
Abstract
Inhibition of the KCa3.1 potassium channel has therapeutic potential in a variety of human diseases, including inflammation-associated disorders and cancers. However, KCa3.1 inhibitors with high therapeutic promise are currently not available. This study aimed to establish a screening assay for identifying inhibitors of KCa3.1 in native cells and from library compounds derived from natural products in Thailand. The screening platform was successfully developed based on a thallium flux assay in intestinal epithelial (T84) cells with a Z′ factor of 0.52. The screening of 1352 compounds and functional validation using electrophysiological analyses identified 8 compounds as novel KCa3.1 inhibitors with IC50 values ranging from 0.14 to 6.57 µM. These results indicate that the assay developed is of excellent quality for high-throughput screening and capable of identifying KCa3.1 inhibitors. This assay may be useful in identifying novel KCa3.1 inhibitors that may have therapeutic potential for inflammation-associated disorders and cancers.
Introduction
The intermediate-conductance Ca2+-activated potassium channel (KCa3.1; also known as KCNN4, IK1, IKCa1, SK4, and Gárdos channel) is ubiquitously expressed in many tissues throughout the body, including cells of hematopoietic lineages, epithelial tissues, vascular endothelia, fibroblasts, and smooth muscle cells. 1 The function of KCa3.1 was first described in human erythrocytes by Gárdos in 1958. 2 The efflux of K+ through this channel alters the osmotic balance and causes shrinkage of erythrocytes, establishing KCa3.1 as a regulator of erythrocyte cell volume. 3 In addition, KCa3.1 is functionally coupled with calcium channels. Calcium influx through the Ca2+ channels elevates the intracellular Ca2+ level, causing KCa3.1 activation. Potassium efflux through KCa3.1 in turn induces membrane hyperpolarization, which counteracts membrane depolarization caused by Ca2+ influx, maintaining the electrical gradient and sustaining Ca2+ channel activity. 4 Due to this role, KCa3.1 is an important player in cellular calcium signaling involved in the activity of various immune cells. 5 The activation of T lymphocytes is associated with the upregulation of KCa3.1, whereas loss of KCa3.1 function is associated with the reduction of activated phenotypes such as cytokine production. 6 Similar to T cells, KCa3.1 is involved in functions of B lymphocytes, macrophages, microglia, immature dendritic cells, mast cells, and neutrophils.5,7 Furthermore, KCa3.1 is expressed in most epithelial cells, 8 where it functions to protect against hypotonic stress9,10 and provide a driving force for anion secretion.11,12
KCa3.1 is regarded as a promising drug target for many diseases. Its role in erythrocyte volume regulation leads to the first potential therapeutic application of KCa3.1 inhibitor in sickle cell disease characterized by deformation of erythrocytes into a sickle-like shape because of self-polymerizing sickle hemoglobin (HbS). This “sickling” process is highly sensitive to intracellular HbS concentration and exacerbated by cell dehydration. 13 The roles of KCa3.1 in immune cell activation established KCa3.1 inhibitors as anti-inflammatory agents. 6 In intestinal epithelial cells, KCa3.1 has been proposed to be a drug target for the treatment of diarrheal diseases caused by excessive Cl− secretion, such as rotavirus-induced diarrheas 14 and cholera.15,16 In addition, inhibition of KCa3.1 has been shown to increase the migration of intestinal epithelial cells in a PI3K/Akt-dependent manner, which may be useful for promoting wound healing. 17 Importantly, several lines of evidence suggest that KCa3.1 inhibitors may be useful in the treatment of cancer. KCa3.1 overexpression is associated with poor prognosis of lung cancer, 18 renal cell carcinoma, 19 and ovarian cancer. 20 KCa3.1 inhibition reduces cancerous phenotypes in disease models of pancreatic cancer, prostate cancer, breast cancer, melanoma, glioblastoma, hepatocellular carcinoma, colorectal cancer, lymphocytic leukemia, and endometrial carcinoma.21–23
Despite these diverse potentials, there is currently no KCa3.1 inhibitor available for therapeutic use. TRAM-34, a KCa3.1 inhibitor widely used in research, has a short half-life and poor oral bioavailability, 1 rendering it unfavorable as a drug candidate. In addition, Senicapoc, another KCa3.1 inhibitor that entered a clinical trial for sickle cell disease, ultimately failed the phase 3 study due to its lack of efficacy in reducing vaso-occlusive crises in patients. 24 In order to identify novel classes of KCa3.1 inhibitors from a large number of molecular scaffolds, the implementation of high-throughput screening technologies is required. The screening method using thallium (Tl+) flux assays in KCa3.1-transfected HEK293 cells has been reported. 25 Since KCa3.1 function depends not only on intracellular Ca2+ but also on posttranslational modifications, including serine 26 and histidine 27 phosphorylation as well as protein trafficking, 28 assays using exogenously introduced KCa3.1 might not fully recapitulate the complex regulatory mechanisms of endogenously expressed KCa3.1. Therefore, an assay with an endogenous expression system would be preferable in this regard. Intestinal epithelial cells have been shown to express KCa3.1, and functional measurement of KCa3.1 activity in these cells is readily achievable by basolateral K+ current analyses. Therefore, this study was aimed at developing a cell-based assay for identifying KCa3.1 inhibitors in an endogenous expression system, that is, intestinal epithelial cell lines, and using this assay for the initial screening of compounds derived from Thai natural products. We have successfully established a Tl+ flux assay in T84 cells and several novel KCa3.1 inhibitors have been identified by this assay.
Materials and Methods
Chemical Reagents
Fetal bovine serum (FBS), culture media, trypsin, penicillin, streptomycin, and FluxOR II Green Potassium Ion Channel Assay were purchased from Thermo Fisher Scientific Inc. (Waltham, MA). Other chemicals were purchased from Sigma-Aldrich (St. Louis, MO). The natural compound library was provided by the Excellent Center for Drug Discovery, Faculty of Science, Mahidol University (Bangkok, Thailand).
Cell Culture
T84 (CCL-248), HT-29 (HTB-38), and HCT 116 (CCL-247) cells were obtained from the American Type Culture Collection (Manassas, VA) (passages 10–30) and cultured in a mixture of Dulbecco’s modified Eagle’s medium and Ham’s F-12 medium (1:1) supplemented with 10% FBS, 100 U/mL penicillin, and 100 μg/mL streptomycin. The cells were grown in a humidified incubator under an atmosphere of 5% CO2 at 37 °C. The culture medium was changed every other day and cells were split every 7 days.
Thallium Flux Assay
The cells were plated at a density of 2.5 × 105 cells/well in black, flat-bottom, 96-well plates (Costar, New York, NY) and cultured overnight until confluence. The cells were then incubated for an hour with FluxOR II reagent dye in loading buffer, followed by washing and 30 min of exposure to 50 µL of assay buffer containing DMSO (vehicle control), TRAM-34 (KCa3.1 inhibitory control), or test compounds. Then, baseline fluorescence intensity was obtained before the injection of 50 µL of stimulus buffer containing NS309 (KCa3.1 activator) and 5 mM Tl2SO4, immediately followed by measurements of fluorescence kinetics for 1–2 min. The fluorescence intensity was measured at excitation/emission wavelengths of 495 nm/519 nm using a microplate reader installed with a dispenser unit for injection of the stimulus buffer (EnVision; PerkinElmer, Waltham, MA). The fluorescence intensity of each well was normalized with an average baseline fluorescence intensity value from 10 repeated measurements prior to stimulus buffer injection. Normalized data points after Tl+ injection were fitted to quadratic regression. If R2 for quadratic regression was more than 0.9, the initial rate of the quadratic curve was used to represent the rate of Tl+ influx. If data fitted poorly to the quadratic curve (R2 < 0.9), simple linear regression would be performed instead, and the slope of the regression line was used to indicate the Tl+ flux rate. All regression models were performed in Microsoft Excel 2019 (Redmond, WA). The KCa3.1 activator and inhibitor control wells were situated as the first and last columns of the 96-well plate with the instrument set to read the signal by row, ensuring the data from control groups were evenly interspersed across the plate during measurements. To calculate the percentage of Tl+ flux inhibition, the Tl+ flux rate of each well was compared against the means of the KCa3.1 activator and KCa3.1 inhibitor control groups from the same plate, using the following equation:
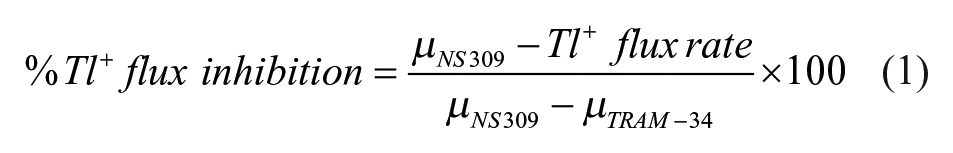
To evaluate the assay quality, the Z′ factor was calculated from data of the KCa3.1 activator and KCa3.1 inhibitor control groups from multiple separated experiments.
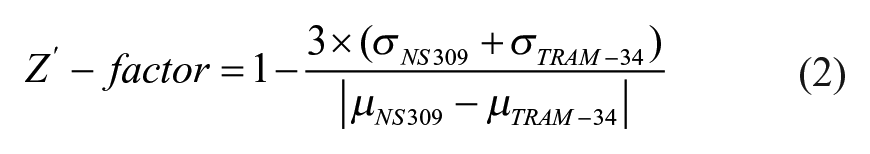
Measurement of Basolateral K+ Current (IK+)
T84 cells were plated on Snapwell polycarbonate inserts (Costar, New York, NY) at a density of 5 × 105 cells/insert. The cells were cultured with a daily replacement of media for 2 weeks until the transepithelial electrical resistance was >1000 Ω.cm2, as measured by a Millicell ERS-2 volt-ohm meter (Merck Ltd., an affiliate of Merck KGaA, Darmstadt, Germany). For basolateral IK+ measurement, solutions with high K+ and low K+ concentrations (pH 7.4) were added into apical and basolateral hemichambers, respectively, to generate an apical-to-basolateral K+ gradient. The apical high K+ solutions contained 145 mM potassium gluconate, 5 mM KCl, 1 mM MgCl2, 1 mM CaCl2, 5 mM HEPES, and 10 mM glucose. In the basolateral low-K+ solution, 145 mM potassium gluconate was replaced with 145 mM sodium gluconate. To induce apical membrane permeabilization, amphotericin B was added into the apical hemichamber 30 min before basolateral IK+ measurement. The solutions were maintained at 37 °C and bubbled with 100% O2. IK+ was recorded using a DVC-1000 voltage-clamp (World Precision Instruments, Sarasota, FL) with Ag/AgCl electrodes and 3M KCl agar bridges.
Statistical Analysis
The statistical difference was tested using Student t tests for pairs of measurements or one-way analysis of variance (ANOVA) followed by Bonferroni’s post hoc test for multiple groups of measurements, with P < 0.05 considered significantly different. GraphPad Prism software (version 5; La Jolla, CA) was used for all statistical analyses.
Results
Development of a Screening Assay Based on the Tl+ Flux Assay
A Tl+ flux potassium channel assay was used in this study. This assay utilized two main components, fluorometric membrane-permeable Tl+ indicator dye and Tl+ ion.
29
Since Tl+ could pass through KCa3.1, the presence of high extracellular [Tl+] would generate a driving force for Tl+ influx into the cells. If the KCa3.1 was opened, Tl+ entered into the cells and intracellular Tl+ interacted with the Tl+ indicator dye, yielding a fluorescence signal (
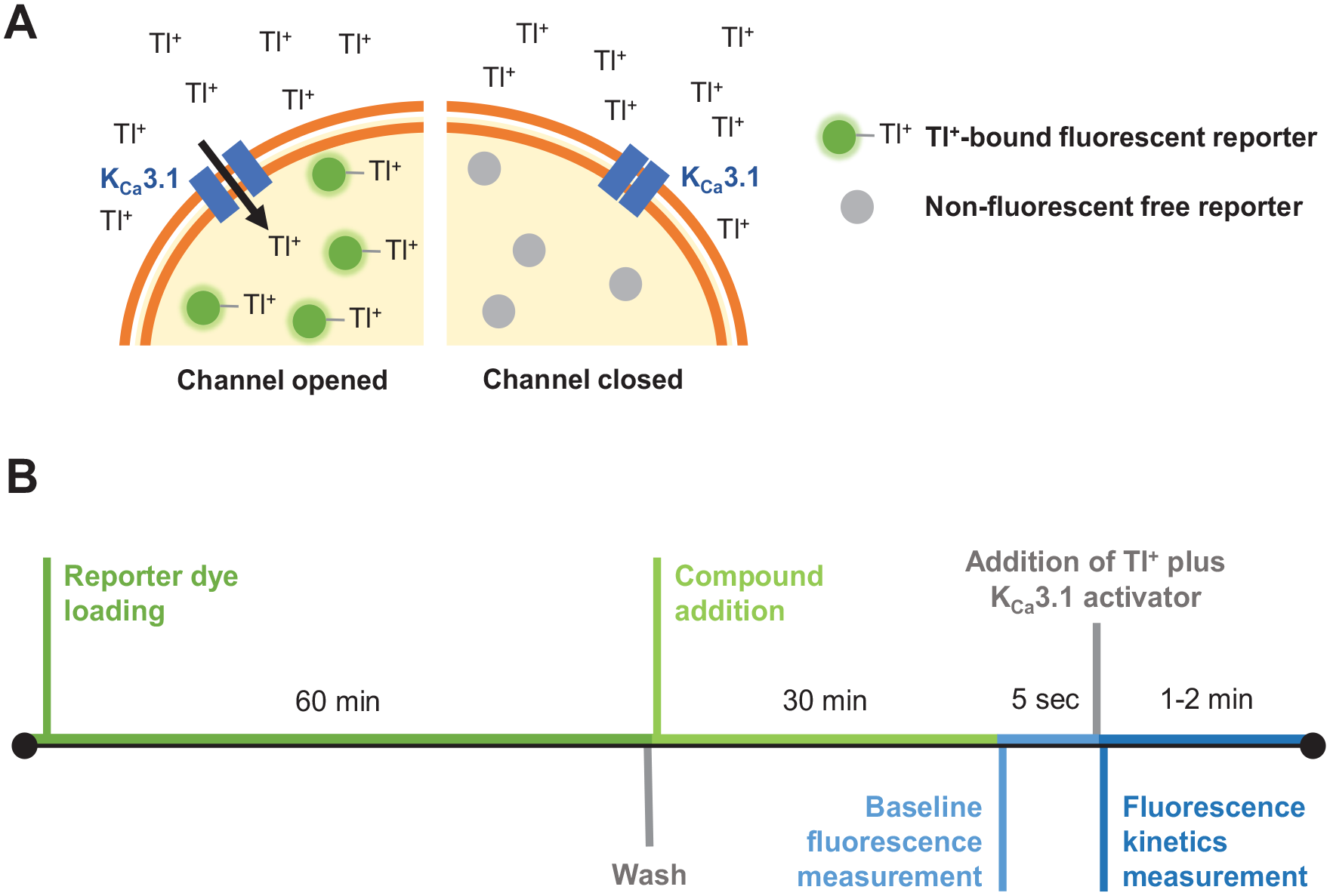
Overview of Tl+ flux assay. (
To find the intestinal epithelial cell line suitable for this assay, we compared KCa3.1-mediated fluorescence signal induced by 10 µM NS309 in intestinal cell lines expressing KCa3.1, including HT-29, T84, and HCT 116. We found that the maximal signal at 1 min after Tl+ addition from HT-29 cells was significantly smaller than those from the other two cell lines. In addition, assays using T84 produced a more consistent signal than those using HCT 116 (
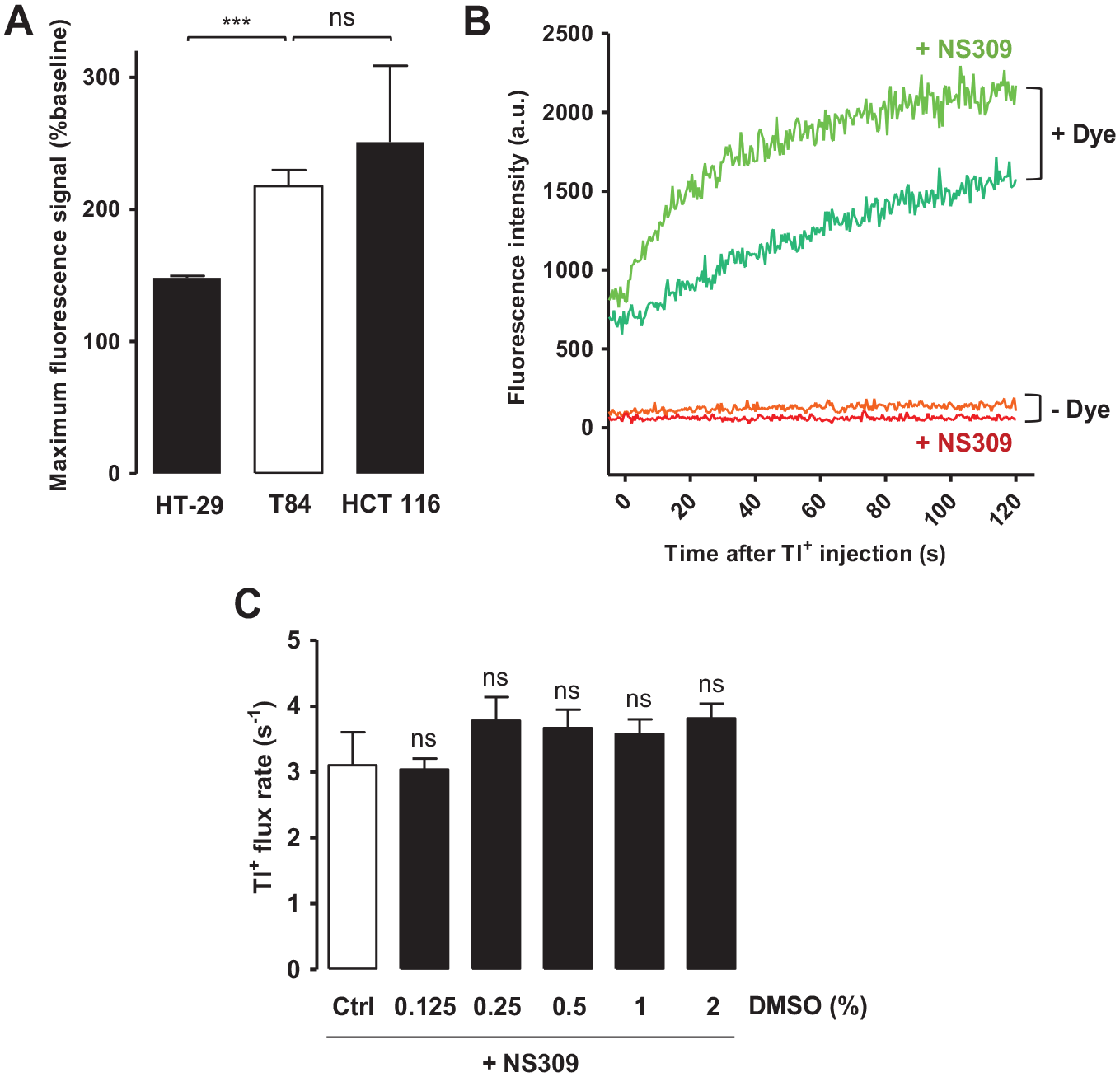
Tl+ flux assay development. (
Validation and Optimization of Tl+ Flux Assay
Since NS309 was previously reported to activate both KCa3.1 and small-conductance KCa2 channels and Tl+ flux can occur through K+ channels other than KCa3.1,30,31 the assay specificity to KCa3.1 activity was determined. A panel of K+ channel inhibitors was used to identify ion channels contributing to NS309-induced Tl+ flux. We found that Tl+ flux was sensitive to only TRAM-34 (KCa3.1 inhibitor) and ouabain (Na+/K+-ATPase inhibitor), but not apamin (KCa2 inhibitor), BaCl2 (KcAMP inhibitor), or bumetanide (Na+-K+-2Cl− co-transporter inhibitor), as shown in
Figure 3A
. These findings indicate that Tl+ flux was mainly mediated by KCa3.1 and the constitutively active Na+/K+-ATPase. The contribution of KCa3.1 and KCa2 to the NS309-stimulated signal was also evaluated using the electrophysiological method. We found that the NS309-induced IK+ was sensitive to TRAM-34 but not apamin, confirming that KCa2 did not contribute to the NS309-induced K+ flux in T84 cells (
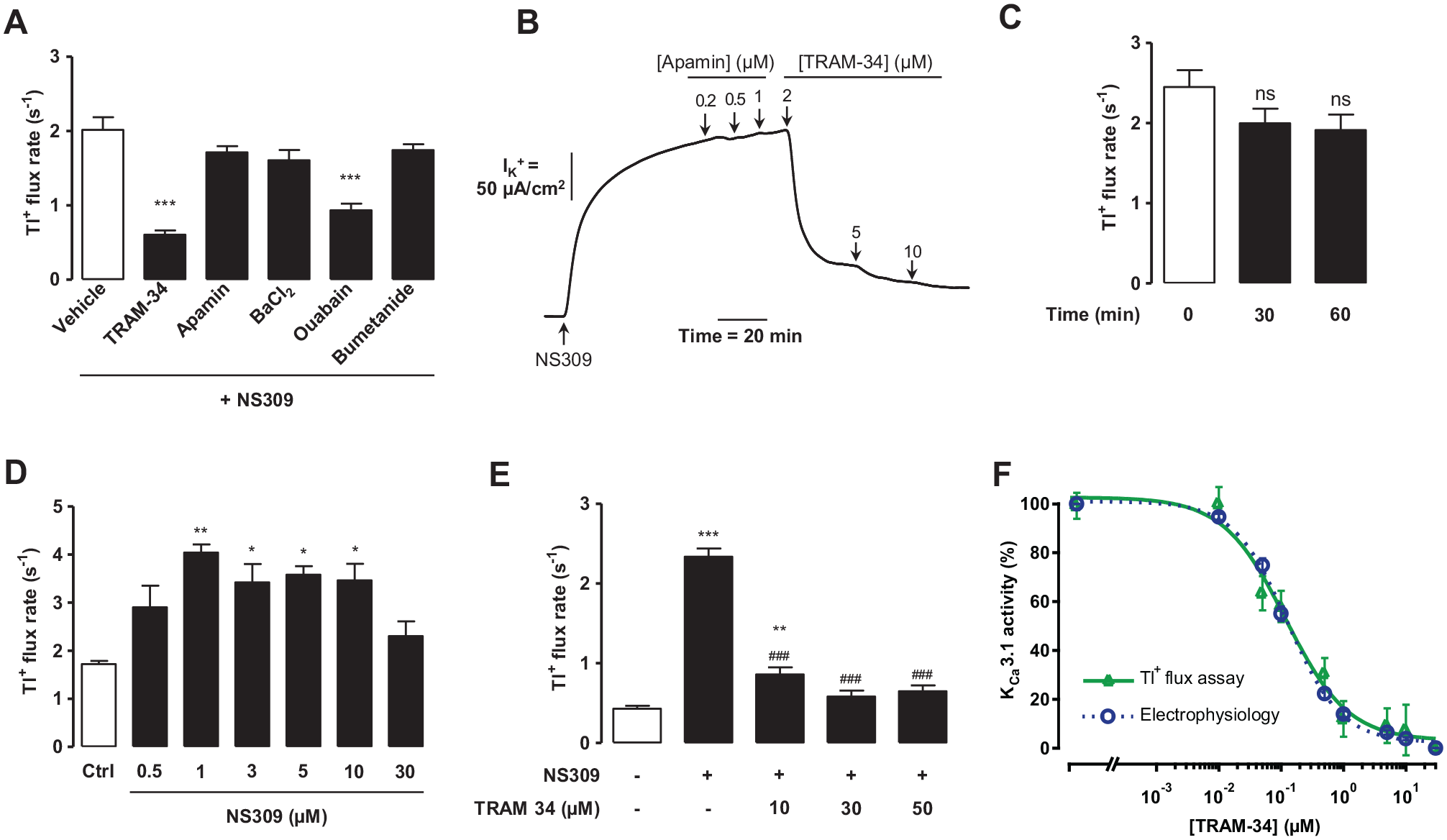
Specificity and optimization of screening assay. (
Subsequently, experimental parameters of our assay were optimized to achieve the best fluorescence signal for the screening. First, the optimal incubation time of NS309 activator was determined. Prolonged NS309 incubation time before kinetics measurement up to 60 min did not affect the Tl+ flux rate (
Evaluation of Assay Quality
To verify the ability of the assay to differentiate the inhibitor control group from the activator control group, changes in fluorescence signal were compared between both groups. Similar to
Figure 2B
, NS309 produced an initial sharp rise in fluorescence signal, while co-treatment with TRAM-34 only allowed a steady slow increase in the signal (
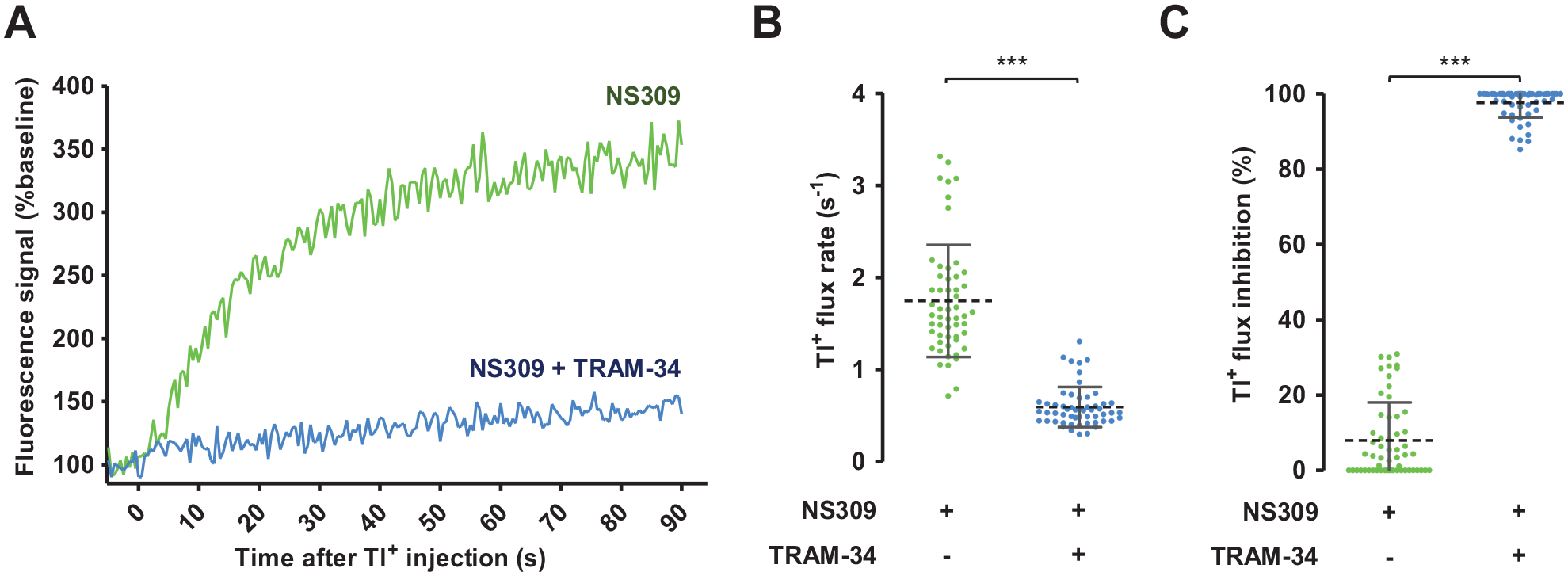
Evaluation of assay quality. (
Screening of the Compound Library
To evaluate the practicality of our assay, the Tl+ flux assay was used to identify potential KCa3.1 inhibitors from the small set of the compound library. The screening library contained 1352 samples of 679 isolated natural compounds, 273 synthetic derivatives, and 400 crude extracts from various plants and fungi in Thailand. Compounds in the library were prepared as 100× working stock at 5 mM in DMSO. Primary screening with Tl+ flux assay was performed at a concentration of 50 µM with a final DMSO concentration of 1.2%, using criteria of more than 80% Tl+ flux inhibition for compound selection. A total of 23 compounds passed the selection criteria of the Tl+ flux assay. Next, KCa3.1 inhibitory properties of compounds obtained from screening were validated by measuring the effect of the compounds on basolateral IK+ stimulated by NS309. A natural compound, resveratrol, was previously reported to inhibit KCa3.1 with an IC50 of approximately 10 µM, 32 which is a relatively low potency compared with other small-molecule KCa3.1 inhibitors with submicromolar IC50. 22 Therefore, a threshold of at least 50% inhibition of KCa3.1-mediated IK+ at 10 µM was used to identify effective inhibitor compounds and to exclude molecules with low potency. With this, we verified eight inhibitors of the KCa3.1 channel to undergo a more detailed concentration–response relationship analysis, summarized in Table 1 . Examples of a concentration–response relationship of compounds N15, N18, and N602 are demonstrated in Figure 5A . Notably, we have identified several compounds with submicromolar IC50 (N134, S267, and S269) and others with low micromolar potency (IC50 ~ 1–6 µM). The preliminary evaluation of potential nonspecific inhibitory effects was also performed. All eight KCa3.1 inhibitor compounds did not significantly alter basal Tl+ flux without NS309 stimulation, indicating a negligible effect on other constitutive K+-permeable channels in T84 cells (data not shown).
Summary of Identified KCa3.1 Inhibitors. a
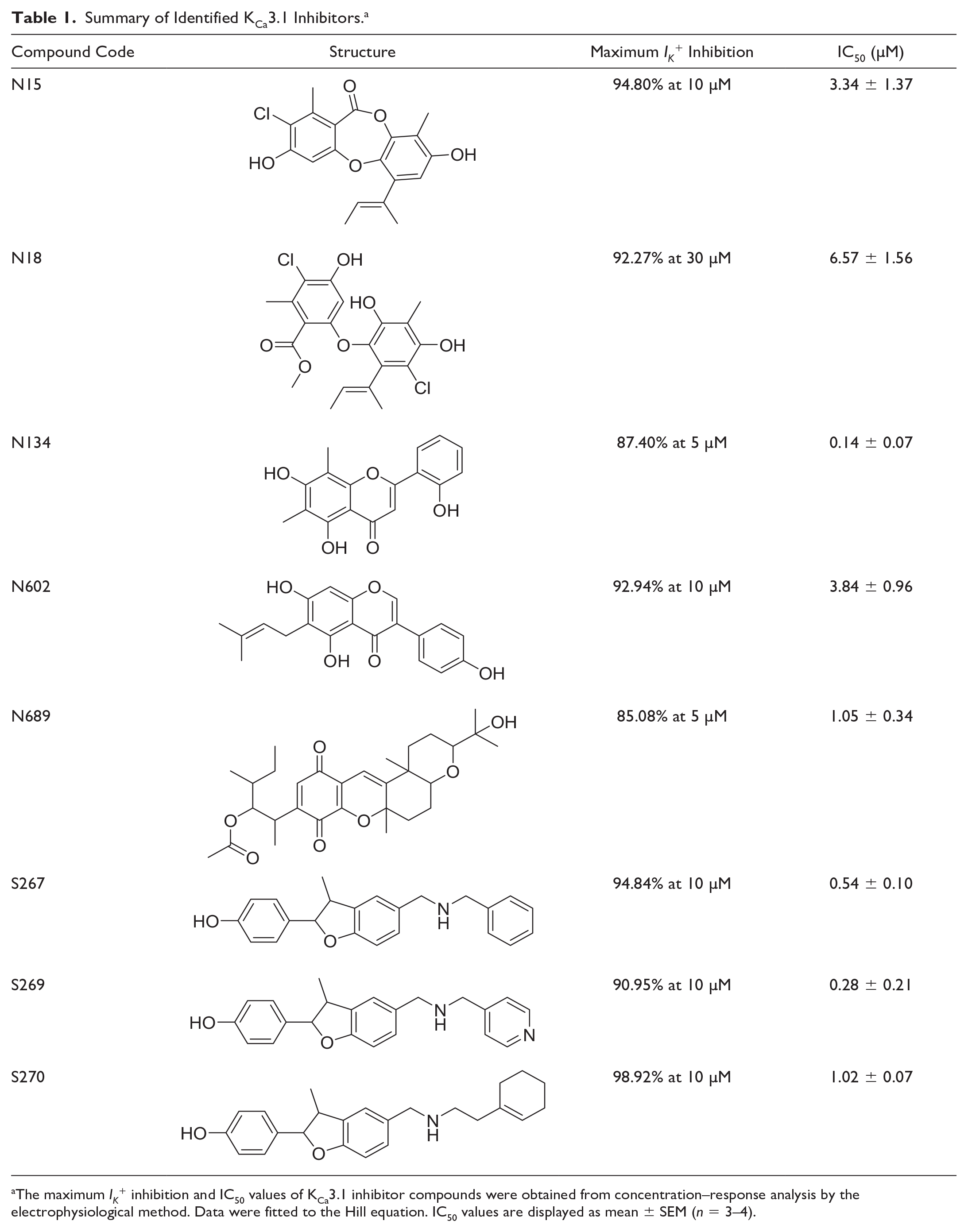
The maximum IK+ inhibition and IC50 values of KCa3.1 inhibitor compounds were obtained from concentration–response analysis by the electrophysiological method. Data were fitted to the Hill equation. IC50 values are displayed as mean ± SEM (n = 3–4).
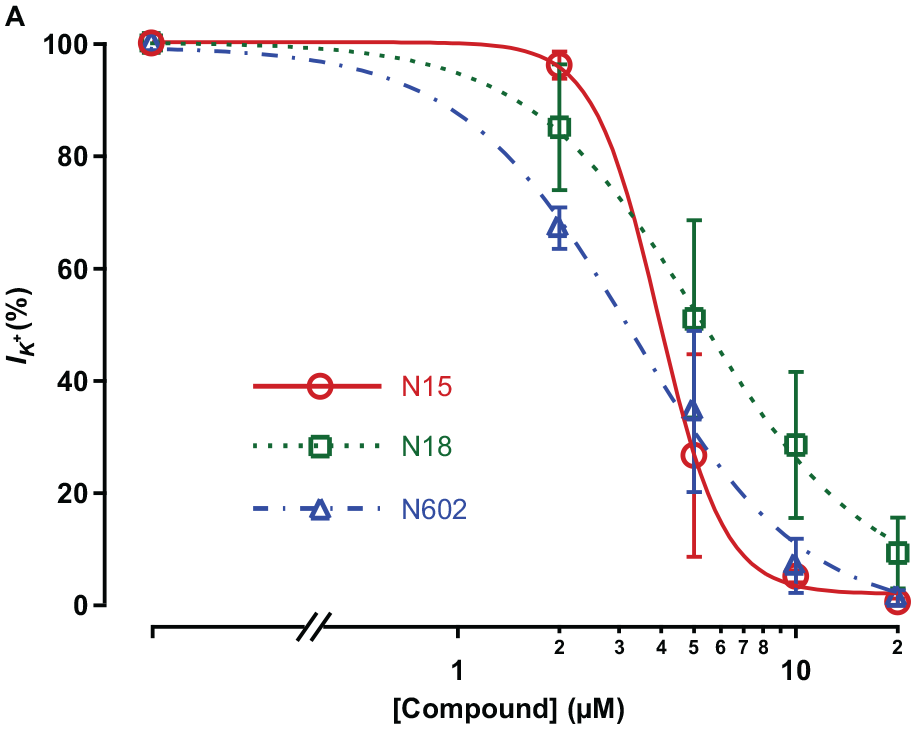
Examples of concentration–response data. (A) Concentration–response relationship of N15, N18, and N602 (n = 3). Basolateral IK+ in the T84 cell monolayer was obtained from Ussing chamber analysis. NS309 (10 µM) was used to stimulate the KCa3.1 current. All compounds were added to both apical and basolateral chambers. Data were fitted to the Hill equation and are displayed as mean ± SEM.
Discussion
To establish a screening assay targeting the KCa3.1 channel, numerous approaches were considered as a basis for its methodology. The nonfunctional ligand-binding assay was excluded since molecular binding does not always confer functional modulation. On the other hand, highly sensitive direct electrophysiological assays were not suitable due to the necessity of specialized equipment and operation skills. Therefore, a fluorescence flux-based Tl+ flux assay was used because it requires only a standard fluorescence plate reader and because of its optimizable components.
During the selection of suitable cell lines, we have observed differential functional expression of KCa3.1 between cell lines. Interestingly, HCT 116 cells showed noticeably higher signal variability between wells, possibly due to greater heterogeneity in cell population compared with the other two cell lines. While it is ideal to activate KCa3.1 via a native mechanism using Ca2+, this method of activation is not specific to KCa3.1 channels. A rise in intracellular Ca2+ would activate not only KCa3.1 but also other Ca2+-activated channels, like large-conductance KCa1/BK. Moreover, NS309 acts by increasing the opening probability of KCa3.1 at a given Ca2+ concentration, 33 allowing the basal intracellular Ca2+ level to stimulate the channels. Therefore, the activation of KCa3.1 by NS309 is still dependent on stimulation by Ca2+. Another thing of note is that even though NS309 could activate both intermediate-conductance KCa3.1 and small-conductance KCa2 channels through interaction with their shared calmodulin-binding domain required in intracellular Ca2+ sensing, 22 we found that NS309 activated only KCa3.1 in T84 cells during both the Tl+ flux assay and electrophysiological measurement. We hypothesized that T84 cells might express a very low level of KCa2 channels, which therefore contribute very little to NS309-stimulated signal. We also found that Tl+ flux was significantly reduced upon administration of ouabain, implicating a role of Na+/K+-ATPase in mediating Tl+ flux. The main function of Na+/K+-ATPase is the active transport of Na+ efflux and K+ influx. Since Tl+ could be transported via any K+-permeable transporters, Na+/K+-ATPase would uptake Tl+ into the cell as well, a phenomenon that has been previously reported in myocardial cells. 30 In addition, Na+/K+-ATPase is responsible for maintaining negative resting membrane potential, and therefore its function would be important in providing the electrical driving force for the entry of positively charged Tl+ into the cells.
Unexpectedly, prolonged exposure or excessive concentration of the KCa3.1 activator NS309 apparently diminished Tl+ flux, an effect not seen with the KCa3.1 inhibitor TRAM-34. This might be due to a desensitization mechanism of the KCa3.1 channel, which thereby prevents overactivation by NS309. Moreover, TRAM-34 showed considerably lower potency (IC50 = 129 nM) in our electrophysiological analysis than previously reported in other publications (IC50 ~ 10–25 nM). 22 However, it is important to note that these studies often use different methodologies or cell models to determine the potency of TRAM-34, which might contribute to this discrepancy. The fluorescence signal pattern elicited by NS309 showed a rapid increase that slowed down as time went on, corresponding to the driving force for Tl+ flux from the concentration gradient. The difference in [Tl+] would be highest immediately after Tl+ addition into the extracellular buffer, resulting in a strong initial Tl+ flux. With the passage of time, Tl+ influx would gradually increase intracellular [Tl+] and reach equilibrium, where the concentration gradient of Tl+ was diminished, eliminating the driving force of the Tl+ flux. This stage would manifest as late steady state of the signal, where the signal retained a constant rate of change, comparable to those without NS309 stimulation or with KCa3.1 inhibited by TRAM-34. Since Na+/K+-ATPase was shown to contribute to Tl+ flux, we hypothesized that this residual baseline Tl+ flux might result from Na+/K+-ATPase-mediated active transport of Tl+. Nevertheless, it is important to emphasize that our experimental design did not exclude the possibility of saturation of the fluorescent reporter dye or precipitation of excess Tl+ with intracellular Cl− as the cause of signal plateau.
Our Tl+ flux screening assay had identified several new KCa3.1 inhibitors validated by electrophysiological measurements from the natural compound library, demonstrating the effectiveness of a screening assay based on endogenous expression of the KCa3.1 channel in the detection of KCa3.1 inhibitors. A total of 23 compounds from a library of 1352 compounds passed the selection criteria of the Tl+ flux assay, and only 8 compounds were classified as KCa3.1 inhibitors after validation by electrophysiology. Therefore, the initial hit rate and validated hit rate for our assay were 1.7% and 0.6%, respectively. Among eight discovered compounds, only the bioactivities of N15 and N602 have been previously reported. A depsidone N15 from marine-derived fungus has been shown to inhibit aromatase and xanthone oxidase enzymes 34 and reduce lipopolysaccharide (LPS)-induced nitric oxide production in vitro. 35 A flavonoid N602 was also reported to have antifungal, 36 antimicrobial, 37 and anticancer38,39 effects. However, none of the eight compounds have been described for their inhibitory effect on the KCa3.1 channel. In addition, none of the identified KCa3.1 inhibitors shared structural similarity with well-known inhibitor scaffolds: triaryl-methanes, phenyl-pyrans, and cyclohexadienes. 22 According to the structural similarity, these compounds could be loosely categorized into four groups: N15 and N18; N134 and N602; N689; and S267, S269, and S270. Among compounds S267, S269, and S270, an amine-attached cyclic hydrocarbon chain seems to greatly affect the potency of the compound in this family. Introduction of nitrogen atom into the end of a benzene ring increased the potency of S269 almost twofold compared with S267. Substitution of a benzyl group in S267 to cyclohexenyl ethyl in S270 reduced its potency by half. Nevertheless, further comprehensive analysis of the structure–activity relationship is required to ascertain these observations. It is important to note that, while we have confirmed the effects of these compounds in KCa3.1-mediated IK+ inhibition, further studies are needed to determine whether they inhibit KCa3.1 via direct interaction with the channel or inhibition of KCa3.1 was a secondary effect from interaction with other target molecules within the regulatory system of the KCa3.1 channel. For instance, the compounds could modulate the function of nucleoside diphosphate kinase B (NDPK-B) and mammalian protein histidine phosphatase (PHPT1), which regulate phosphorylation at the histidine 358 residue of KCa3.1 that is required for channel activation.40,41 In addition, a test of specificity against related channels within the KCa family and other critical ion channels such as the cardiac human ether-à-gogo-related gene (hERG) is still needed to ensure the specificity and safety of these KCa3.1 inhibitors. Moreover, detailed pharmacokinetics and pharmacodynamics data of these compounds are necessary for further in vivo testing in disease models to progress further toward therapeutic agents in humans.
In summary, a high-throughput screening assay targeting the KCa3.1 channel inhibitor was successfully established based on the Tl+ flux assay in the T84 intestinal epithelial cell line. This assay was capable of identifying potent KCa3.1 inhibitors verified by the electrophysiological method from the natural compound library. Further refinement and upscaling of our assay may result in a streamlined high-throughput screening process for rapid drug discovery. Further study of these newly discovered inhibitory compounds might lead to a new class of KCa3.1 inhibitors applicable for human use.
Footnotes
Declaration of Conflicting Interests
The authors declared no potential conflicts of interest with respect to the research, authorship, and/or publication of this article.
Funding
The authors received the following financial support for the research, authorship, and/or publication of this article: This work was supported by the Thailand Research Fund (DBG5980001, DBG6180029); Faculty of Science, Mahidol University; and Medical Scholars Program, Mahidol University.