For Open Access articles published under a Creative Commons License, all supplemental material carries the same license as the article it is associated with.
For non-Open Access articles published, all supplemental material carries a non-exclusive license, and permission requests for re-use of supplemental material or any part of supplemental material shall be sent directly to the copyright owner as specified in the copyright notice associated with the article.