
Abstract
Pseudomonas aeruginosa is a multidrug-resistant (MDR) pathogen and a causative agent of both nosocomial and community-acquired infections. The genes (tyrS and tyrZ) encoding both forms of P. aeruginosa tyrosyl-tRNA synthetase (TyrRS-S and TyrRS-Z) were cloned and the resulting proteins purified. TyrRS-S and TyrRS-Z were kinetically evaluated and the Km values for interaction with Tyr, ATP, and tRNATyr were 172, 204, and 1.5 μM and 29, 496, and 1.9 μM, respectively. The kcatobs values for interaction with Tyr, ATP, and tRNATyr were calculated to be 3.8, 1.0, and 0.2 s−1 and 3.1, 3.8, and 1.9 s−1, respectively. Using scintillation proximity assay (SPA) technology, a druglike 2000-compound library was screened to identify inhibitors of the enzymes. Four compounds (BCD37H06, BCD38C11, BCD49D09, and BCD54B04) were identified with inhibitory activity against TyrRS-S. BCD38C11 also inhibited TyrRS-Z. The IC50 values for BCD37H06, BCD38C11, BCD49D09, and BCD54B04 against TyrRS-S were 24, 71, 65, and 50 μM, respectively, while the IC50 value for BCD38C11 against TyrRS-Z was 241 μM. Minimum inhibitory concentrations (MICs) were determined against a panel of clinically important pathogens. All four compounds were observed to inhibit the growth of cultures of both Gram-positive and Gram-negative bacteria organisms with a bacteriostatic mode of action. When tested against human cell cultures, none of the compounds were toxic at concentrations up to 400 μg/mL. In mechanism of inhibition studies, BCD38C11 and BCD49D09 selectively inhibited TyrRS activity by competing with ATP for binding. BCD37H06 and BCD54B04 inhibited TyrRS activity by a mechanism other than substrate competition.
Keywords
Introduction
In recent years, infectious diseases have become increasingly difficult to treat, as many bacteria are becoming increasingly resistant to therapeutic remedies, which has attracted significant media attention. The Centers for Disease Control and Prevention (CDC) estimated in 2013 that at least 23,000 individuals in the United States die each year from pathogens exhibiting resistance to various antibiotics. At the molecular level, bacteria can develop resistance through both intrinsic and acquired mechanisms. Due to the exponential nature of bacterial reproduction, these processes can occur and lead to resistance very quickly. At the societal level, reasons for increased resistance include the misuse and overuse of antibacterial agents in the clinical and community setting, inappropriate prescribing, the availability of nonprescription antibiotics in countries outside of the United States, the widespread use of drugs in livestock directly consumed by the human population, and the availability of few new antibiotics. 1 Certain bacteria have developed resistance to more than one antibiotic, resulting in multidrug-resistant (MDR) bacteria, a class of “superbugs” that have no known drugs to treat them.
The Gram-negative aerobic bacterium Pseudomonas aeruginosa, a common cause of healthcare-associated infections, including pneumonia, urinary tract infections, and bloodstream infections, is included in the class of superbugs. The World Health Organization (WHO) listed P. aeruginosa as “critical” and classified it as one of the top three most threatening bacteria. 2 In P. aeruginosa, intrinsic resistance is the result of multiple factors, including the increasing prevalence of efflux pumps, the reduced permeability of the bacterial cell membrane, and target site alteration, which occurs through mutations in the binding site of the drug. Resistance is further increased by the pathogen’s remarkable ability to acquire resistance mechanisms through the uptake of resistance genes via horizontal gene transfer. 3 Proteomic investigations have also shown that P. aeruginosa may become resistant via adaptive changes, which involve the bacterium becoming conditioned to antibiotics after repeated use, and may result from antibiotic treatment of chronic infections. 4 In addition, P. aeruginosa has the ability to form resilient biofilms on implanted medical devices, like catheters, as well as hospital surfaces and water sources, which exacerbates the problem because bacterial cells composing the biofilms have 10–1000 times greater potential to become resistant compared with stand-alone bacteria. 5 P. aeruginosa is especially life threatening to immunocompromised individuals due to the high mortality rate associated with this bacterium. For example, cystic fibrosis (CF) patients are at increased risk from this pathogen as it forms biofilms in the lungs of these individuals, leading to chronic pulmonary infections, and eventually resulting in the death of the patients.
In bacteria, there are two different types of tyrosyl-tRNA synthetases (TyrRS), TyrRZ and TyrRS. 6 The bacteria containing one or the other TyrRS genes are not split along taxonomic lines and members of various taxa can contain either gene. 7 P. aeruginosa, unlike most bacteria, contain both synthetases, one encoded by the tyrZ gene and one encoded by the tyrS gene (TyrRS-Z and TyrRS-S). These two enzymes share only 27% sequence conservation, but both are classified as class I aminoacyl-tRNA synthetases (aaRS). The class I aaRS are characterized by the presence of two consensus sequences in the active-site region, HIGH and KMSKS, and contain a Rossman fold catalytic region that binds ATP and tyrosine (Tyr). TyrRS and TrpRS are both further classified as class Ic enzymes due to their similar subunit structure, and both are functional as α2 dimers, unlike most class I tRNA synthetases. TyrRS recognition of the cognate tRNA (tRNATyr) is also unusual, as TyrRS binds the tRNA from the major groove side of the acceptor stem, as do class II aaRSs, and binds the tRNA across both of the TyrRS subunits in the α2 dimer. TyrRS binds the cognate tRNA between the long variable arm and the anticodon stem of the tRNA. This specificity of tRNA binding, along with anticodon recognition, allows discrimination against the binding of noncognate tRNAs. 8
The attachment of the correct amino acids to the cognate tRNAs is essential for the fidelity of protein synthesis. This process is known as tRNA identity and aaRS have several fidelity mechanisms to ensure accuracy. Unlike some other tRNA synthetases, TyrRS has no independent internal editing function to aid in the discrimination of near-cognate amino acids. Instead, an induced-fit binding mechanism for the binding specificity of Tyr by TyrRS is promoted by conformational changes that occur in the active site as a result of ATP binding. The aminoacyl adenylate is then stabilized by hydrogen bonding with amino acid residues in the active site. 9
The genes encoding both forms of TyrRS from P. aeruginosa were cloned, the resulting overexpressed proteins were purified, and the kinetic parameters governing the interaction with ATP, Tyr, and tRNATyr (KM, Vmax, and kcatobs) were determined for both enzymes. A high-throughput screening platform was then developed and optimized to screen for potential inhibitors of the activity of both forms of TyrRS using scintillation proximity assay (SPA) technology. Out of ~2000 chemical compounds, 4 compounds were identified that inhibited the activity of P. aeruginosa TyrRS (BCD37H06, BCD38C11, BCD49D09, and BCD54B04). All four compounds inhibited the activity of TyrRS-S, but only BCD38C11 inhibited the activity of TyrRS-Z. All four compounds were characterized for their effect on enzymatic activity, bacterial growth, and human cell cultures as well as assessing their effect on ATP and Tyr binding.
Materials and Methods
Materials
All chemicals were obtained from Fisher Scientific (Pittsburgh, PA). DNA oligonucleotides were from Integrated DNA Technologies (Coralville, IA). DNA sequencing was performed by Functional Bioscience (Madison, WI). Radioactive isotopes, SPA beads, and 96-well screening plates were from PerkinElmer (Waltham, MA). The Soluble Diversity chemical compound library was from ChemDiv, Inc. (San Diego, CA). Compounds stocks were dissolved in DMSO to a concentration of 10 mM, stored at –20 °C, and thawed immediately before analysis. The compounds had a minimum purity of at least 90%.
Cloning and Purification
The gene encoding P. aeruginosa TyrRS-Z was amplified using PCR (MJ Mini Thermo Cycler; Bio-Rad, Hercules, CA) using P. aeruginosa PAO1 genomic DNA (ATCC 47085) as a template. A forward primer (5′-TAACGCTAGCAAATCGGTTGAAGAGCAGCT-3′) and a reverse primer (5′-CTCTAAGCTTTTCAGCTTTCAGCGTGATG-3′) were used to add Nhe1 and HindIII restriction sites to the ends of the gene. The amplified gene was inserted into pET24b(+) plasmid (Novagen; Sigma-Aldrich, St. Louis, MO), which was also digested with Nhe1/HindIII restriction enzymes forming pET-PaTyrZ. pET-PaTyrZ was transformed into Escherichia coli Rosetta 2 (DE3) Singles Competent Cells, and bacterial cultures were grown in Terrific Broth (TB) containing 50 µg/mL of kanamycin and 25 µg/mL of chloramphenicol. The cultures were grown at 37 °C to an optical density (A600) of 0.6–0.8. Overexpression of TyrRS-Z was induced by addition of isopropyl β-
Amplification of TyrRS-S by PCR was initially unsuccessful. Therefore, the gene sequence was optimized for codon expression in E. coli and for decreased G/C content, the sequences encoding NheI/HindIII restriction sites were added to each end of the gene as described above, and the construct was synthetically synthesized by Integrated DNA Technologies (Coralville, IA). The synthesized construct was inserted into the plasmid pUCIDT-KAN. The resulting plasmid containing the synthesized tyrS gene was digested with NheI and HindIII and the fragment was inserted into the pET24b(+) plasmid (Novagen) also digested with NheI/HindIII restriction enzymes, forming pET-PaTyrS. pET-PaTyrS plasmids were transformed into E. coli Rosetta 2 (DE3) Singles Competent Cells, and bacterial cultures were grown in Luria–Bertani (LB) broth containing 50 µg/mL of kanamycin and 25 µg/mL of chloramphenicol at 25 °C until an optical density (A600) of 0.6–0.8 was reached. Overexpression of TyrRS-S was induced by addition of IPTG to 25 µM. The culture was grown for 5 h postinduction at 25 °C, and the bacteria were harvested by centrifugation (10,000g, 4 °C, 45 min).
Fraction I lysates from each bacterial growth were prepared as previously described. 10 The initial step in purification of P. aeruginosa TyrRS-Z was precipitation of the protein from a solution containing 50% saturated ammonium sulfate (AS), while P. aeruginosa TyrRS-S was precipitated from a 60% AS solution. Both PaTyrRS-Z and PaTyrRS-S were purified to greater than 95% homogeneity using nickel nitrilotriacetic acid (NTA) affinity chromatography (Perfect Pro, 5 Prime), followed by dialysis two times against a buffer containing 20 mM HEPES-KOH (pH 7.0), 40 mM KCl, 1 mM MgCl2, 0.1 mM EDTA, and 10% glycerol. Purified proteins were flash frozen in liquid nitrogen and stored at –80 °C.
Gel Electrophoresis and Protein Analysis
For sodium dodecyl sulfate–polyacrylamide gel electrophoresis (SDS-PAGE), 4%–12% polyacrylamide precast gradient gels (Novex NuPAGE; Invitrogen, Waltham, MA) were used with 3-(N-morpholino)propanesulfonic acid (MOPS) as the running buffer (Invitrogen). The gels were stained with Simply Blue Safe Stain (Invitrogen) to visualize the proteins. The protein standard was EZ-Run Rec Ladder (Fisher Scientific). Coomassie Protein Assay Reagent (Thermo Scientific, Waltham, MA) was used to determine protein concentrations with bovine serum albumin as the standard.
ATP:PPi Exchange Reactions
ATP:PPi exchange reactions (100 μL) were carried out at 37 °C for 20 min in 50 mM Tris-HCl (pH 7.5), 10 mM KF, 8 mM MgCl2, 1 mM dithiothreitol (DTT), 2 mM [32P]PPi (50 cpm/pmol), and 0.4 μM of either TyrRS-Z or TyrRS-S and varying concentrations of ATP and Tyr as described. 11 In reactions in which the concentration of ATP was varied (50, 100, 200, 300, 400, 500, and 600 µM), the amino acid (Tyr) concentration was held constant at 2 mM; alternatively, when the concentration of Tyr was varied (12.5, 25, 50, 100, and 200 uM), the ATP concentration was held at 2 mM. The exchange of PPi in these reactions was measured between 1 and 5 min. Initial velocities for exchange of PPi were determined and the kinetic parameters (KM, Vmax, and kcat) for the interactions of TyrRS-Z and TyrRS-S with ATP and Tyr were determined by plotting the initial velocities against substrate concentration and fit to the Michaelis–Menten steady-state model using XLfit (IDBS, Boston, MA).
Aminoacylation Assays
Timed aminoacylation reactions were measured using SPA technology as described. 12 Briefly, reactions (50 µL) were carried out in 96-well plates (costar). A master mix (35 μL) contained components to result in final concentrations of 50 mM Tris-HCl (pH 7.5), 10 mM MgCl2, 2.5 mM ATP, 0.5 mM spermine, 75 µM [3H] Tyr, 1 mM DTT, and either 0.0015 µM PaTyrRS-Z or 0.05 µM PaTyrRS-S. Reactions were initiated by addition of tRNA (15 μL) to the master mix and incubated at 37 °C. In different assays, tRNA concentrations were varied at 10, 20, 30, 40, 50, 60, 70 and 80 µM total tRNA (0.16, 0.32, 0.48, 0.64, 0.8, 0.96, 1.12, and 1.28 µM tRNATyr). Reactions were stopped at 1, 2, 3, 4, and 5 min intervals by the addition of 5 µL of 0.5 M EDTA. From these assays, initial velocities were measured and the kinetic parameters (KM, Vmax, and kcat) were determined by fitting the data to the Michaelis–Menten steady-state model using XLfit (IBDS).
Chemical Compound Screening
The tRNA aminoacylation assay was developed into a screening platform for identification of inhibitory compounds using SPA technology, as described. 11 Briefly, screening reactions were carried out in 96-well microtiter plates. Test compounds, dissolved in 100% DMSO (2 μL of compound; 3.3 mM), were equilibrated by addition of 33 μL of the previously described master mixture, containing 0.1 μM of either TyrRS-Z or TyrRS-S, and incubated at ambient temperature for 15 min. Control reactions contained DMSO in the absence of compound or 10 μL of 0.5 M EDTA. Reactions were initiated by the addition of 15 μL of E. coli tRNA (~50 μM tRNA and 0.8 μM tRNATyr) and incubated for 1 h at 37 °C. In assays to determine IC50 values, the same assays were used, except the enzyme concentration was 0.05 μM and the tRNA concentration was 35 μM. In these assays, test compounds were serially diluted to result in final concentrations from 400 to 0.4 µM.
Microbiological Assays
Broth microdilution minimum inhibitory concentration (MIC) testing was carried out according to Clinical and Laboratory Standards Institute (CLSI) guidelines M7-A7. MIC values were determined against a panel of bacteria that included E. coli (ATCC 25922), E. coli TolC mutant, Enterococcus faecalis (ATCC 29212), Haemophilus influenzae (ATCC 49766), Moraxella catarrhalis (ATCC 25238), P. aeruginosa (ATCC 47085), P. aeruginosa PAO200 (efflux-pump mutant), P. aeruginosa hypersensitive strain (ATCC 35151), Staphylococcus aureus (ATCC 29213), and Streptococcus pneumoniae (ATCC 49619).
Time-kill studies were performed using M. catarrhalis and S. aureus, based on the MIC assay results, according to the CLSI document M26-A. Growth media was Brain Heart Infusion and Trypticase Soy Broth from Remel (Lenexa, KS). The compound concentration was four times the MIC.
In Vitro Cytotoxicity Test
To determine the toxicity of each compound on the growth of human cell cultures, human embryonic kidney 293 cells (HEK293) were used as described. 11 The Trevigen TACS MTT Cell Proliferation Assay Kit (Gaithersburg, MD) was used to assess impacts on human cell viability. MTT assays were carried out in triplicate at each compound concentration (25–400 μg/mL). Student’s two-tiered t test was utilized to assess statistical significance.
Binding Mode Assay
To determine if the mechanism of inhibition by the hit compounds was by competition with either ATP or Tyr for enzyme binding, IC50 values for each compound were determined in aminoacylation assays as described. 13 The concentration of TyrRS-S was set at 0.05 μM. For competition with ATP, IC50 values were determined in reactions containing varying ATP concentrations (25, 50, 100, 250, 500, and 1000 μM) while holding the amino acid concentration at 75 μM. To determine competition with Tyr, the same assays were used with varying concentrations of the amino acid (25, 50, 100, 200, and 300 µM) as the concentration of ATP was held at 2.5 mM. Compounds were serially diluted in IC50 assays from 200 to 0.4 µM. Background amounts of free [3H]Tyr in the absence of TyrRS were subtracted from each assay for baseline development.
Results
Cloning, Expression, and Purification
The gene encoding the P. aeruginosa TyrRS-Z protein, tyrZ, was cloned using the standard procedures described under Materials and Methods. However, numerous attempts to PCR amplify the tyrS gene, which encodes the P. aeruginosa TyrRS-S protein, from genomic P. aeruginosa DNA were unsuccessful. Although multiple sets of primers were designed, complementary not only to the gene but also to various regions upstream and downstream of the gene, amplification of this region in the genome by PCR was unproductive. Finally, after grudgingly admitting PCR defeat, a decision was made to synthetically synthesize the gene. A modified tyrS gene sequence was designed that was optimized for codon expression and decreased G/C content ( Fig. 1 ). A modified gene was synthesized by Integrated DNA Technologies (Coralville, IA) and inserted into a proprietary plasmid, pUCIDT-KAN. This tyrS gene was then transferred to the pET24b(+) plasmid, which had also been used to express the tyrZ gene. The alterations of the tyrS gene sequence were chosen so as not to affect the amino acid sequence of the P. aeruginosa TyrRS-S protein. Both the TyrRS-Z and TyrRS-S proteins were able to be overexpressed in E. coli and purified to greater than 95% homogeneity, as visualized by SDS-PAGE ( Suppl. Fig. S1 ).
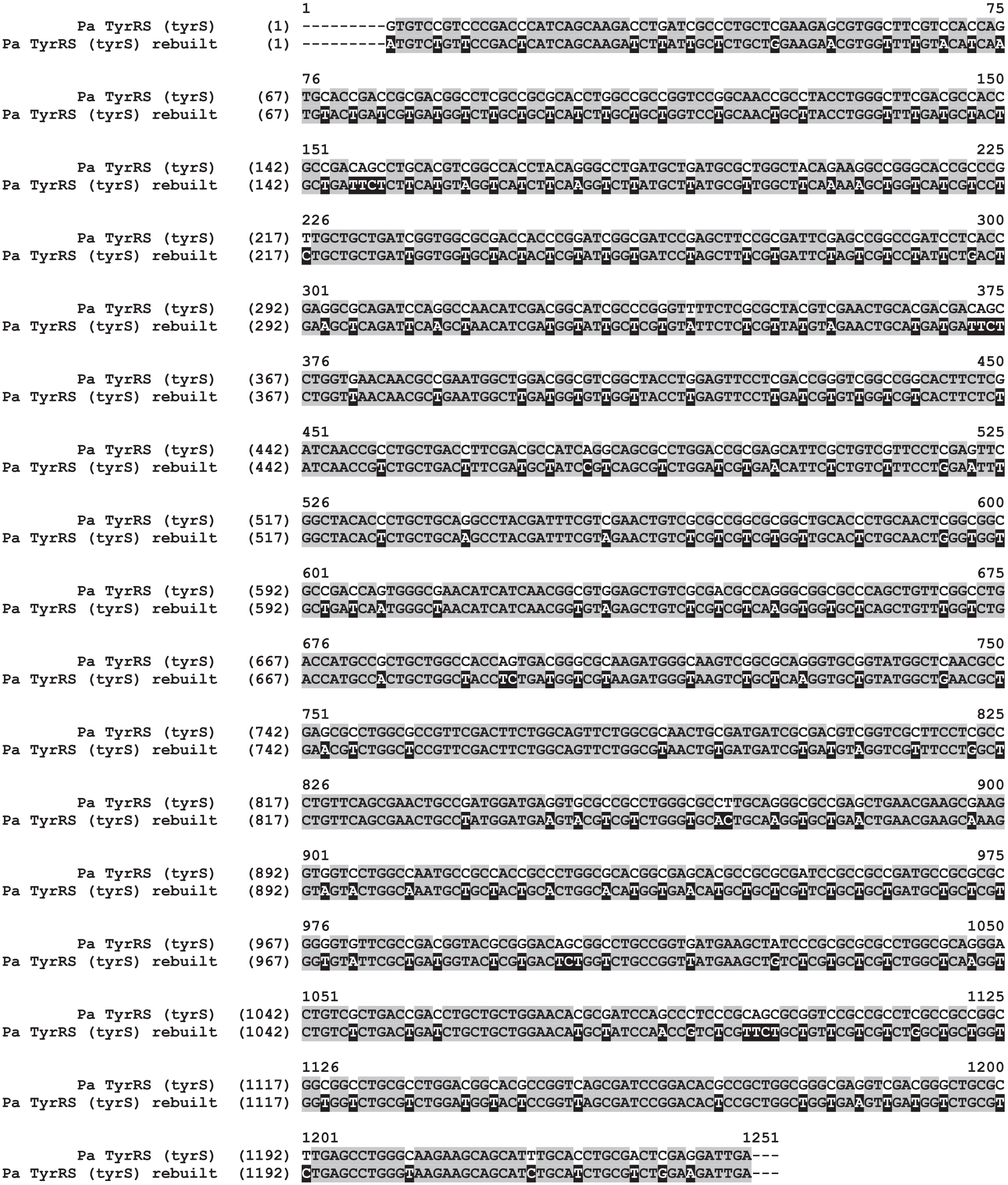
Rebuilding of the P. aeruginosa tyrS gene. The P. aeruginosa tyrS gene was synthesized after the codons encoding specific amino acids were optimized for expression and to reduce the G/C content. The native and rebuilt sequences of the tyrS gene are shown. Identical (unchanged) nucleotides are shown as black letters on a gray background. The nucleotides that were changed in the rebuilt version of the gene are shown as white letters on a black background, and the nucleotides that were replaced in the native sequence are shown as black letters on a white background.
Enzymatic Characterization
To determine the ability of the two forms of TyrRS to aminoacylate cognate tRNA, each of the purified enzymes was titrated into aminoacylation assays at varying concentrations (
Suppl. Fig. S2
). The result of the aminoacylation reaction is the covalent attachment of a specific amino acid, in this case Tyr, to the cognate tRNA, and this process occurs via two distinct steps for most aaRS enzymes. The first step results in the condensation of the amino acid and ATP into an aminoacyl adenylate intermediate, in which the β and γ phosphates of the ATP are released as an inorganic pyrophosphate. With all but three aaRS enzymes, this reaction is reversible when a cognate tRNA is not present and allows the parameters governing the kinetic interaction of the aaRS with the amino acid and ATP substrates to be determined. The kinetic parameters (KM, kcatobs, and kcatobs/KM) for the interaction of both forms of TyrRS with both Tyr and ATP were determined by measuring the initial rate of the ATP:PPi exchange and fitting the data to the Michaelis–Menten steady-state model. To determine the interaction of TyrRS with ATP, the concentration of Tyr was held constant (2 mM) while concentrations of ATP were varied (100–600 µM) (
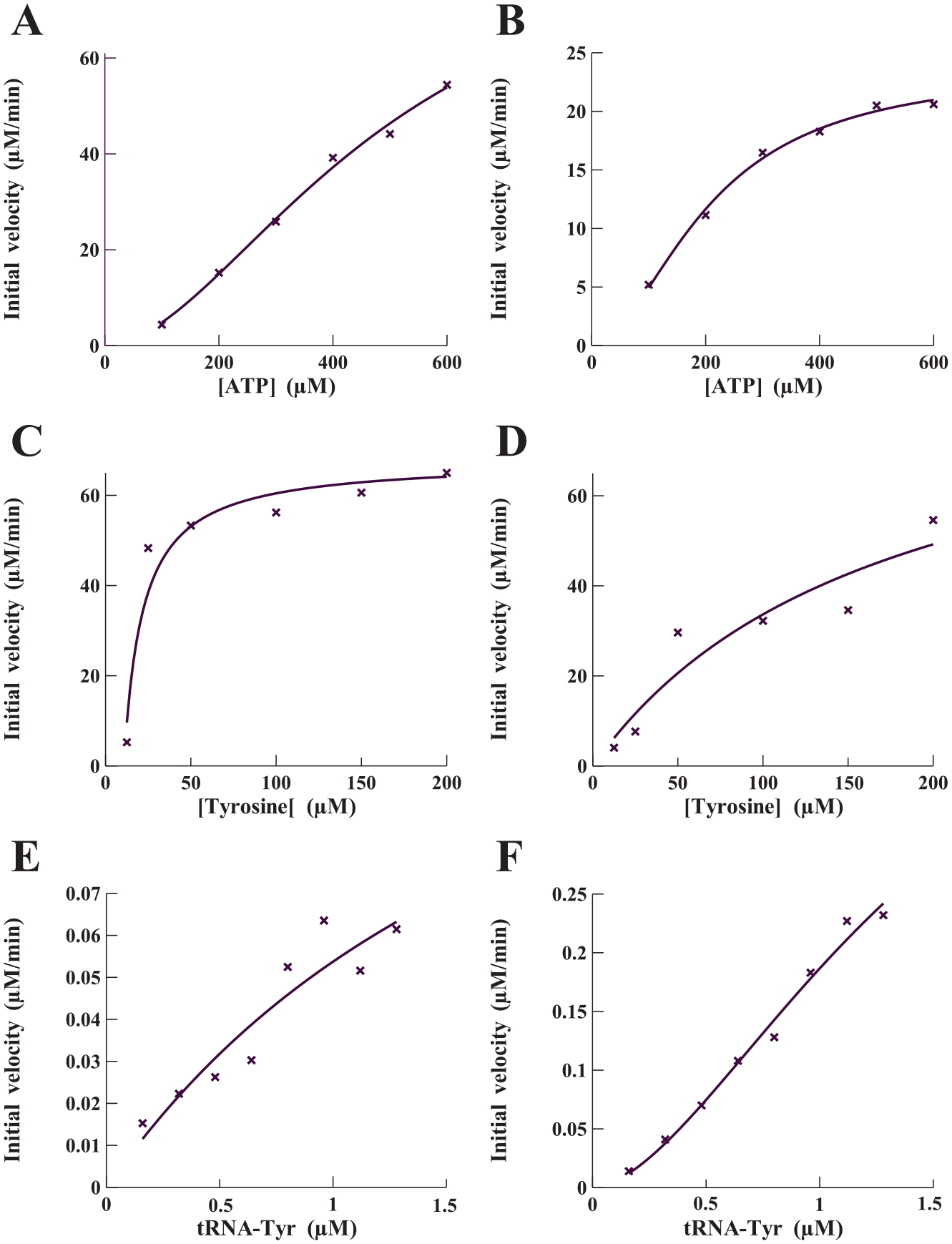
Determination of the kinetic parameters governing interactions of P. aeruginosa TyrRS-Z and TyrRS-S with their three substrates: ATP, Tyr, and tRNATyr. Initial velocities for the interaction of TyrRS-Z with ATP (
The second step in the aminoacylation process involves the formation of an ester bond between the hydroxyl group of the 3′-terminal adenosine of tRNA and the carboxylic acid functional group of the amino acid with the subsequent release of the AMP. The ability of both P. aeruginosa TyrRS-S and TyrRS-Z to aminoacylate tRNATyr was determined at various concentrations of tRNATyr (0.15–1.3 μM) while maintaining the concentrations of ATP and Tyr constant at 2.5 mM and 75 μM, respectively (
Amino Acid Sequence Analysis of Both Forms of TyrRS
Most bacterial species contain either TyrRS-Z or TyrRS-S, but typically not both forms of the enzyme. The type of TyrRS expressed by any particular bacterium does not seem to be split along taxonomic lines, and members of various taxa can contain either of the proteins 7 ( Suppl. Fig. S3A ). In an alignment of 37 TyrRS enzymes from bacteria across the eubacteria phyla, there was an overall conservation of certain amino acids observed among all TyrRS enzymes, but there were also subtle variations noted between the TyrRS-Z and TyrRS-S proteins ( Suppl. Fig. S3B ). When all the enzymes were aligned, there was 42% consensus of conserved amino acids. However, when the TyrRS-Z or TyrRS-S proteins were aligned separately, the consensus increased to 58% and 61%, respectively, indicating two significantly different types of enzymes. P. aeruginosa is an exception to the normal and, unlike most bacteria, expresses both forms of TyrRS. An alignment of just TyrRS-Z and TyrRS-S from P. aeruginosa shows that the proteins are 41% conserved and 27% identical ( Suppl. Fig. S4 ). As can be seen in the alignment in Supplemental Figure S3B , there are a few other bacteria that have both forms of TyrRS. However, the only previous comparison has been of TyrRS-Z and TyrRS-S from Bacillus subtilis. B. subtilis produces the toxic D-tyrosine when in the stationary growth phase, and this work showed that TyrRS-Z had increased selectivity for ʟ-tyrosine, thus preventing misincorporation of the D-tyrosine during protein synthesis while the bacterium was in the nongrowth phase. However, when expressed in an E. coli temperature-sensitive tyrS mutant, either form of the B. subtilis TyrRS complemented aminoacylation of tRNATyr. 14
The three-dimensional crystal structures of both TyrRS-S and TyrRS-Z have been determined. The structure of TyrRS-S from E. coli TyrRS bound to both Tyr and a Tyr-adenylate analog has been solved to 2.0 and 2.7 Å resolutions, respectively. 15 The x-ray crystal structure of TyrRS-Z from Thermus thermophilus (Tth) bound to its cognate tRNATyr has been solved to a 2.9 Å resolution. 8 In lieu of a crystal structure of P. aeruginosa TyrRS-S or TyrRS-Z, sequence analysis enables comparison of this active-site structure and tRNA binding regions with that of the homolog from E. coli, as well as with the homology from Tth TyrRS, in which the structures have been solved ( Fig. 3 ). Also included in the alignment are the two forms of TyrRS from B. subtilis to aid in highlighting the distinct forms of the enzyme.

Alignment of the amino acid sequence of both forms of P. aeruginosa TyrRS with homologs. Both forms of TyrRS from B. subtilis and P. aeruginosa are aligned with E. coli TyrRS-S and Tth TyrRS-Z, whose crystal structures have been solved. The accession numbers for the protein sequences of TyrRS from E. coli and Tth are KXL21237 and BAD71222, respectively. The accession numbers for P. aeruginosa TyrRS-Z and TyrRS-S are NP_249359 and NP_252827, respectively, and the accession numbers for B. subtilis TyrRS-Z and TyrRS-S are AOA56631 and NP_390845, respectively. Sequence alignments were performed using Vector NTI Advance 11.5.4 (Invitrogen). Identical residues are indicated by white letters on a black background, while similar sequences are shown by black letters on a gray background. The HIGH and KMSKS structural motifs are boxed. Amino acids that interact with ATP (■), Tyr (♦), the acceptor stem of tRNATyr (•), and the anticodon stem/loop of tRNATyr (
As noted above, TyrRS contains no independent editing function to ensure that the cognate amino acid is attached to tRNATyr. Instead, an induced-fit binding mechanism for the binding specificity is promoted by conformational changes that occur in the active site as a result of ATP binding. A comparison of the amino acid residues Thr43, Gly50, His51, Arg89, Gly198, Ser199, Asp200, Leu227, and Ile228 (E. coli numbering) that recognize and form hydrogen bonds with ATP shows that they are either strictly conserved or similar (
Fig. 3
). The amino acid residues that form hydrogen bonds with Tyr, Tyr37, Thr76, Asp81, Tyr175, Gln179, Asp182, and Gln201 show that they are strictly conserved, except at position Tyr37. At this position, there is a Tyr residue in all of the TyrRS-S proteins, while at this position in all of the TyrRS-Z proteins, the amino acid residue is a lysine (
The amino acid residues that interact with the acceptor stem of tRNATyr in the active site of the enzyme are conserved/similar in the TyrRS-S or TyrRS-Z proteins but differ between the two forms. The acceptor stem of the tRNA binds across the dimer interface into the active site of the opposing TyrRS subunit of the α2 dimer. 16 The acceptor end of the tRNA makes contact at the interface with Thr148, Gln151, and Glu154 (Tth numbering), 8 which are conserved in the TyrRS-Z enzymes but are replaced with Phe, Asp, and Ile/Leu, respectively, in TyrRS-S enzymes ( Fig. 3 ). The amino acid residues Arg198, Leu202, Arg205, Glu206, and Arg209 appear to interact with the acceptor stem of the tRNA. Arg198 is not conserved in TyrRS-Z but is conserved as a Trp in TyrRS-S, while Leu202 and Arg205 are conserved in the TyrRS-Z but not the TyrRS-S proteins. Glu206 and Arg209 are charged residues in both forms of TyrRS. Val23, which is similar in both forms of TyrRS, packs against the ribose of nucleotide G1 at the double-stranded end of the acceptor stem of the tRNA. Glu156 in Tth plays a discriminatory role by rejection of noncognate tRNA and corresponds to Glu155 in E. coli ( Fig. 3 ). This residue is either a Glu or an Asp in all TyrRS enzymes analyzed ( Suppl. Fig. S3B ). Taken together, these amino acids are more or less similar between the two forms of TyrRS and potentially interact with the acceptor stem similarly.
Amino acids that interact with other parts of the tRNA vary in percent conservation. Residues Trp370, Arg373, and Gln409 in Tth TyrRS form hydrogen bonds with nucleotides in the variable loop of the tRNA. There does not appear to be any conservation of these residues in other TyrRS proteins analyzed, likely because there is a wide variation in size and nucleotide sequence in the variable arm of the various tRNATyr forms. The other residues noted in Figure 3 recognize the nucleotides composing the anticodon itself and tend to be conserved in one form of TyrRS or the other. Overall, the primary sequence of the C-terminal of TyrRS is much less conserved than the catalytic domain and, with the diversity of shapes, sequences, and base modifications of tRNATyr across the various phyla, has evolved to recognize cognate tRNAs from species to species. This is not to say that TyrRS from one species of bacteria will not aminoacylate tRNATyr across the species, which we know occurs, 16 but the efficiencies are not known.
Screening Chemical Compounds against the Activity of P. aeruginosa TyrRS
The two forms of TyrRS are split across the bacterial phyla, and most strains express just one of them. However, P. aeruginosa contains both forms of the enzyme. In the studies with the two forms of TyrRS in Bacillus, the tyrS gene was shown to be expressed under normal growth conditions, while the tyrZ gene was expressed during the nongrowth phase and during biofilm production. It may be that this is also the case for the expression of the two forms of TyrRS in P. aeruginosa. The rationale for targeting two forms of the same enzyme was that identification of a dual inhibitor may enhance the chance of killing persister cells and prevent the formation of biofilms during P. aeruginosa chronic infections. In addition, identification of chemical compounds that only inhibited one or the other of the two forms would still have broad-spectrum activity against a large number of bacteria pathogens.
In the development of screening platforms for both forms of TyrRS, to achieve the maximum signal in the SPAs the nonenzymatic components of the aminoacylation reaction were individually titrated into the assay to determine a saturating concentration of each ( Suppl. Fig. S5 ). Next, tRNA was titrated into the assay to ensure that the assay was within the linear region of the reaction detection time and to determine the concentration of tRNATyr to be used in the screening assays ( Suppl. Fig. S6A,B ). Chemical compounds were dissolved in 100% DMSO, resulting in a final DMSO concentration in screening assays of 4%. Therefore, the ability of P. aeruginosa TyrRS to function in the presence of increasing amounts of DMSO was determined ( Suppl. Fig. S6C,D ). There was no decrease of activity observed in aminoacylation assays containing up to 10% DMSO. Finally, TyrRS was titrated into the optimized assay to determine the enzyme concentration yielding maximum sensitivity to enzymatic inhibition ( Suppl. Fig. 2S ). From these data the concentration of both forms of P. aeruginosa TyrRS was set at 0.1 μM, which is at the inflection point in the titration curve and allowed for the detection of any decrease of activity in the screening reactions.
Using this optimized assay, the activity of both forms of TyrRS in the aminoacylation assay was screened against the Soluble Diversity-2000 chemical compound library from ChemDiv, Inc. (San Diego, CA). This chemical compound library contained 2000 compounds predicted to have high aqueous solubility, high diversity, and druglike properties with a molecular mass less than 450 Da and a low number of rotatable bonds. Initial screening assays contained chemical compounds at a concentration of 132 μM and were carried out as single-point assays. Compounds observed to inhibit at least 50% of enzymatic activity were reassayed in triplicate. From the initial screens, 16 compounds were identified that inhibited TyrRS-Z and 30 compounds were identified that inhibited TyrRS-S ( Suppl. Fig. S7 ). In confirmation assays carried out in triplicate, one compound was confirmed that inhibited the activity of TyrRS-Z and 10 of the initial hit compounds were confirmed to inhibit the aminoacylation activity of TyrRS-S. Structural analysis was performed, and of these 10 compounds, 4 were identified for additional analysis (BCD37H06, BCD38C11, BCD49D09, and BCD54B04) ( Fig. 4 ). The compound BCD38C11, which inhibited the enzymatic activity of TyrRS-S, was also a confirmed hit compound of TyrRS-Z. The concentration of each compound that inhibited 50% of the enzymatic activity (IC50) of TyrRS-S was determined using SPA-based aminoacylation assays for all four hit compounds. In addition, the IC50 of BCD38C11 was determined similarly for inhibition of TyrRS-Z. In these assays to determine IC50 values, the inhibitory compounds were titrated from 400 to 0.4 μM. The IC50 values for BCD37H06, BCD38C11, BCD49D09, and BCD54B04 against TyrRS-S were 24, 71, 65, and 50 μM, respectively, while the IC50 for BCD38C11 against TyrRS-Z was 241 μM ( Fig. 5 ).
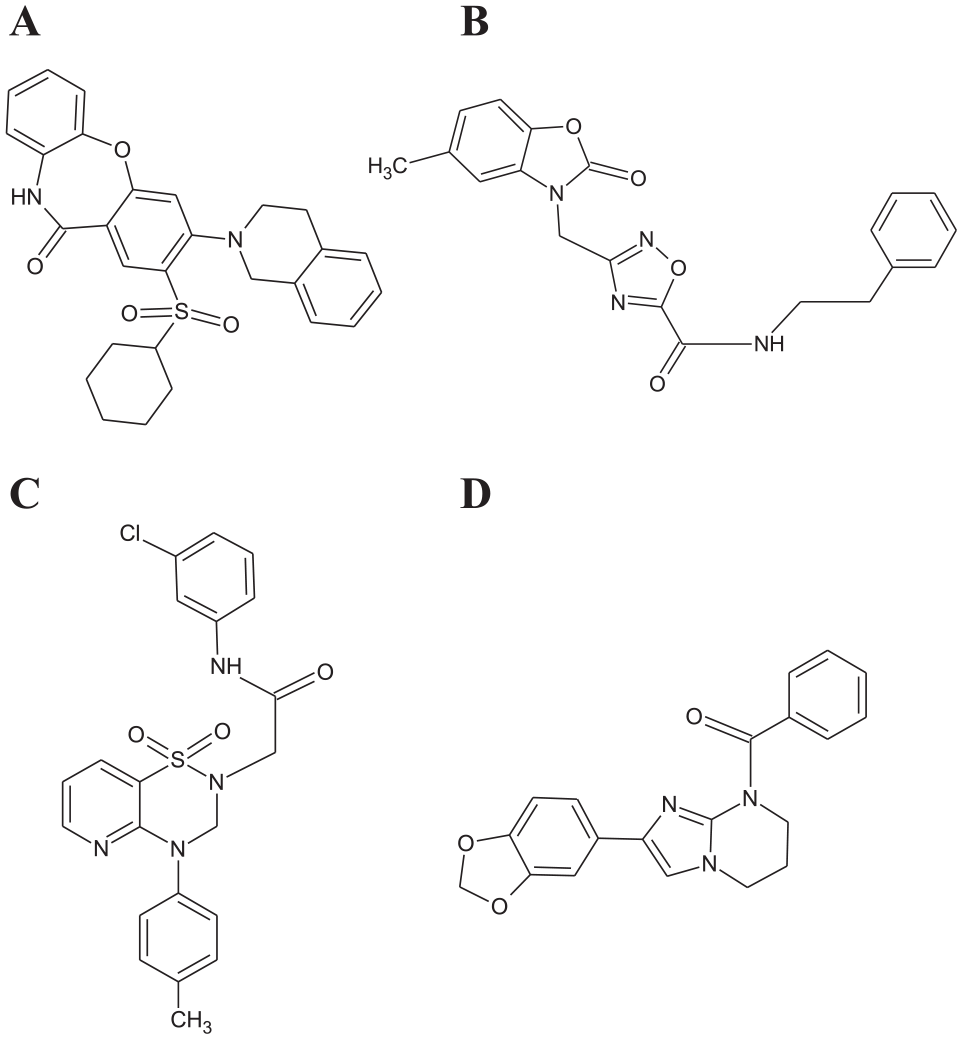
The chemical structure of the hit compounds: (
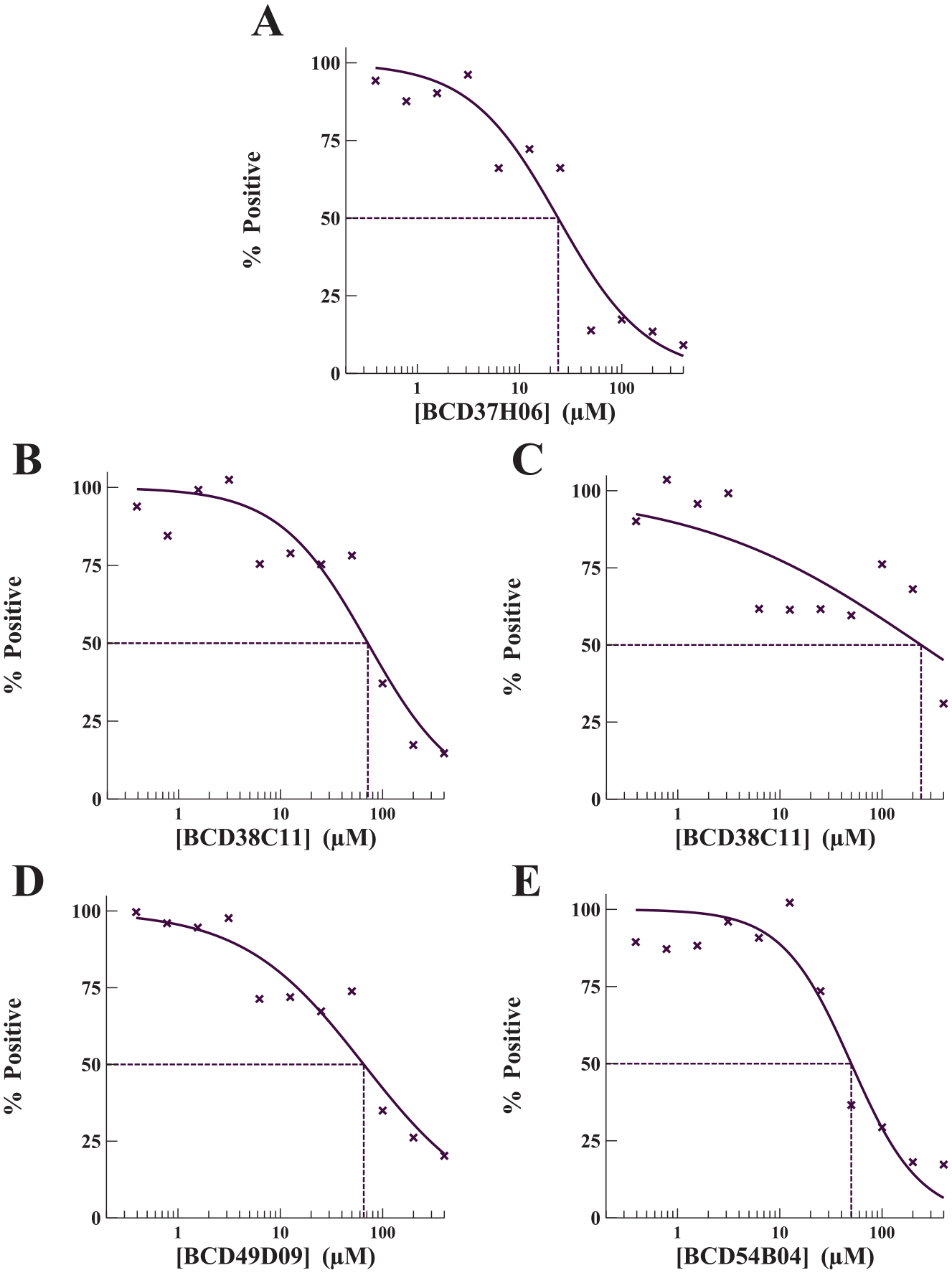
Determination of the IC50 values of the hit compounds. IC50 values for the inhibitory potency of (
Microbiological Assays
The four hit compounds were tested in broth microdilution assays to determine MICs against a panel of 10 pathogenic bacteria, including efflux-pump mutants of E. coli and P. aeruginosa and a hypersensitive strain of P. aeruginosa ( Suppl. Table S2 ). The four compounds were effective in inhibiting the growth of cultures of all Gram-positive bacteria. The compounds BCD37H06, BCD38C11, BCD49D09, and BCD54B04 all inhibited cultures of the Gram-negative M. catarrhalis. However, only BCD37H06 and BCD54B04 were effective against cultures of the Gram-negative respiratory pathogen H. influenzae. All four compounds inhibited growth of cultures of the efflux-pump mutant forms of both E. coli and P. aeruginosa. However, none of the compounds inhibited the wild-type strains of either E. coli or P. aeruginosa. This indicates that the lack of activity against wild-type strains of E. coli and P. aeruginosa is likely to be efflux mediated.
Next, time-kill studies were performed using the four compounds to determine whether they were bacteriostatic or bactericidal. Based on the MIC results, all four compounds were most effective against the Gram-negative M. catarrhalis and the Gram-positive S. aureus. Therefore, the compounds were tested against these bacteria in time-kill studies at four times the MIC, and between 0 and 24 h. All four compounds were observed to be bacteriostatic against both pathogens. In the presence of the compounds, the bacterial cultures had constant growth, but there was a decrease in colony forming units (CFU) of 2 to 4 log10 compared with the control during the first 6 h tested (
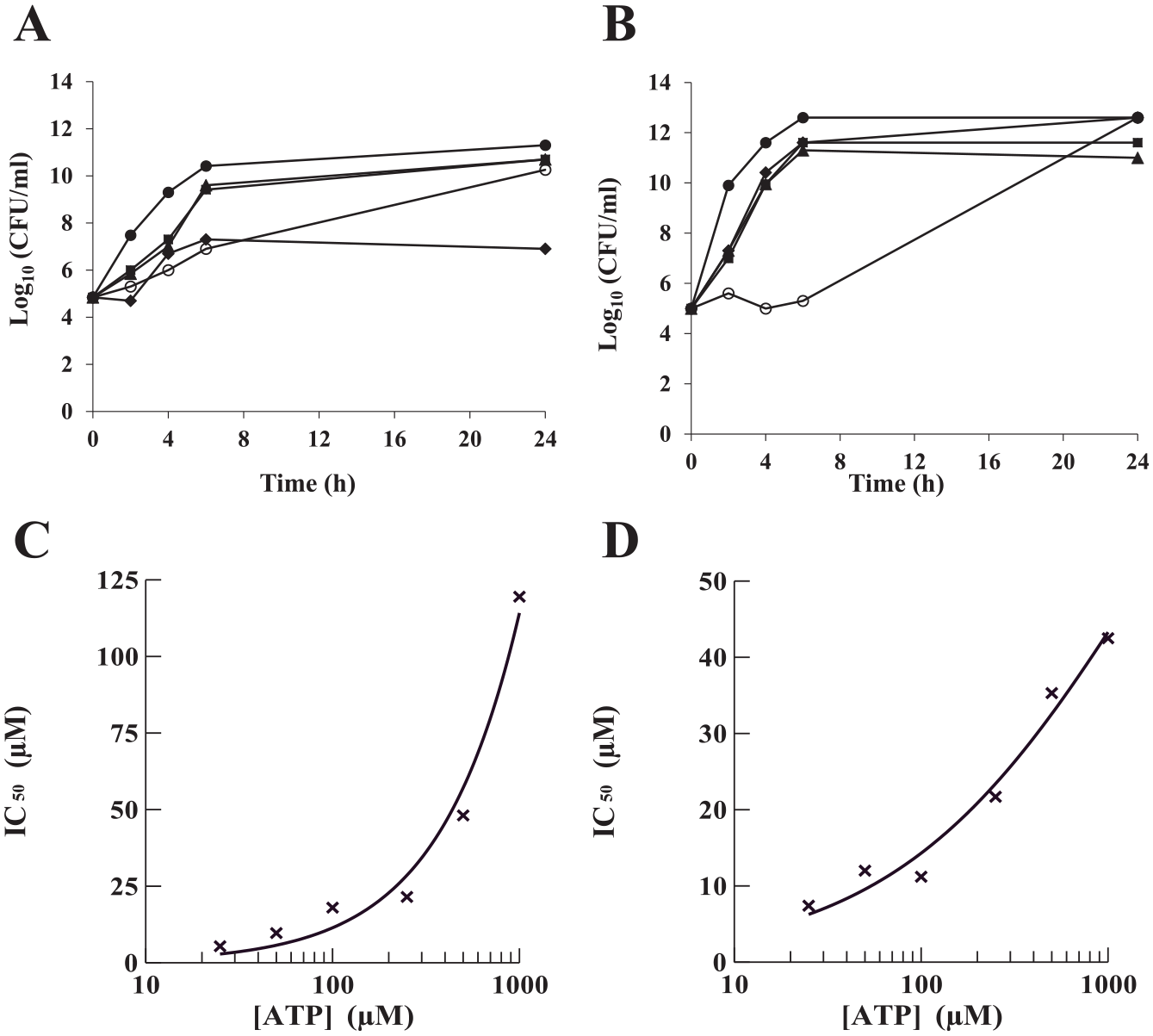
Time-kill kinetic and ATP competition assays. The activities of the hit compounds against the growth of cultures containing (
Antibiotic therapies that specifically target bacterial translation could potentially be toxic to mammalian cells, given that both cytoplasmic and mitochondria forms of TyrRS are contained within the eukaryotic cell. To determine the effect of the compounds on the growth of human cell cultures, the compounds were tested using MTT assays. HEK293 cells were treated with 25–400 µg/mL of each of the compounds for 24 h under standard tissue culture conditions in triplicate. The compounds were not observed to be toxic to cells at any concentration tested. In the absence of any cytotoxic effect, these compounds have the potential to be amenable as therapeutics.
Binding Mode Assay
aaRS in general have three substrates: tRNA, ATP, and the amino acid. ATP and amino acids are small molecules similar in size to the compounds tested, and both have binding sites in the active-site region of the synthetase. Competition of the hit compounds with either ATP or Tyr is a potential mechanism of action for inhibition. To determine if a hit compound competed for binding with either substrate, the IC50 was determined in varying concentrations of either ATP or Tyr. A shift in the IC50 as the concentration of the substrate increased is indicative of competition. In these assays, the ability of a compound to compete with substrate binding by TyrRS-S was determined using the tRNA aminoacylation assay. A shift in the IC50 with respect to ATP competition was determined at various ATP concentrations (25, 50, 100, 250, 500, and 1000 µM) while holding the concentration of the amino acid at 100 μM. These concentrations ranged from fourfold below to fourfold above the KM for ATP. To determine a shift in the IC50 with respect to the amino acid, the same assay was used, except that ATP was held constant at a saturating concentration (2.5 mM) and the IC50 was determined at different concentrations of Tyr (25, 50, 100, 200, and 300 µM), which ranged from fourfold below to twofold above the KM. In assays containing BCD37H06 or BCD54B04, there was no shift in the IC50 detected at any concentration of either ATP or Tyr, indicating that competition for binding with these substrates was not a mechanism of action of these compounds. In assays containing BCD38C11 and BCD49D09 in increasing concentrations of Tyr, there was no change in the IC50 values observed either, also indicating that these compounds did not compete with the amino acid for binding. However, the IC50 was observed to increase as the concentration of ATP was increased (
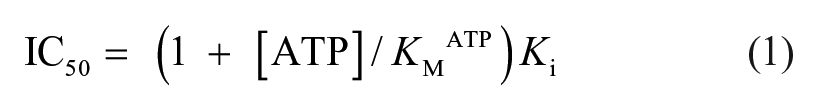
This equation was used to calculate Ki for BCD38C11 and BCD49D09 with IC50 values determined at 100 μM Tyr and 2.5 mM ATP. After multiple determinations, BCD38C11 and BCD49D093 had mean Ki values of 5.28 μM and 4.91 μM when tested against P. aeruginosa TyrRS-S.
Discussion
As resistance to antimicrobial agents in bacteria continues to increase, there is a growing need for the discovery of new antibacterial agents with modes of action different than the currently marketed antibiotics. The aaRS are ideal targets for the development of new antibacterial agents for several reasons. They play a critical role in protein biosynthesis and thus are essential for cell viability and growth. Next, the tRNA synthetases from prokaryotic origins tend to be conserved in primary structures, yet they are sufficiently divergent from the eukaryotic forms of the enzymes to make them fitting targets for the development of broad-spectrum antibacterial agents. Finally, the crystal structures of most aaRS have been solved and provide useful information for rational drug design.
In bacteria there are two forms of TyrRS (TyrRS-S and TyrRS-Z), which tend to be distributed across the eubacterial phyla without an apparent pattern. These two forms of TyrRS appear to vary considerably at the amino acid sequence level, yet the two forms of the enzyme complete identical functions. Most bacteria contain one form of the enzyme or the other. However, there are a small number of bacteria that contain both forms of TyrRS. P. aeruginosa, as one such bacteria, contains both TyrRS-S and TyrRS-Z. The structures of E. coli TyrRS-S and Tth TyrRS-Z were previously solved, and when the amino acid sequence from these enzymes was compared with the compatible form of P. aeruginosa TyrRS, the critical amino acid residues were found to be strictly conserved, indicating likely conservation of structure as well as function. In the present work, we have cloned, expressed, and purified both forms of TyrRS (TyrRS-S and TyrRS-Z) from P. aeruginosa. Both forms were then kinetically characterized.
SPA technology was used to develop a screen for inhibitors of both forms of P. aeruginosa TyrRS. The screening assays were robust and resulted in Z′ factors for TyrRS-Z/TyrRS-S of 0.44/0.41 and Z factors of approximately 0.36/0.27, respectively, across all plates. The signal-to-background ratio of the DMSO positive controls to negative controls for TyrRS-S/TyrRS-Z were approximately 3:1/2.5:1, respectively. From 2000 compounds, 4 compounds (BCD37H06, BCD38C11, BCD49D09, and BCD54B04) were identified that inhibited the activity of P. aeruginosa TyrRS-S. One of these compounds, BCD38C11, also inhibited the activity of TyrRS-Z. BCD37H06, BCD38C11, BCD49D09, and BCD54B04 inhibited the activity of TyrRS-S with IC50 values of 24, 71, 65, and 50 μM. BCD38C11 displayed an IC50 value against TyrRS-Z at a higher concentration of 241 μM.
All four compounds exhibited moderate MICs against both Gram-positive and Gram-negative bacteria. MIC determination also indicated that the compounds had little inhibitory effect against cultures of wild-type E. coli and P. aeruginosa, but all of the compounds did have an effect on the growth of cultures containing the efflux-pump mutants of these bacteria. It is likely that the compounds can enter the cells of these pathogens but are efficiently removed before they can inhibit growth. This is reinforced by the fact that all the compounds inhibited the growth of cultures of M. catarrhalis, another Gram-negative bacterium, which is more susceptible to many inhibitors because of less robust efflux systems. Since wild-type forms of E. coli and P. aeruginosa are notoriously hard to kill, expectations for very low MIC values are never too high. The lower MIC observed against the efflux-pump mutants would be expected. One reason that those values were not lower is that these efflux-pump mutants only have one or a few efflux systems knocked out, and either of these bacteria contain numerous efflux systems (in the case of P. aeruginosa, more than 50 different efflux systems) that are still functional, and some may potentially affect the concentration of the compound inside the cell. Also, these bacteria have multiple ways besides efflux in which they can inactivate inhibitors that may be at play here. The modest reduction of MIC against the efflux-pump mutants was somewhat encouraging, indicating that efflux likely is the reason that the wild-type forms of the bacteria are less affected by the compounds, and that during structure–activity relationship (SAR) studies it may be possible to increase the effectiveness of these compounds. Also, the fact that the three compounds that were not dual inhibitors still inhibited the growth of the efflux-pump mutants during MIC testing supports the idea that TyrRS-S is functional during normal growth conditions and that P. aeruginosa TyrRS-Z may likely be expressed only during times of biofilm production. Alternatively, the much higher concentration of compound used in MIC testing as compared with the screening assay may have resulted in inhibition of both forms of the enzyme by all of the hit compounds.
Time-kill studies indicated that all four compounds were bacteriostatic. This would be the expected mode of action for compounds that inhibit the activity of an aaRS, since inhibition of the aminoacylation activity mimics starvation for amino acids by lowering the ratio of charged to uncharged tRNA, and induces the stringent response resulting in static bacterial growth.
Information for BCD37H06 (3-(3,4-dihydroisoquinolin-2(1H)-yl)-2-(piperidin-1-ylsulfonyl) dibenzo[b,f][1,4]oxazepin-11(10H)-one) ( Fig. 4A ) is available in the PubChem Substance and Compound database through the unique chemical structure identifier CID 53097039. There was no information that BCD37H06 or compounds with a similar structure had been tested in bioassays. BCD38C11 (1-acetyl-5-bromo-N-[2-(2-methylpiperidin-1-yl)ethyl]indoline-6-sulfonamide) has the chemical structure identifier CID 53130510 ( Fig. 4B ). Compounds with similar structures were reported to have been tested in bioassays to identify small molecules that disrupt the protein–protein interaction of G alpha-i with GIV (G alpha-Interacting Vesicle-associated protein), which are nonreceptor activators of heterotrimeric G proteins found in human cancer cells. However, these compounds with similar structures were inactive in those assays. BCD49D09 (N1-(3-chlorophenyl)-2-[4-(4-methylphenyl)-1,1-dioxo-3,4-dihydro-1λ6-pyrido[2,3-e][1,2,4]thiadiazin-2(1H)-yl]acetamide) has the chemical structure identifier CID 49667507 ( Fig. 4C ). Compounds with similar structures to BCD49D09 have also been tested against the protein–protein interaction of G alpha-i with GIV and found to be inactive. Other similar compounds have been tested to determine inhibition in various sets of cancer-related protein–protein interaction bioassays and were found to be inactive in all of these assays. BCD54B04 ([2-(1,3-benzodioxol-5-yl)-6,7-dihydroimidazo[1,2-a]pyrimidin-8(5H)-yl](phenyl)methanone) has the chemical structure identifier CID 53183345 ( Fig. 4D ). The only bioassay information for BCD54B04 and similar compounds was the profiling for toxicity in HEK-293 and NIH-3T3 cell lines using quantitative high-throughput screening, and in these studies they were found to be nontoxic.
Recently, a number of bacterial TyrRS inhibitors have been described. SB-219383, isolated from Micromonospora species, was identified as an inhibitor of bacterial S. aureus TyrRS with an IC50 in the low nanomolar range. 18 In structure studies, SB-219383 and analogs were found to bind in the active-site region of S. aureus TyrRS, potentially competing with ATP or Tyr. 19 Several series of compounds based on arylfuran scaffolds also inhibited TyrRS. A series of compounds were developed and tested against TyrRS from S. aureus and were observed to have low micromolar IC50 values and activity against cultures of S. aureus. 20 More recently, adenosine has been attached to the arylfuran scaffold to mimic the Tyr aminoacyl-adenylate. 21 These compounds were also observed to have IC50 values in the low micromolar range against TyrRS, and moderate antibacterial activity was extended to E. coli and P. aeruginosa. From this research group, a series of metronidazole-pyrazole derivatives were identified that have shown good inhibitory activity against TyrRS, again with IC50 values in the nanomolar range. This series was also shown to have good antibacterial activity against both Gram-positive and Gram-negative pathogens. However, how much of the antibacterial activity can be attributed to inhibition of TyrRS in vivo is uncertain, since metronidazole is a known antibiotic that works by inhibiting nucleic acid synthesis by binding to cellular DNA. Studies on the effect of natural products on TyrRS have identified epigallocatechin, gallate, acacetin, kaempferide, and chrysin as having inhibitory effects. 22 There is no information available as to the current status of these potential antimicrobial agents in development as a marketable antibiotic.
Small chemical compounds primarily used in drug screening have a molecular mass that is similar to those of ATP and Tyr, which have binding sites in the Rossman fold active-site region of TyrRS. We therefore determined if the mechanism of action for the hit compounds may be derived from interference with the binding of one or both of these substrates. In competition assays, BCD37H06, BCD38C11, BCD49D09, and BCD54B04 were shown to be noncompetitive with Tyr. In the assays containing BCD37H06 or BCD54B04 there was also no competition observed in the presence of increasing concentrations of ATP. However, in assays containing BCD38C11 and BCD49D09 as the concentration of ATP was increased, in a dose-dependent manner, the IC50 values increased, indicating competition with ATP for TyrRS binding. ATP utilization as a substrate is prevalent in both bacterial and eukaryotic cells, and general inhibition resulting from ATP competition might decrease the utility of these compounds as a potential drug candidate. However, if the competition with ATP for binding is specific for TyrRS, this would increase the potential of a compound as a therapeutic agent. An example is the specificity of cladosporin as a potent ATP competitive inhibitor of the eukaryotic LysRS. 23 We also recently showed that bilirubin was a specific inhibitor of ATP binding LysRS. 24 In support of ATP competition specificity by these compounds in TyrRS, this same chemical compound library has been screened in our laboratory against several other aaRS enzymes (PheRS, GluRS, MetRS, GlnRS, LeuRS, LysRS, ProRS, ArgRS, AspRS, and HisRS) from P. aeruginosa, all of which have ATP as a substrate. In no instance in these screens were the TyrRS hit compounds observed to inhibit the activity of these additional aaRS enzymes.
The use of cell systems in drug discovery is an alternative to whole animal systems for predicting toxic potentials of chemicals. This is a first step in drug hit-to-lead selection and downstream development. The hit compounds identified here, BCD37H06, BCD38C11, BCD49D09, and BCD54B04, were tested for cytotoxicity in human HEK-293 cell cultures. All four of the compounds failed to exhibit toxic effects at any concentrations tested. This absence of toxicity at an early stage is certainly encouraging for drug development. These results indicate that the hit compounds identified here may be amenable for further development as therapeutics against bacterial infections.
In an era of the emergence of MDR superbugs, new antibacterials with a different mechanism of action would be highly beneficial in the fight to control bacterial infections. Since the 1960s, only one novel series of antibiotics has been discovered, the oxazolidinones, also protein synthesis inhibitors. Recently, there has been an increase in media attention to the issues created by MDR pathogens. Even with increasing number of deaths in the United States attributed to MDR infections, and the increased cost of treating MDR infections, there has been little increase in research in this area. Outside of mupirocin, there are no marketed antibiotics that target the aaRS, even though a number of inhibitors of various aaRS enzymes have been identified. 25 Four compounds were identified as inhibitors of TyrRS in this study. To enhance the inhibitory potency of the compounds that will be required for development as therapeutic agents, additional SAR studies will be required.
Supplemental Material
JMB_TyrRS_manuscript-Supplemental – Supplemental material for Two Forms of Tyrosyl-tRNA Synthetase from Pseudomonas aeruginosa: Characterization and Discovery of Inhibitory Compounds
Supplemental material, JMB_TyrRS_manuscript-Supplemental for Two Forms of Tyrosyl-tRNA Synthetase from Pseudomonas aeruginosa: Characterization and Discovery of Inhibitory Compounds by Casey A. Hughes, Varesh Gorabi, Yaritza Escamilla, Frank B. Dean and James M. Bullard in SLAS Discovery
Footnotes
Supplemental material is available online with this article.
Authors’ Note
The authors disclose that portions of the Materials and Methods section were previously described in the following articles and are described here for clarity and descriptive purposes: Balboa et al.,
24
Hu et al.,
11
Pena et al.,
12
Escamilla et al. (Glutaminyl-tRNA Synthetase from Pseudomonas aeruginosa: Characterization, Structure, and Development as a Screening Platform. Protein Sci.
Declaration of Conflicting Interests
The authors declared no potential conflicts of interest with respect to the research, authorship, and/or publication of this article.
Funding
The authors received the following financial support for the research, authorship, and/or publication of this article: The authors are grateful for the financial support provided by the National Institutes of Health (grant no. SC3GM098173). The contents of this article are solely the responsibility of the authors and do not necessarily represent the official views of the National Institutes of Health. Partial undergraduate student support was from the NIH UTRGV RISE program (grant no. 1R25GM100866). A portion of graduate student support was from a departmental grant from the Robert A. Welch Foundation (grant no. BG-0017).
References
Supplementary Material
Please find the following supplemental material available below.
For Open Access articles published under a Creative Commons License, all supplemental material carries the same license as the article it is associated with.
For non-Open Access articles published, all supplemental material carries a non-exclusive license, and permission requests for re-use of supplemental material or any part of supplemental material shall be sent directly to the copyright owner as specified in the copyright notice associated with the article.