
Abstract
MRG15 is a transcription factor containing the methyl-lysine reader chromodomain. Despite its involvement in different physiological and pathological states, to date the role of this protein has not been fully elucidated due to the lack of a specific and potent chemical probe.
In this work, we report the development of a microscale thermophoresis (MST)-based assay for the study of MRG15–ligand binding interactions. After the development, the assay was validated using a small focused library and UNC1215 as the reference compound, to yield the identification of 10 MRG15 ligands with affinities ranging from 37.8 nM to 59.1 µM.
Hence, our method is robust, convenient, and fast and could be applied to other methylation reader domain-containing proteins for the identification of new chemical probes.
Keywords
Introduction
A variety of fundamental processes in cells are regulated, either directly or indirectly, by posttranslational modifications occurring on histone proteins. The indirect regulation is mediated by specialized “reader” domains that can identify and bind specific covalent modifications such as, for example, lysine methylation.1,2 A misregulation of such chromatin-reading capability often occurs in various diseases, including cancer, and as a consequence, methylation reader proteins have gained growing interest in recent years as major targets for drug development.2,3 One of the most interesting methyl-lysine reader proteins is MRG15 (MORF-related gene on chromosome 15), a chromodomain-containing transcription factor that plays a vital role in embryonic development, cell proliferation, and cellular senescence.
4
It is expressed in a wide variety of human tissues, and its homologs have been identified in many other eukaryotes.
5
Human MRG15 consists of a putative chromodomain (N-terminal residues 1–85) and a conserved MRG domain (C-terminal residues 151–323), which are linked together by a flexible region (residues 86–150). The MRG domain is highly conserved among all MRG proteins and the crystal structure of the MRG domain of human MRG15 was determined in 2006 (
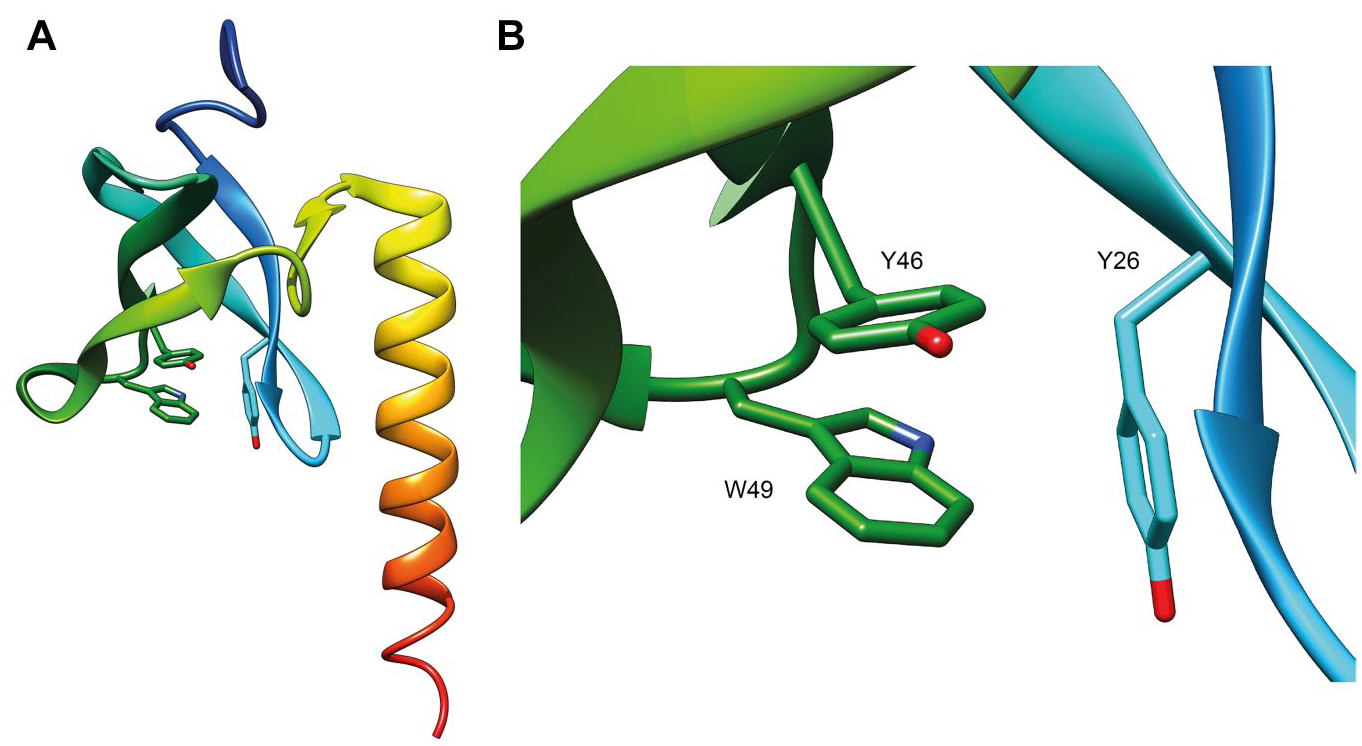
(
The relatively low availability of chemical probes is a feature of this class of epigenetic proteins, and it strictly depends on the mechanism of action of readers. Differently from extensively studied writer and eraser enzymes, the inhibition of which represents a rather canonical medicinal chemistry problem, the study of reader proteins is much more complicated because of the weak interaction with native substrates and the superficial binding sites that make the development of selective inhibitors challenging.2,3,11–14 Moreover, the discovery of new modulators for reader proteins requires the use of techniques suitable for the study of protein–small-molecule interactions. In this regard, many biophysical methods are commonly used in drug discovery programs for the identification of new ligands (each of them with advantages and drawbacks), including differential scanning fluorimetry (DSF), surface plasmon resonance (SPR), ITC, and microscale thermophoresis (MST).
DSF is based on the well-established thermodynamic principle that the thermal stability of a protein, often quantified as the midpoint of thermal denaturation or the melting point (Tm), can be altered by a binding ligand.15,16 DSF is indeed a useful screening method, and provides an easy optimization of assay conditions and fast assay preparation and data analysis.15,17 Nevertheless, this method suffers from sensitivity problems, as low-affinity ligands are usually not detected, and furthermore, it is a qualitative method that generally cannot give an indication about the KD. Moreover, it is prone to give false negatives, as not all ligands that bind will also lead to a change in the thermal stability, and consequently, a few binders could be missed.
SPR allows the real-time high-sensitivity measurement of interactions by flowing a solution of analyte over the surface of target biomolecules immobilized on a sensor chip.18–20 Since first introduced in the early 1990s, SPR has been proven to be one of the most powerful technologies to determine specificity, affinity, and kinetic parameters of analyte–ligand interactions. However, the need to immobilize one of the binding partners on the surface of a sensor chip can modify the binding ability of the ligand. 21
ITC is a robust technique that can measure a wide range of binding affinities. 22 Using this technology, thermodynamic parameters (enthalpy and entropy), as well as binding constants and stoichiometry, can be obtained from a single titration. The major drawback is the requirement of a high quantity of protein for each measurement, meaning that ITC cannot be used for the screening of a large number of compounds. 23
Among all the reported techniques, MST is the most recently introduced one, even if it is based on the principle of thermophoresis that was already observed and reported by Carl Ludwig in 1856 and further understood by Charles Soret in 1879.24,25 MST is a biophysical technique that allows us to measure the strength of the interaction between two molecules. It does that by measuring the changes in fluorescence as a result of an infrared laser-induced temperature change. The fluorescence detected can be the intrinsic fluorescence from tryptophan or, most commonly, from a fluorophore attached to the molecule. There are two components that contribute to the fluorescence change that is measured. One component is thermophoresis, which is the directed movement of molecules within a temperature gradient. Typically, molecules move away from a heat source, but it can also happen in the opposite direction. As molecules move away from the point where the heat is applied, a change in the intensity of the fluorescence is recorded. 26 The other is the major component of the MST signal detected; this is called temperature-related intensity change (TRIC), an effect where the fluorescence intensity of a fluorophore is temperature dependent. This temperature dependency is also related to changes in the local microenvironment of the fluorophore, which can be strongly affected by the binding of a ligand to the fluorescent target molecule. 27
From different points of view, MST technology is superior to other methods in determining the parameters of molecular interactions; indeed, it requires low sample consumption, it allows a rapid analysis, no surface immobilization is required, and it allows the measurement of interactions in close-to-native protein conditions. In addition, it enables a live detection of sample aggregation, sticking, and precipitation effects. 25
Herein, we report the development and optimization of a convenient MST assay for the study of MRG15–ligand binding interactions and its feasibility as a robust tool for the identification of new ligands.
Materials and Methods
MRG15 Protein Expression and Purification
GST-MRG15 protein (amino acids 1–120) was expressed in BLR(DE3)pLysS cells in lysogeny broth (LB) in the presence of 100 μg/mL ampicillin. Cells were grown at 37 °C to an OD600 of 0.8, induced by isopropyl-1-thio-D-galactopyranoside (IPTG; 100 μM), and incubated overnight at 37 °C. Cells harboring the expressed protein were pelleted at 5000g for 15 min at 4 °C, washed twice with phosphate-buffered saline (PBS), and frozen at −80 °C. For protein purification, the pellet was thawed on ice and suspended in 40 mL of lysis buffer (PBS buffer, pH 7.4) supplemented with a protease inhibitor cocktail. Cells were lysed using a sonicator (Vibra-Cells, Sonics, Newtown, CT; amplitude 30%) for 20 min on ice, and cell debris was pelleted at 10,000g for 30 min at 4 °C. The clarified lysate was filtered using a 0.45 μm syringe filter and loaded onto a 1 mL GSTrap HP column (Cytiva, formerly GE Healthcare Life Sciences, Milan, Italy) at a flow rate of 1 mL/min, previously conditioned with the binding buffer (PBS buffer, pH 7.4). The protein was eluted in isocratic mode using the elution buffer (50 mM Tris-HCl, 10 mM reduced glutathione, pH 8.0). Fractions containing the protein were confirmed by sodium dodecyl sulfate–polyacrylamide gel electrophoresis (SDS-PAGE), pooled, and dialyzed overnight at 4 °C to remove glutathione.
Protein Labeling
Fluorescence labeling of MRG15 was performed following the protocol for N-hydroxysuccinimide (NHS) coupling of the RED-NHS second-generation dye (MO-L011; NanoTemper Technologies, Munich, Germany) to lysine residues. Briefly, 100 μL of a 20 μM solution of MRG15 protein in labeling buffer (130 mM NaHCO3, 50 mM NaCl, pH 8.2) was mixed with 100 μL of 60 μM second-generation dye in labeling buffer and incubated for 30 min at room temperature (r.t.). Unbound fluorophores were removed by size-exclusion chromatography with MST buffer as running buffer. The degree of labeling (DOL) was determined using extinction coefficients ε = 65,507 M–1 cm–1 for the protein and ε = 195,000 M–1 cm–1 for the dye, with a correction factor (CF) of 0.04 at 280 nm and a path length (d) of 1 cm, using the following equations:
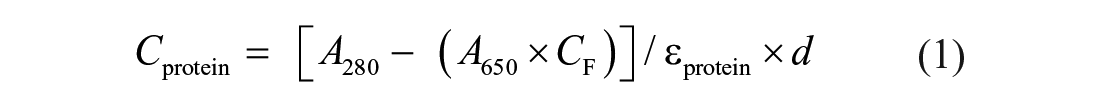
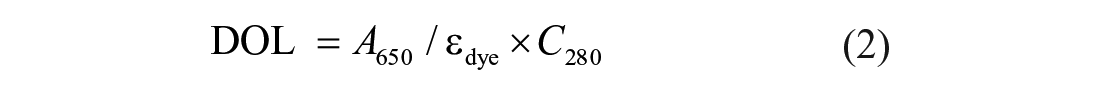
where Cprotein = protein concentration, A280 = protein absorbance at 280 nm, A650 = dye absorbance at its maximum λ, εprotein = extinction coefficient of the protein, εdye = extinction coefficient of the dye, and d = path length of the spectrophotometer.
For storage, MRG15 was frozen in 10 μL aliquots at −80 °C. Prior to MST experiments, aliquots of MRG15 were thawed on ice and centrifuged for 15 min at 4 °C and 12,000g to remove protein aggregates.
Protein Thermal Stability Measurements
To verify if the labeling of MRG15 with the dye altered the protein integrity, thermal unfolding profiles of both proteins were recorded using Tycho NT.6 (NanoTemper Technologies Munich, Germany). This test was performed using 3 µM protein diluted in MST buffer. Thermal unfolding profiles were recorded from 35 to 95 °C with a thermal ramp of 30 °C/min. The inflection temperature and a profile similarity factor were directly obtained from the Tycho NT.6 instrument.
Assay Development for MST Screening
The MST experiments were performed on the Monolith NT.115Pico instrument (NanoTemper Technologies) using 5 nM protein concentration, premium coated capillaries, and 20% LED power. Data were analyzed at medium MST power. Briefly, all small molecules were stored at 50 mM in pure DMSO at −20 °C. For small molecules, a 16-step serial dilution was prepared. For this, 10 µL of assay buffer was added to Eppendorf tubes 2–16. Twenty microliters of the highest small-molecule concentration (200 µM, 1% DMSO) was transferred into tube 1. Then 10 µL was transferred from tube 1 to tube 2 and mixed by pipetting up and down. Next, 10 µL was transferred from tube 2 to tube 3 and again mixed by pipetting up and down. This step was repeated for all remaining tubes. Ten microliters was discarded from tube 16. In a next step, 10 µL of the labeled MRG15 (10 nM) was added to each tube of the dilution series. Samples were mixed by pipetting up and down. The reaction mixtures were incubated for 10 min at r.t. and loaded into premium coated capillaries. Data were acquired using the MO.Control software in Binding Check, Binding Affinity, or Expert mode. MST measurements were performed at 25 °C for 20 s.
KD values were calculated from compound concentration-dependent changes in normalized fluorescence (Fnorm) of MRG15 after 2.5 s of MST on-time. Each binding experiment was performed in triplicate and KD values obtained are the mean of these three independent measurements. Data were analyzed using MO Affinity Analysis software (NanoTemper Technologies).
The Z′ factor 28 to estimate the assay quality was calculated using the following equation:
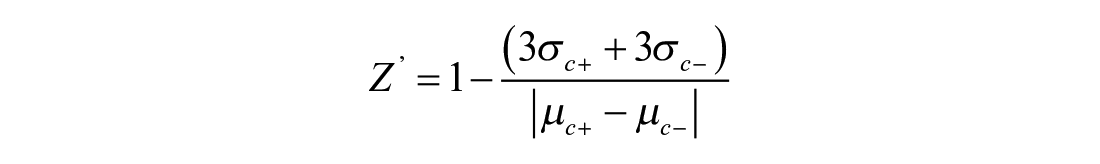
where σc+ and σc– are standard deviations of the positive and negative control measurements, respectively, and μc+ and μc– are the mean of the respective positive and negative signal controls.
SPR Experiments
SPR experiments were performed on a Biacore 3000 optical biosensor (Cytiva, formerly GE Healthcare Life Sciences) equipped with a research-grade CM5 sensor chip. GST-MRG15 was immobilized (50 μg/mL in 10 mM sodium acetate, pH 4.5) on the sensor chip surface at a flow rate of 10 μL/min by using standard amine-coupling protocols to obtain densities of 10 kRU. One flow cell was left empty for background subtractions.
The compounds were diluted in PBS supplemented with 0.005% surfactant P20, keeping a final 1% DMSO concentration, and they were tested starting from 200 µM as the maximum concentration. Binding experiments were performed at 25 °C by using a flow rate of 30 μL/min, with 60 s of association monitoring and 150 s of dissociation monitoring, both with and without 0.5 mg/mL bovine serum albumin (BSA). Regeneration of the surfaces was performed, when necessary, by a 10 s injection of 5 mM NaOH.
The simple 1:1 Langmuir binding fit model of the BIAevaluation software was used for determining equilibrium dissociation constants (KD) and kinetic dissociation (kd) and association (ka) constants by using eqs 3 and 4:
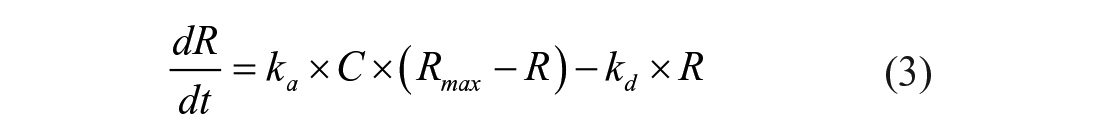
where R represents the response unit and C is the concentration of the analyte, and

Results and Discussion
The development of our MST assay started with the choice of the detection method. 29 In this regard, there are several possibilities: for example, the intrinsic fluorescence of the protein (given by aromatic aminoacids) can be exploited using an UV detector; otherwise, a labeling reaction between the protein and one of the available dyes can be performed, which can give a fluorescence signal in one of the three different channels (blue, green, and red). Among all these possible approaches, we preferred the use of a red dye that is one of the most reported strategies. This choice relies on the evidence that many screening compounds and reference molecules interfere at UV and blue wavelengths.30,31
In particular, we selected the NanoTemper proprietary second-generation red dye (Monolith Protein Labeling Kit RED-NHS 2nd Generation, NanoTemper Technologies) that carries a reactive NHS–ester group that reacts with primary amines of lysine residues to form a covalent bond. It is reported that the second-generation dye gives enhanced binding amplitudes and higher signal-to-noise (S/N) ratios with respect to the first-generation dye. In addition, data can be analyzed from measurements with lower MST power and shorter MST on-times. 32
The labeling reaction was sequentially optimized, performing three different approaches (
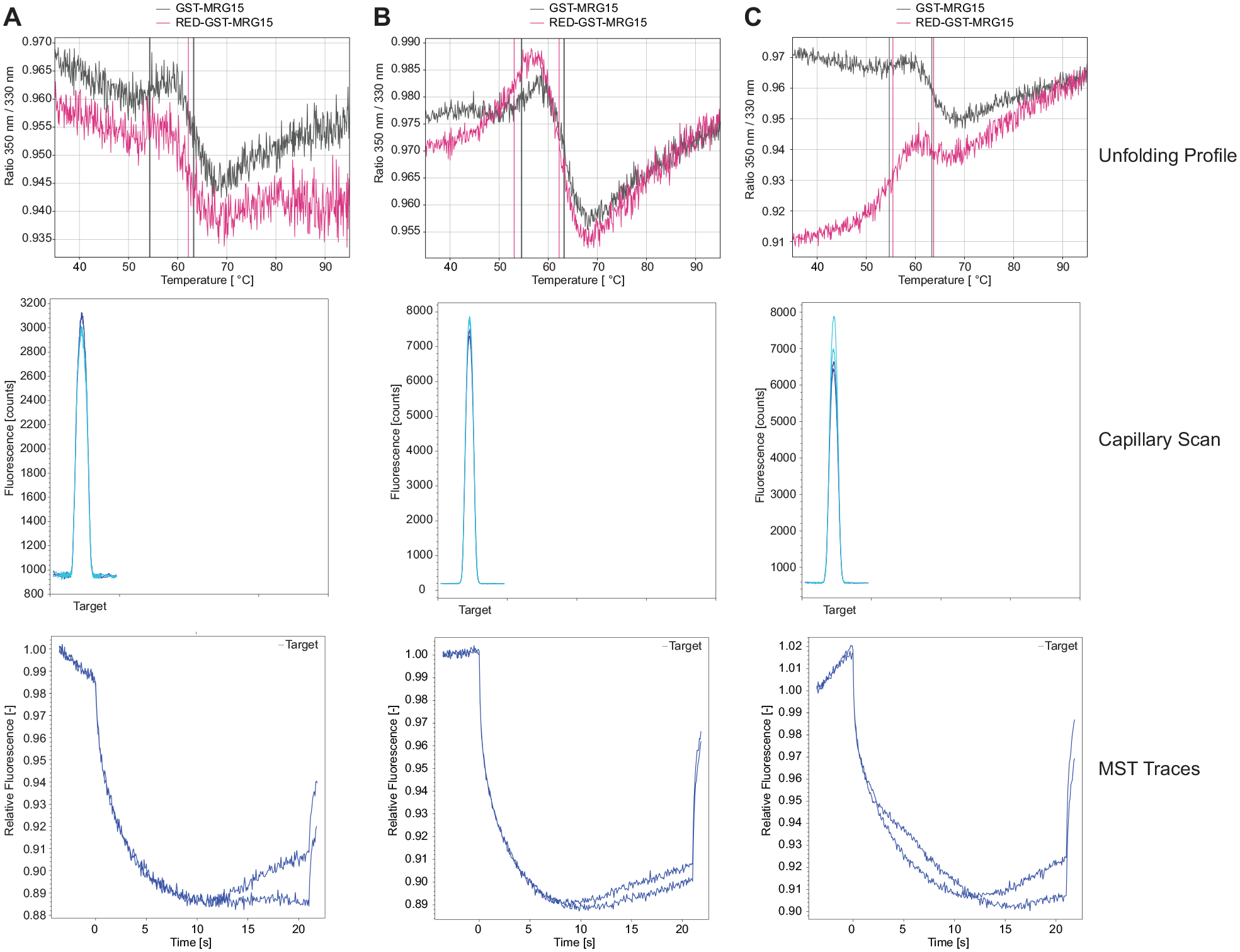
Labeling reactions performed for MRG15. Tycho NT.6 unfolding profile, capillary scan, and MST traces of MRG15 unlabeled (black) and labeled (pink) using (
The following step was the optimization of the assay buffer. In general, the buffer composition plays a fundamental role for both the detection of binding interactions and the assessment of enzymatic activity. 33 Each protein has individual properties and requirements, such as specific pH and ionic strength values and the need for cofactors or coenzymes. All these reagents should be added to the assay buffer, and all protein requirements should be taken into account to create the most suitable environment for the protein. In addition, it is important to optimize the concentration of each reagent to be used in the buffer, as it will influence the protein behavior. Additionally, another important parameter to consider is the type of assay that has been selected to test the protein. Indeed, MST measurements are performed in small capillaries and the use of detergents such as Tween 20 or Pluronic F127 is highly recommended to avoid protein adsorption to the capillary walls. 34
Therefore, being interested in the identification of optimal interaction conditions for the binding of MRG15 to small molecules, we started with the buffer optimization using the reference compound UNC1215 (
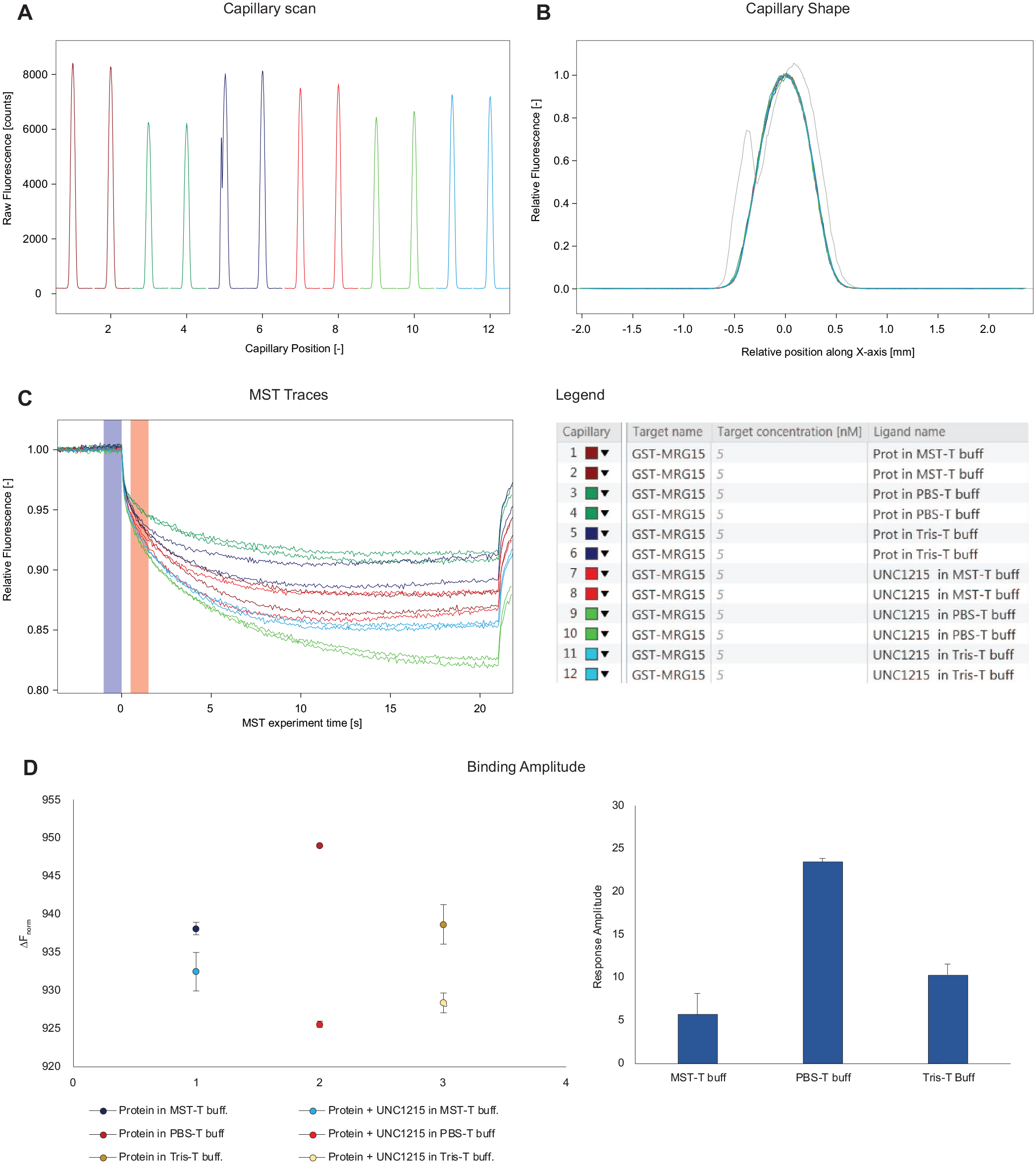
Buffer optimization. Three different buffers, MST buffer (50 mM Tris-HCl, pH 7.4, 150 mM NaCl, 10 mM MgCl2), PBS buffer, and Tris buffer (50 mM Tris-HCl, pH 8.0, 50 mM NaCl), all supplemented with 0.05% Tween 20, were tested in a single-point screening, comparing the protein alone with the protein in the presence of the reference compound (UNC1215). Four parameters were evaluated: (
Regarding the fluorescence intensity, the capillary scan (
As depicted in Figure 3D , the use of PBS buffer resulted in the highest amplitude value and in the lowest noise level. Hence, considering the high amplitude, the good reproducibility of MST traces, and the well-behaved capillary shapes, PBS buffer was selected as the optimal buffer to be used for the following binding experiments.
To further optimize the buffer composition, we evaluated the eventual effect of BSA on the MST signal, as it has been reported that 0.5 mg/mL BSA can stabilize proteins in solution. 35
In fact, BSA is often used in MST assays to reduce nonspecific binding (NBS) on plasticware and capillary tubes, preventing the loss of material and promoting the interaction of the protein with the compounds.
36
Indeed, the addition of 0.5 mg/mL BSA caused a reduction of the amplitude (amplitude value of 16.5 without BSA, amplitude value of 11 with BSA), but of note, it resulted in a meaningful increase of the S/N ratio, thus behaving as a stabilizing agent for the ligand–protein interaction (
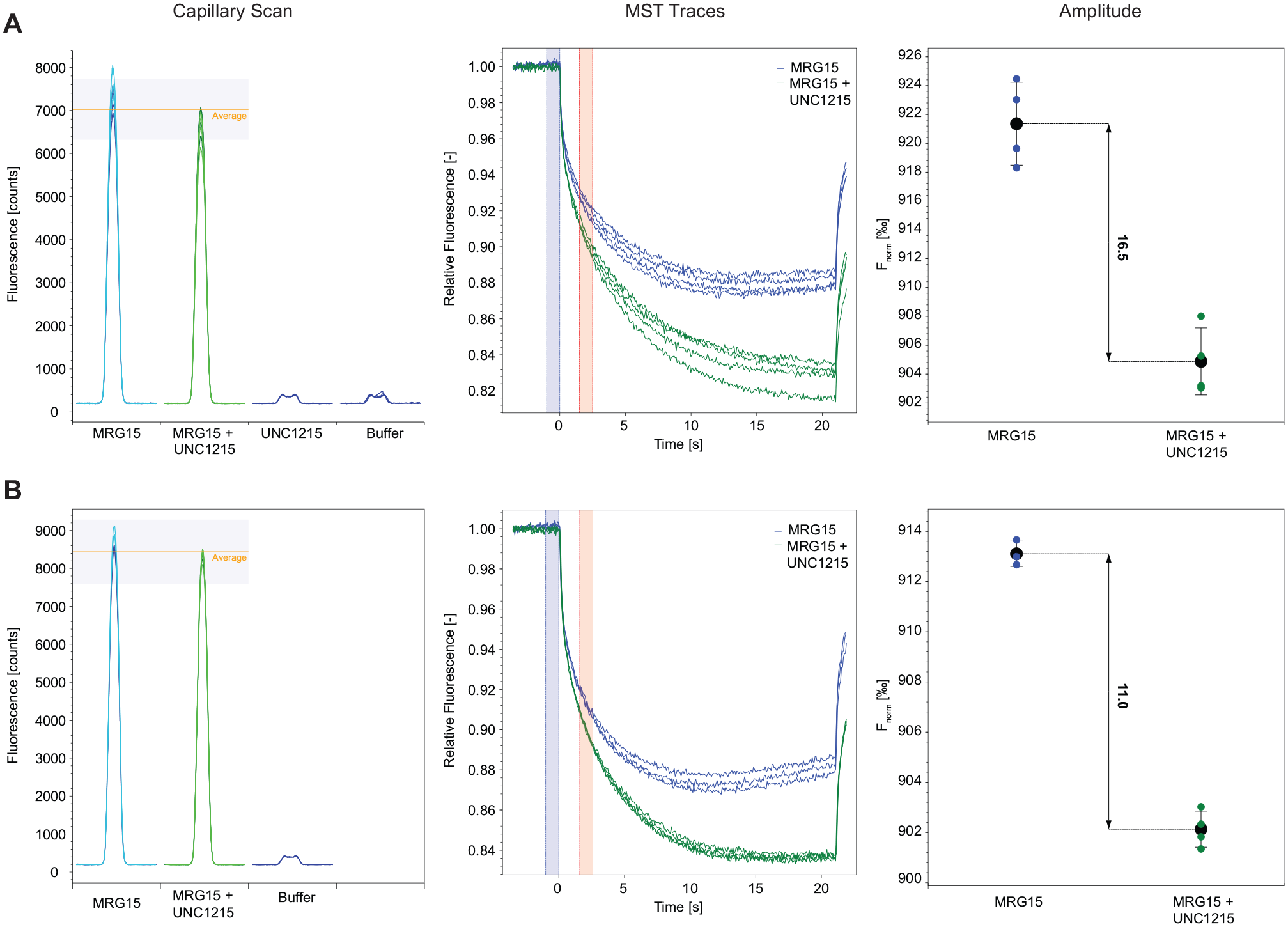
Effect of BSA on MST signal. Binding check experiment performed in (
Generally, an important step in the setup of a screening approach is the establishment of the interaction of a reference molecule with the target protein in order to validate the assay selected for the screening of compounds. In our case, we used the compound UNC1215 as a reference, since it is the only reported compound capable of interacting with the protein MRG15. Regarding this compound, the KD reported in the literature (determined by ITC)
10
for UNC1215 is 29.1 µM, while in our assay we interestingly obtained a KD of 43.1 nM (
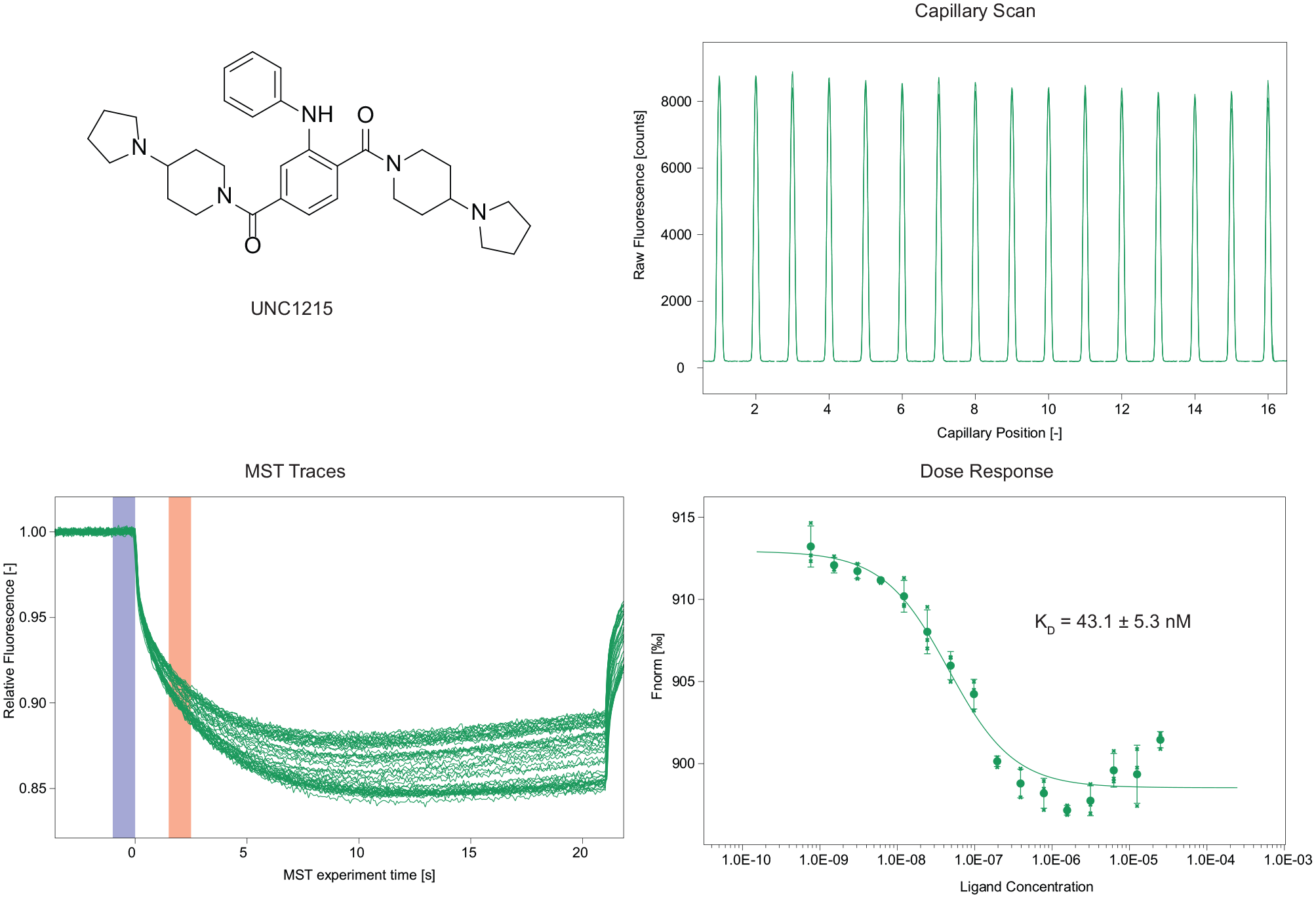
Binding experiment with the reference compound UNC1215.
After the optimization of the experimental conditions and the validation of our MST screening method by the use of a reference compound, we decided to test a small selection of compounds (
The screening of the compounds was performed starting with a single-point screening (
In
KD Values Obtained for Each Compound Using MST. a
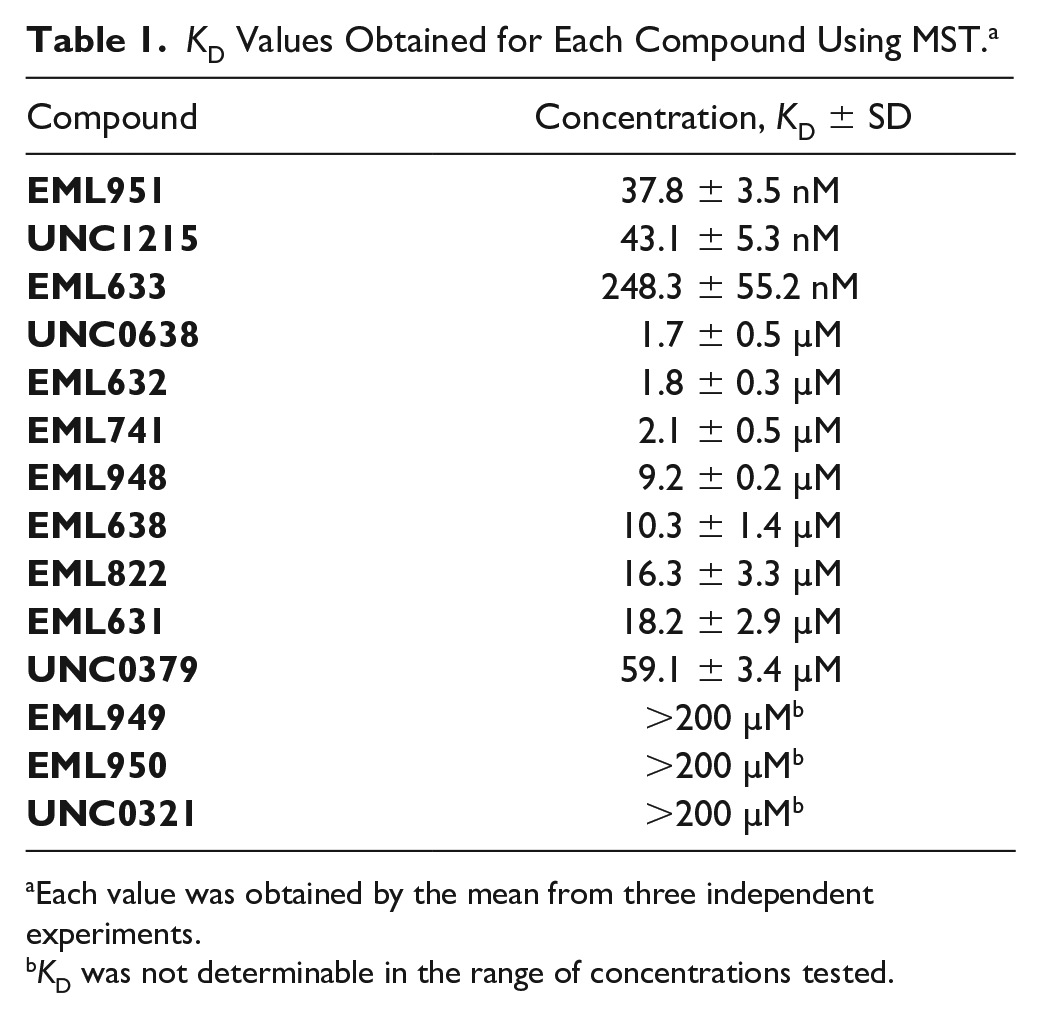
Each value was obtained by the mean from three independent experiments.
KD was not determinable in the range of concentrations tested.
To validate the robustness of our MST method in the identification of MRG15 binders, we decided to repeat the binding experiments using SPR as an orthogonal assay. To this aim, MRG15 protein was immobilized on the surface of a CM5 sensor chip and the interaction of the protein was evaluated with both the reference UNC1215 and the best compound in our library, EML951. Compound UNC0321 (which was identified as a nonbinder in the previous MST single-point screening) was used as a negative control. Since BSA (0.5 mg/mL) has a crucial role in our MST assay in increasing the S/N ratio, we decided to perform SPR experiments both in the absence and in the presence of BSA, in order to investigate the eventual effects of the latter on the binding of the compounds. We found that, in the absence of BSA, both UNC1215 and EML951 bind MRG15 with KD values of 143.5 ± 6.3 µM and 137 ± 15 µM, respectively (
In conclusion, we developed an MST-based method that allowed the identification and subsequent KD determination of 10 small-molecule binders for MRG15. The method is robust and convenient, especially considering its speed and low material requirements, and could be extended to other methyl-lysine or methylarginine reader proteins. Therefore, it could be a useful tool for the identification of new chemical probes to further investigate the physiopathological role of these proteins.
Supplemental Material
Supplemental_material_revised_20200721 – Supplemental material for Development of a Microscale Thermophoresis-Based Method for Screening and Characterizing Inhibitors of the Methyl-Lysine Reader Protein MRG15
Supplemental material, Supplemental_material_revised_20200721 for Development of a Microscale Thermophoresis-Based Method for Screening and Characterizing Inhibitors of the Methyl-Lysine Reader Protein MRG15 by Alessandra Feoli, Vincenzo Pisapia, Monica Viviano, Sabrina Castellano, Tanja Bartoschik and Gianluca Sbardella in SLAS Discovery
Footnotes
Supplemental material is available online with this article.
Declaration of Conflicting Interests
The authors declared the following potential conflicts of interest with respect to the research, authorship, and/or publication of this article: Tanja Bartoschik is an employee of NanoTemper Technologies GmbH. Vincenzo Pisapia spent part of his PhD working in NanoTemper Technologies GmbH laboratories in Munich, and he is currently an employee of NanoTemper Technologies GmbH. The other authors declared no potential conflicts of interest with respect to the research, authorship, and/or publication of this article.
Funding
The authors disclosed receipt of the following financial support for the research, authorship, and/or publication of this article: The Sbardella laboratory’s work on epigenetics has received funding from the Italian Ministero dell’Istruzione, dell’Università e della Ricerca (MIUR), Progetti di Ricerca di Interesse Nazionale (PRIN 20152TE5PK); from the University of Salerno (FARB grant); and from Regione Campania (Italy) grant “Combattere la resistenza tumorale: piattaforma integrata multidisciplinare per un approccio tecnologico innovativo alle oncoterapie—CAMPANIA ONCOTERAPIE” (project no. B61G18000470007). Vincenzo Pisapia was funded by a PhD fellowship from the Italian Minis-tero dell’Istruzione, dell’Università e della Ricerca (MIUR), Programma Operativo Nazionale Ricerca e Innovazione 2014–2020, Fondo Sociale Europeo, Azione I.1 “Dottorati Innovativi con caratterizzazione Industriale.”
References
Supplementary Material
Please find the following supplemental material available below.
For Open Access articles published under a Creative Commons License, all supplemental material carries the same license as the article it is associated with.
For non-Open Access articles published, all supplemental material carries a non-exclusive license, and permission requests for re-use of supplemental material or any part of supplemental material shall be sent directly to the copyright owner as specified in the copyright notice associated with the article.