
Abstract
CRISPR/Cas9 is increasingly being used as a tool to prosecute functional genomic screens. However, it is not yet possible to apply the approach at scale across a full breadth of cell types and endpoints. In order to address this, we developed a novel and robust workflow for array-based lentiviral CRISPR/Cas9 screening. We utilized a β-lactamase reporter gene assay to investigate mediators of TNF-α-mediated NF-κB signaling. The system was adapted for CRISPR/Cas9 through the development of a cell line stably expressing Cas9 and application of a lentiviral gRNA library comprising mixtures of four gRNAs per gene. We screened a 743-gene kinome library whereupon hits were independently ranked by percent inhibition, Z′ score, strictly standardized mean difference, and T statistic. A consolidated and optimized ranking was generated using Borda-based methods. Screening data quality was above acceptable limits (Z′ ≥ 0.5). In order to determine the contribution of individual gRNAs and to better understand false positives and negatives, a subset of gRNAs, against 152 genes, were profiled in singlicate format. We highlight the use of known reference genes and high-throughput, next-generation amplicon and RNA sequencing to assess screen data quality. Screening with singlicate gRNAs was more successful than screening with mixtures at identifying genes with known regulatory roles in TNF-α-mediated NF-κB signaling and was found to be superior to previous RNAi-based methods. These results add to the available data on TNF-α-mediated NF-κB signaling and establish a high-throughput functional genomic screening approach, utilizing a vector-based arrayed gRNA library, applicable across a wide variety of endpoints and cell types at a genome-wide scale.
Introduction
The type II clustered regularly interspaced short palindromic repeats (CRISPR)/Cas9 system from the bacterium Streptococcus pyogenes has been modified to enable precise editing of mammalian genomes and is today a powerful method to systematically disrupt gene function.1 –7 The modified two-component system used in functional genomic applications is composed of a noncoding single guide RNA (gRNA) containing crRNA and tracrRNA regions with a protospacer-adjacent motif (PAM) that serves to direct the second component, Cas9 endonuclease, to a specific genomic target sequence. The Cas9 nuclease induces a double-strand break (DSB) at this target site, which can result in gene inactivation. Whole-genome CRISPR/Cas9 gRNA pools have been used to perform loss-of-function screens with demonstrated success.8 –11 Such screens are dependent on assays where the desired phenotype is selectable and, as such, there has been a need to invest in the development of arrayed gRNA libraries, which can be used to interrogate more diverse phenotypes. 12 Arrayed gRNA libraries have recently been developed in viral vector-based, 13 chemically synthesized, 14 and plasmid-based formats.15,16 Some of these libraries have then been validated for loss-of-function screens using gRNA libraries targeting up to 200 genes,13,17 –19 640 genes,14,20 –23 and 1514 genes,15,16 respectively. A number of challenges for successful prosecution at scale persist and include the requirement to deliver gRNAs to hard-to-transfect cells at a large/gene family/whole-genome scale in a robust fashion. Nevertheless, the prospect of whole-genome-scale arrayed CRISPR/Cas9-based screening is an attractive one as an alternative or complementary approach to RNAi or cDNA functional genomic arrayed screens to systematically map gene function.
Nuclear factor-κB (NF-κB) signaling is central to a number of physiological responses, and its dysregulation is associated with many diseases, including chronic inflammatory diseases and cancer progression, as well as resistance to standard chemotherapeutic agents.24,25 A complete understanding of the signaling mechanisms of NF-κB could enable selective targeting in order to minimize systemic toxicity, avoid broad suppression of innate immunity, and avoid the potential for enhancement of tumorigenesis.26,27 The NF-κB transcription factor proteins comprise several members of the structurally related Rel family and exist as inactive dimers in the cytoplasm under normal physiological conditions through association with inhibitor of κB (IκB) family members. NF-κB dimers are activated by IKK-mediated phosphorylation of IκB via either canonical or noncanonical signaling. The major and most well-studied canonical pathway can be activated by several ligands of the tumor necrosis factor (TNF) family, including TNF-α binding to the cell surface receptor TNFR1. This binding leads to formation of oligomeric receptor complexes and recruitment of adaptors to the cytoplasmic domain of the receptor. The adapters, collectively known as complex I (TNFR1-associated death domain protein [TRADD], TNF receptor-associated factor [TRAF2], receptor interacting protein [RIP1/RIPK1], cIAP1/2, and members of the LUBAC complex), facilitate recruitment and activation of the trimeric IKK complex, consisting of the catalytic subunits IKKα (IKK1/CHUK) and IKKß (IKK2/IKBKB) and the regulatory scaffold subunit IKKγ (NEMO/IKBKG). Additional upstream kinases are recruited: NF-κB inducing kinase (NIK/MAP3K14) and transforming growth factor-β activated kinase (TAK1/MAP3K7), which serve to initiate traditional kinase cascades within and outside the NF-κB pathway. The activated IKK complex preferentially phosphorylates one of eight IκB family members, IκBα, ultimately leading to its degradation. The canonical pathway activates NF-κB dimers mainly comprising RelA/p65:p50, c-Rel:p50, and RelB:p50, which upon activation translocate to the nucleus and bind to promoter and enhancer regions containing κB sites, whereupon NF-κB can regulate the expression of a wide variety of target genes, including providing context-specific feedback regulation. 26 Several of the key regulators involved in classical TNF-α/NF-κB signaling are members of the protein kinase family 28 and therefore inherently druggable. In many cases, their role and diversity of effect in any given disease setting is little understood.29,30 In recent years, a number of studies have employed functional screening approaches in mammalian cells using either small molecules, 31 cDNA, 32 RNAi,33–35 or miRNA 36 to interrogate TNFα-mediated NF-κB signaling in an attempt to derive further insight, but there is limited concordance between datasets, and although useful, each screen reveals only partial insight to signaling. These divergent data are a result of cell line-specific differences or either technical differences (i.e., analysis, library design, and phenotypic readout). In general, poor overlap across different RNAi studies is observed and has been attributed at least in part to a lack of efficiency and specificity. 37 A new differentiated approach is required in order to overcome the challenges associated with previous studies. Systematic assessment of TNF-α-mediated NF-κB signaling via CRISPR/Cas9 could confer two benefits: (1) gene targeting with increased efficiency and specificity and (2) the ability to knock out rather than knock down a target. Such a capability has the potential to assign genes to pathways with increased confidence and to uncover genes with roles in signaling where full knockout is required (e.g., residual expression of 10% is often sufficient for kinase activity). Ultimately, the development of new high-throughput functional genomic approaches with improved specificity and efficiency will allow full exploration of signaling to uncover novel points for therapeutic intervention.
We developed and optimized a vector-based arrayed CRISPR screening platform capable of the high-throughput profiling of signaling networks. Vector-based arrayed libraries offer particular utility for screening at scale in hard-to-transfect cell types. Furthermore, the libraries allow for the selection of edited cells, making their use compatible with extended assay timelines. The platform was successfully used at the kinome scale to identify the dependence of TNF-α-mediated NF-κB signaling in ME180 cervical cancer cells on a number of canonical regulators of the pathway. These data demonstrate for the first time that vector-based arrayed screening reagents can be supplied at scale and with sufficient consistency to obtain meaningful CRISPR/Cas9 screening results at at least the kinome scale. gRNAs were ranked using a stochastic rank aggregation method, and as part of this, an objective data-driven approach was used to select hits. We further investigated the relative contribution of individual gRNAs to hits identified through a 152-gene subset screen. High-throughput, next-generation amplicon sequencing was used to assess editing efficiency across this 152-gene subset. Over 83% genes had at least one gRNA with specific cleavage efficiency of >70%, and 75% genes had at least one gRNA with >90% editing efficiency. Gene expression levels for the 152 genes were also determined by RNA sequencing. Screening in singlicate format was most successful at identifying the majority of kinase genes with known regulatory roles in TNF-α-mediated NF-κB signaling and at a higher success rate than observed with previous RNAi-based studies. The dataset was found to be largely concordant with a previous pooled CRISPR screen for TNF-α-mediated p65 translocation 38 and served to corroborate some novel findings from prior RNAi studies. Our study not only adds to the available data on TNF-α-mediated NF-κB signaling but also highlights the use of known reference genes and gene expression analysis to experimentally assess screening quality. Important aspects of a number of workflow elements, including library preparation, screening controls, and data analysis, are exemplified. The application of arrayed lentiviral CRISPR/Cas9 gRNA libraries to cell biology developed here offers a new approach to the interrogation of cellular signaling networks that is extendable to the whole-genome scale and using a wide range of phenotypic readout and cell types.
Materials and Methods
Tissue Culture and Cell Line Generation
The CellSensor NF-κB-bla ME180 cell line, which stably expresses a β-lactamase (bla) reporter gene under the control of the NF-κB response element, was obtained from Thermo Fisher Scientific (Carlsbad, CA, cat. K1667). NF-κB-bla ME180 cells were cultured in “growth medium,” comprising Dulbecco’s modified Eagle’s medium (DMEM; cat. 31966021) supplemented with 10% dialyzed fetal bovine serum (dFBS; cat. 26400044), 0.1 mM nonessential amino acids (NEAA; cat. 11140050), 25 mM N-2-hydroxyethylpiperazine-N′-2-ethane sulfonic acid (HEPES; cat. 15630080), 100 U/mL penicillin and 100 µg/mL streptomycin (cat. 15140122), and 5 µg/mL blasticidin (cat. R21001). All cell culture reagents were obtained from Thermo Fisher Scientific.
A stable pool of bla ME180 cells expressing Cas9 was generated using Cas9 lentiviral particles produced using the pLenti-Cas9-TO-hygro construct (Thermo Fisher Scientific) and the Virapower Lentiviral Expression system (Thermo Fisher Scientific). Briefly, NF-κB-bla ME180 cells were treated in dose–response with the antibiotic selection agent, hygromycin, and cell viability was measured using PrestoBlue (Thermo Fisher Scientific, cat. A13261). The lowest concentration of hygromycin required to kill nontransduced cells was selected for further use (600 µg/mL). NF-κB-bla ME180 cells were plated at 60%−70% confluency in growth medium, and after overnight incubation, cells were transduced with Cas9 lentivirus, at a multiplicity of infection (MOI) of 1, in the presence of 8 µg/mL polybrene. After 48 h, the medium was changed to growth medium supplemented with 600 µg/mL hygromycin. The stable pool of NF-κB-bla ME180 cells expressing Cas9 was maintained under selection and expanded for 2 weeks. Cells were then sorted into single-cell clones using a BD FACS Aria flow cytometer (BD Biosciences, San Jose, CA). The clones were expanded for 2–3 weeks and approximately 10 clones were further validated for Cas9 expression, genetic cleavage detection analysis, and β-lactamase activity. Clone N13 was selected for further use (NF-κB-bla ME180-Cas9 cells). Short tandem repeat (STR) profiling of NF-κB-bla ME180-Cas9 cells resulted in an identity matching score of >80%, indicating that the cells were consistent with the ME-180 cell line of origin.
Western Blot Confirmation of Cas9 Expression
Cells were washed once with ice-cold phosphate-buffered saline (PBS) and lysed by addition of ice-cold RIPA buffer (Thermo Fisher Scientific, cat. 89900) and 1:100 of protease and phosphatase inhibitor cocktails (MilliporeSigma, Burlington, MA). The lysate was cleared by centrifugation and an equal amount of total protein was loaded onto each lane for sodium-dodecyl sulfate–polyacrylamide gel electrophoresis (SDS-PAGE; 10% Bis-Tris NuPAGE gels [Thermo Fisher Scientific, cat. NP0301]). The proteins were transferred onto nitrocellulose membrane using the iBlot 2 dry blotting system (Thermo Fisher Scientific) and the membranes blocked and incubated with a primary antibody against Cas9 (Diagenode, cat. C15200203) or β-actin (Santa Cruz Biotechnologies, cat. sc-47778), followed by incubation with alkaline phosphatase-conjugated secondary antibody. The blot was developed using the chromogenic BCIP/NBT substrate (WesternBreeze chromogenic kit, Thermo Fisher Scientific).
Genomic Cleavage Detection Assay
Cleavage efficiency was measured using a GeneArt Genomic Cleavage Detection kit according to the manufacturer’s guidelines (Thermo Fisher Scientific, cat. A24372). Cells were analyzed at 72 h posttransfection. The cleavage efficiencies were calculated based on the relative agarose gel band intensity, which was quantified using AlphaView version 3.4.0.0 (Protein Simple, San Jose, CA).
ToxBLAzer DualScreen NF-κB β-Lactamase Reporter Gene Assay
Regulation of the NF-κB signal transduction pathway was investigated using the ToxBLAzer DualScreen, based on GeneBLAzer β-lactamase reporter technology (Thermo Fisher Scientific, cat. K1139). The fluorescence resonance energy transfer (FRET) assay generates a ratiometric reporter response from stimulated and unstimulated cells and a multiplexed cytotoxicity readout in the same assay. 39 In our studies, NF-κB-bla ME180 and NF-κB-bla ME180-Cas9 cells were transduced with a CRISPR/Cas9 arrayed lentiviral kinome library. Cells were treated with 2.2 ng/mL TNF-α (a concentration equivalent to EC90, Sigma-Aldrich, cat. SRP3177) and, after between 5 and 6 h incubation at 37 °C, loaded with the ToxBLAzer reagent (8 µL, 6× substrate loading solution). The reagent was added to each microplate of the screening batch at 5 min intervals between 5 and 6 h after treatment with TNF-α. Each plate was then incubated for 2 h at 37 °C and read from underneath in a fluorescent plate reader (EnVision 2102 Multilabel Reader, PerkinElmer, Waltham, MA). Each plate takes approximately 5 min to load, read, and unload, but by staggering the time of addition of ToxBLAzer reagent it was possible to ensure that each plate was incubated with the substrate for the same length of time. We found this to be an important step in the generation of robust data from this live cell assay. The ToxBLAzer reagent contains both a LiveBLAzer-FRET Blue/Green (B/G) substrate and a cytotoxic indicator, both of which are readily internalized into cells where they are retained through the activity of endogenous esterases. In the absence of β-lactamase activity, excitation of the coumarin in the intact LiveBLAzer-FRET B/G substrate results in FRET to the fluorescein and the emission of a green fluorescence signal at 520 nm. Following TNF-α stimulation, NF-κB activation, and subsequent β-lactamase expression, enzymatic cleavage of the substrate disrupts FRET, leading to the emission of a blue fluorescent signal at 450 nm ( Fig. 1A ). The cytotoxic indicator leads to the emission of a red fluorescent signal (650 nm) from viable cells.
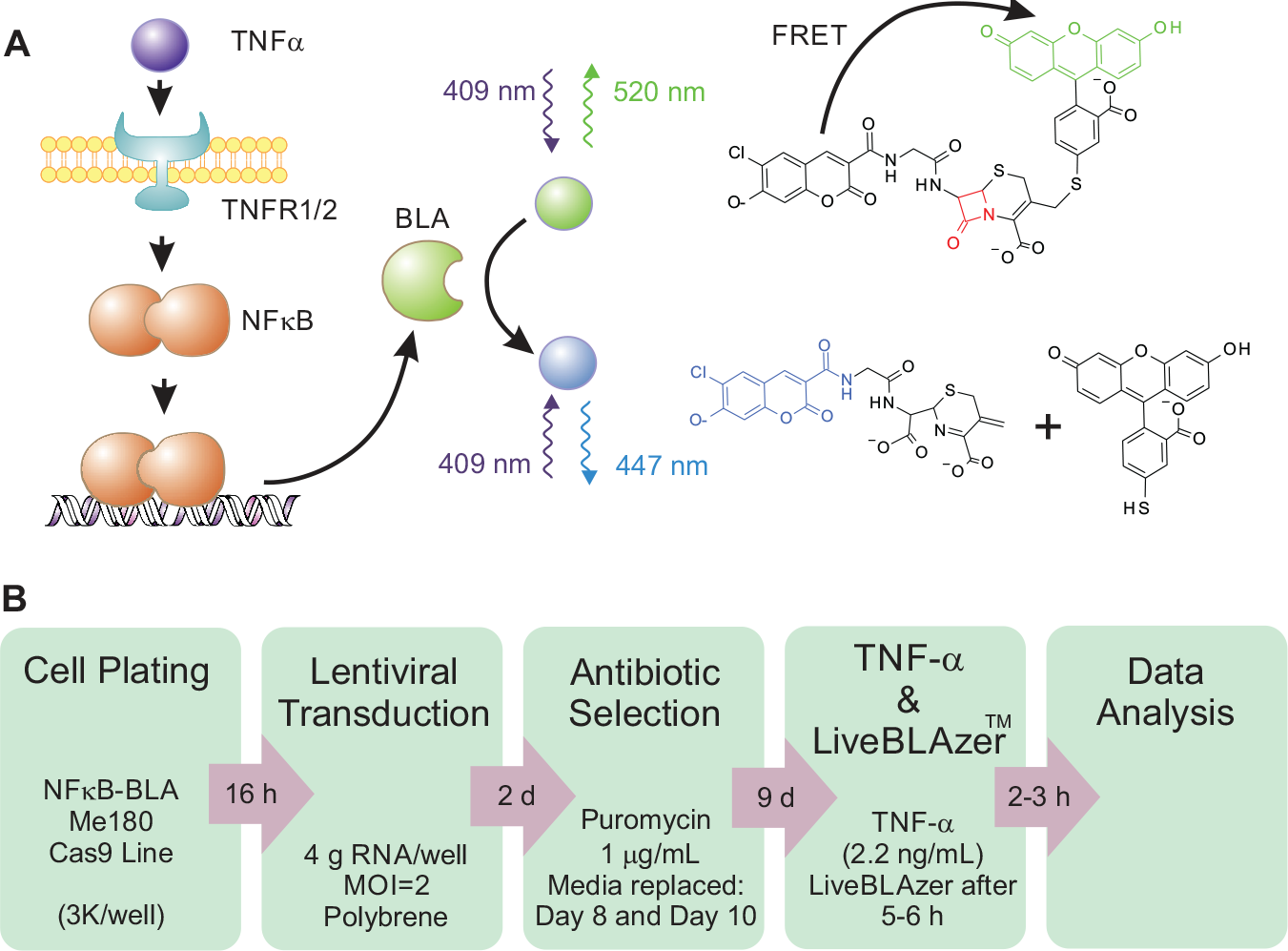
High-throughput assay to identify modulators of TNF-α-mediated NF-κB signaling. (
CRISPR/Cas9 gRNA Arrayed Library and Screening
Arrayed CRISPR screening was completed using a beta-test version of the Invitrogen LentiArray CRISPR Human Kinase Library (Thermo Fisher Scientific) targeting 743 genes with three or four gRNAs per gene. Briefly, the guides were designed to achieve maximal editing efficiency and specificity using a proprietary design tool (Thermo Fisher Scientific) that optimized guides of 18 base pairs (bp) for GC content, proximity to PAM sites, and factors to minimize off-target activity. The guides were selected to knock out all known isoforms of the target gene. Lentiviruses containing the gRNAs were generated using the pLENTI6.4-gRNA-Puro expression vector and an automated high-throughput process for cloning and virus production. The functional titers of the viruses were within the range of 2 × 107 to 2 × 108 TU/mL. The Invitrogen LentiArray CRISPR gRNAs used as controls were to the nontargeting control (NTC), LACTB2 (which targets the β-lactamase enzyme), TNFRSF1A, TRADD, CHUK, BRAF, EGFR, FOS, HPRT, MAPK1, and MAPK8. The Invitrogen LentiArray CRISPR gRNAs used for detailed characterization of individual gRNA effects were supplied as one gRNA per gene target per tube. These LentiArray gRNA particles were supplied as high-titer lentivirus preparations with a titer of >1 × 106 PFU/mL, with the exception of 18 of the individual gRNAs supplied for detailed characterization, which were lower than the viral titer limit. These data were excluded from data analysis. NF-κB-bla ME180-Cas9 cells were seeded in growth media into 384-well black tissue culture-treated microplates (Greiner, UK, cat. 781090) at 3000 cells/well and incubated for 16 h. All incubation steps were at 37 °C in a 5% CO2/95% air humidified atmosphere. After overnight incubation (day 2), growth media was replaced with a transduction media of DMEM, 3% dFBS, 1 mM sodium pyruvate, 0.1 mM NEAA, and 8 µg/mL polybrene (Sigma-Aldrich, UK, cat. H9268), and cells were infected with lentiviral gRNAs at an MOI of 2, in biological triplicate, and returned to incubation. Two columns (columns 1 and 24) of each screening microplate were left without cells and untreated for later background fluorescence subtraction. In addition, 18 wells of each screening microplate were transduced with NTC lentiviral particles, and another 6 wells were infected with β-lactamase control (LACTB2) gRNA lentiviral particles. On day 4, transduction media was replaced with a selection media of DMEM, 10% dFBS, 1 mM sodium pyruvate, 0.1 mM NEAA, 25 mM HEPES buffer (pH 7.3), and 1 µg/mL puromycin (Thermo Fisher Scientific, UK, cat. A1113802). Selection media was further replaced on day 8 and again on day 10. On day 11, six wells of each microplate that were previously infected with NTC lentiviral particles were treated with 10 µM of the selective IκB kinase inhibitor,40,41 2-amino-6-[2-(cyclopropylmethoxy)-6-hydroxyphenyl]-4-(4-piperidinyl)-3-pyridinecarbonitrile (ACHP), and incubated for 1 h at 37 °C. Cells were then stimulated with 2.2 ng/mL TNF-α and the ToxBLAzer DualScreen NF-κB β-lactamase reporter gene assay was completed.
Next-Generation Amplicon Sequence-Based Quantification of Genome Editing Efficiency
ToxBLAzer DualScreen CRISPR/Cas9 gRNA-treated samples were washed with PBS and lysed with lysis buffer (10 μL/well, Roche, cat. 07248431001) for 15 min at room temperature followed by addition of deionized water (30 μL/well). A two-step PCR protocol was used to generate ready-to-sequence amplicons from the samples. First-step PCR primers were designed using an automated platform based on Primer3 to amplify a 250–300 bp region surrounding the CRISPR target site. Primers were supplied as SePOP desalting purified at 100 nmol scale in 384-well microtiter plates (Eurogentec, Belgium). First-step PCR amplifications were performed with Phusion Flash High-Fidelity PCR master mix (4 μL, Thermo Fisher Scientific) combined with 1 μL of sample and 50 nL of 100 nmol PCR primers on the C-1000 Touch (Bio-Rad, CA) with the following conditions: 98 °C for 5 min; 5 cycles at 98 °C for 5 s, 72 °C to 1.5 °C/cycle for 10 s, 72 °C for 30 s; 20 cycles at 98 °C for 5 s, 60 °C for 10 s, 72 °C for 30 s; 72 °C for 2 min; 65 °C for 15 min. At the end of the reaction, unincorporated primers were removed by addition of exonuclease I (NEB M0293) and incubation at 37 °C for 30 min and inactivation at 80 °C for 15 min. The second-step PCR was prepared and run in the same way but using Nextera XT indexing primers containing the Illumina (San Diego, CA) adaptors required for sequencing (25 nL at 100 nmol/sample, Eurogentec) and without the final 65 °C lentiviral inactivation step. These samples were pooled, lyophilized, and resuspended in IDTE buffer (100 μL) and purified using HighPrep PCR (MagBio, US) prior to excision and extraction (QIAEX II gel extraction kit, QIAGEN) of desired bands between 100 and 500 bp in length on a 2% agarose gel. DNA was quantified (Qubit, Thermo Fisher Scientific) and loaded onto a NextSeq cartridge mid-output kit 300 cycles (FC404-2003, Illumina) for next-generation sequencing of all amplicons. Sequencing was performed on a NextSeq 500 (Illumina). The amplicon analysis of each sample first merged the paired reads using FLASH 42 and mapped the resulting single reads to the human genome (version hg19/GRCh37) using the Burrows–Wheeler aligner. 43 The Sequence Alignment Map files were processed to determine the insertion/deletion of bases at the relevant gRNA target site and output was recorded as a percentage per sample using a custom Perl script. The insertions/deletions contributed to the final percentage per sample if the inserted base or bases immediately preceding the insertion/deletion had a minimum quality of 25 and if the insertion/deletion was present in at least 1% (minimum of 10) of reads.
RNA Sequencing for Expression Analysis
NF-κB-bla ME180-Cas9 cells were seeded in growth media into a six-well tissue culture-treated microplate (Corning, UK) at 1 × 106 cells/well and incubated for 24 h at 37 °C in a 5% CO2/95% air humidified atmosphere. Following overnight incubation, cells were lysed and RNA was isolated using the RNeasy Plus Mini kit (QIAGEN, UK, cat. 74134), according to the manufacturer’s instructions. The quality and concentration of the RNA was assessed by a fragment analyzer using the standard sensitivity RNA analyzer kit (Agilent Technologies, Santa Clara, CA). Samples with an RNA integrity number >10 were used for library preparation. One microgram of total RNA was used for each library. An Illumina TruSeq Stranded mRNA Sample Prep kit was used to construct poly(A) selected paired-end sequencing libraries according to the TruSeq Stranded mRNA Sample Preparation Guide (Illumina). All libraries were quantified with the Fragment Analyzer using the standard sensitivity next-generation sequencing kit (Agilent Technologies). Libraries were pooled and quantified using the Qubit instrument, DNA HS kit (Thermo Fisher Scientific), and the library pool was further diluted to 1.8 pM. Sequencing was performed using a high-output kit v2 (75 cycles) on an Illumina NextSeq500. RNA-Seq data were processed with “bcbio-nextgen” (https://github.com/bcbio/bcbio-nextgen) using salmon 44 to quantify the expression values.
Data Analysis
Fluorescence intensity data captured at 450 and 520 nm emission using an EnVision plate reader (EnVision 2102 Multilabel Reader, PerkinElmer) were expressed as a response ratio (450/520 nm) and transformed on a log2 scale. Fluorescence intensity data captured at 650 nm emission were normalized by division to the mode of all samples across the screen. Samples exhibiting significant cell loss were identified as those beyond the lower hinge of the box plot of the normalized 650 nm data (<0.3). These samples were excluded from further analysis. Log2(response ratio) data were normalized by dividing the ratios by the median response ratio of the screen. These data were expressed as robust Z score (RZ′), strictly standardized mean difference (SSMD), and a T statistic with the associated Holm-adjusted p value. Calculations were as follows: Z′ score = D/(median of absolute deviation [MAD] of D); SSMD = mean of D/variance D + variance of negative control; T test = mean of D/(variance of D/√n + variance of negative control/√n), where D denotes the difference between the measured activity of the investigated gRNA or small molecule and the median value of the negative controls for that respective plate and n denotes number of replicates. Log2(response ratio) data were also expressed as percent inhibition normalized to within plate controls: % inhibition = [(median of negative controls – X)/(median of negative controls – median positive controls {LACTB2})]*100, where X denotes gRNA per small-molecule test. Hits were independently ranked by median percent inhibition, median Z′ score, SSMD, and T statistic. A stochastic rank aggregation method, the Borda score (median method) within TopKLists, was then applied to combine these individual rankings. 45 The hit cutoff was defined by observation of a cessation of incremental growth of the Borda score with compound rank.
Results
Optimization of a TNF-α-Mediated NF-κB Signaling Pathway Reporter Gene Assay for High-Throughput CRISPR/Cas9-Based Arrayed Screening
In order to conduct an assessment of the feasibility and validity of large-scale arrayed gRNA CRISPR-based screens for functional genomics, we utilized an NF-κB β-lactamase reporter gene assay31,46 for the identification of mediators of the TNF-α-mediated NF-κB signaling pathway (
Fig. 1A
). Several aspects of the system, which was originally designed for small-molecule screening, were modified to enable CRISPR/Cas9 screening (
Fig. 1B
). We initially attempted to utilize a lentivirus to transduce the cells with Cas9 immediately prior to cell plating and subsequent transduction with lentiviruses containing the gRNAs. This would have avoided the need to generate a cell line expressing Cas9, an attractive possibility were the screening system ever to be used for primary cells. However, we found that editing efficiency with this approach was suboptimal (data not shown). Therefore, we generated a CellSensor NF-κB-bla ME-180 cell line stably expressing Cas9 nuclease under control of an EFS promoter. In order to do this, the CellSensor NF-κB-bla ME-180 cell line was transduced with lentiviral particles at a low MOI of 1, followed by hygromycin selection to obtain a pool of stable Cas9-expressing cells (
Fig. 2A
). The editing efficiencies of clones isolated from this pool were assessed by genomic cleavage detection after delivery of gRNAs designed to the genes HPRT, TNFRSF1, and TRADD (
Fig. 2B
). The level of Cas9 expression across the clones was compared by Western blot (
Fig. 2C
). Clones N13 and N27 were selected for further characterization based on the high editing efficiencies achieved and cellular morphology compared with the parental cell line (data not shown), and to enable profiling of the effect of high and medium expression levels of Cas9, respectively. TNF-α treatment of the clones resulted in comparable EC50 values to the parental cell line (
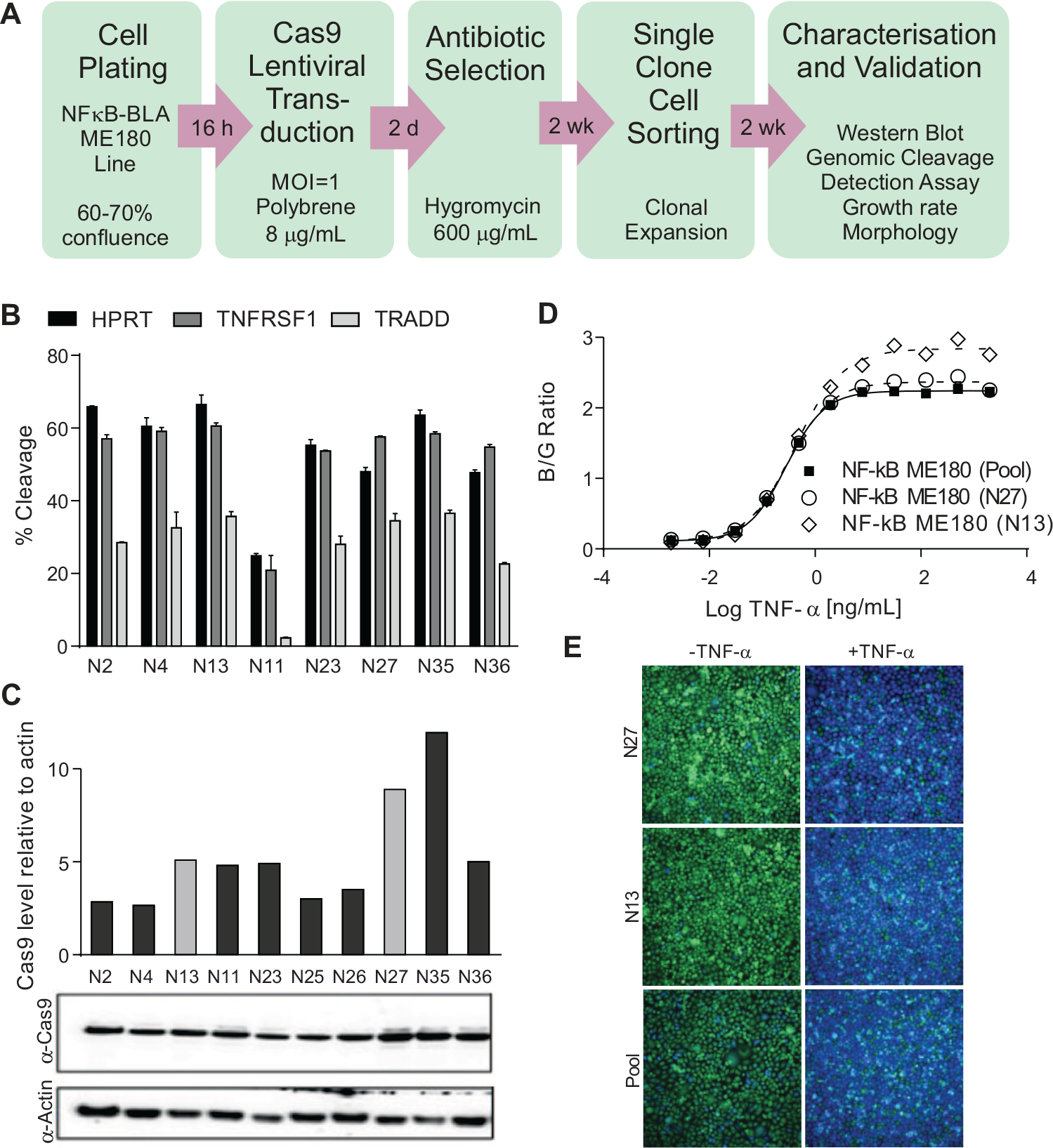
Generation and validation of NF-κB-bla ME180-Cas9 cell line. (
The Invitrogen LentiArray gRNAs are supplied as four predesigned gRNAs per gene amalgamated in a single well per gene. Lentiviruses containing the gRNAs were generated using the pLENTI6.4-gRNA-Puro expression vector with expression of a puromycin resistance gene driven by an EF1 promoter. In the assay, cells were transduced with lentiviruses containing gRNA and placed under puromycin selection for 10 days. Only those cells transduced with the lentivirus survive selection. Nontransduced cells undergo complete cell death. Furthermore, this lentiviral-based method of gRNA delivery results in constitutive expression throughout the time-course of the assay. This ability to select only those cells transduced with gRNA coupled with their constitutive expression is a key differentiator versus lipid-mediated delivery of chemically synthesized gRNAs and likely to maximize the phenotypic effect by maximizing the likelihood of complete loss of protein product following gene knockout in every cell in any given well. In our study, a lentivirally delivered gRNA to the LACTB2 gene alongside a nontargeting (NTC) gRNA was used to optimize the β-lactamase reporter assay workflow to CRISPR/Cas9. The experimental factors that were optimized were cell seeding density, transduction conditions, MOI, antibiotic selection, and assay duration. Thus, the assay was tailored to the broadly applicable CRISPR/Cas9 lentiviral screening system. Functional genomic assay validation was performed using a number of Invitrogen LentiArray gRNAs to genes with expected effects on the pathway alongside a number of negative control genes. The Invitrogen LentiArray gRNAs are supplied as four predesigned gRNAs per gene amalgamated in a single well per gene. These gRNAs were profiled in triplicate across three plates with 10 replicates within each plate, all within a single screening occasion. Of these, gRNAs to genes TNFRSF1A, TRADD, and CHUK abolished signaling to a level comparable to genetic loss of the LACTB2 gene (>90% loss of activity) ( Fig. 3A ). Representative images from wells treated with and without TNF-α in the presence of NTC, LACTB2, or CHUK gRNAs are depicted in Figure 3B . gRNAs to genes BRAF, EGFR, FOS, HPRT, MAPK1, and MAPK8 were included as negative controls, and these, as anticipated, did not result in any abolition of signaling ( Fig. 3A ). These data agree with known biology and confirm the ability of this assay to detect genetic regulators of TNFα-mediated NF-κB signaling. Assessment of the assay window by RZ′ factor (RZ′ ≥ 0.7) indicated suitability of the developed assay for large-scale screening.
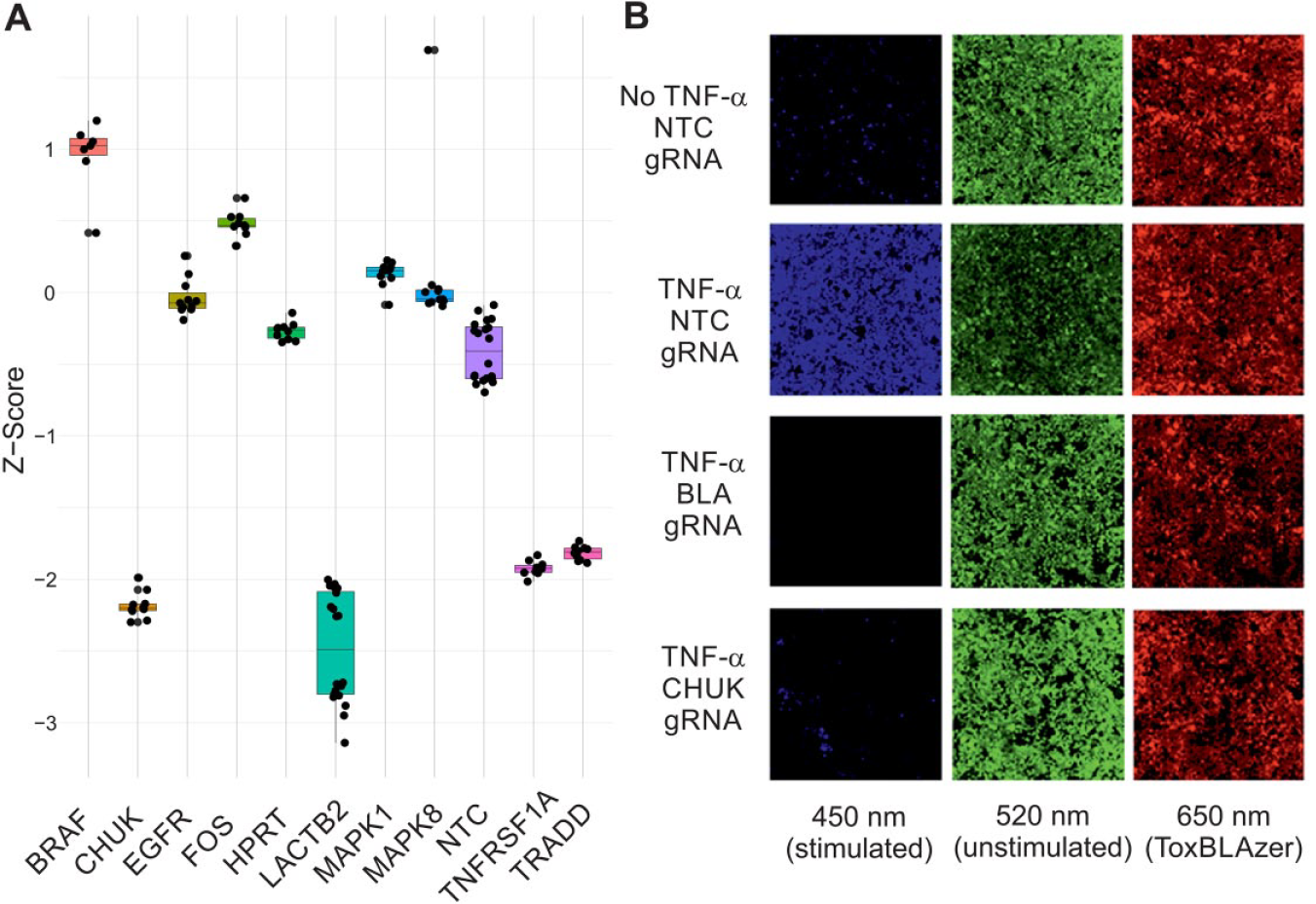
Validation of a TNF-α-mediated NF-κB signaling pathway reporter gene assay for high-throughput CRISPR/Cas9-based arrayed screening. (
Arrayed CRISPR/Cas9 Screen to Identify Mediators of TNF-α-Mediated NF-κB Activity
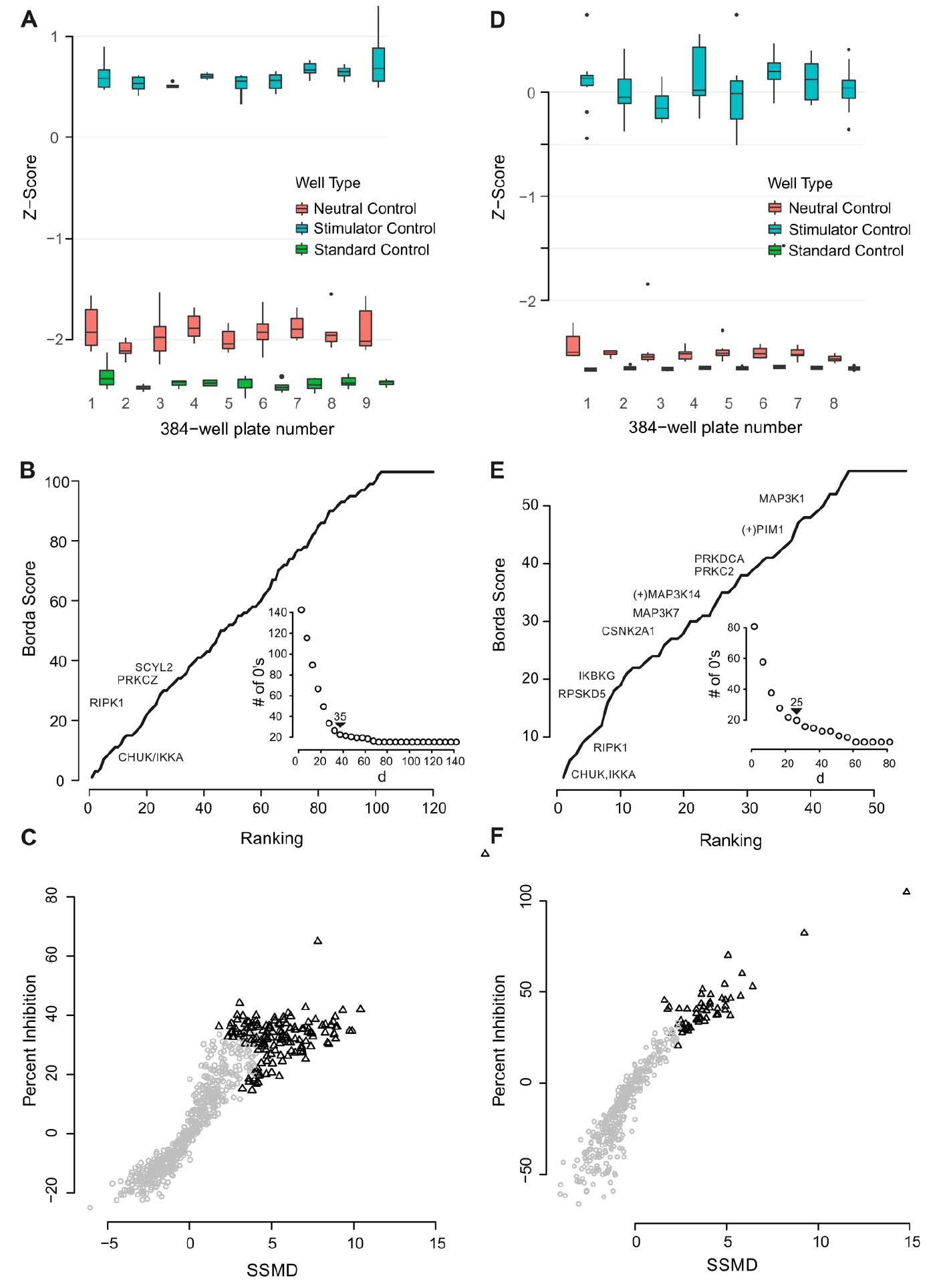
A beta-version of the Invitrogen LentiArray human kinase library was used to screen for genes with a role in TNFα-mediated NF-κB transcriptional activity. Genes were screened in triplicate across different plates within a single screening occasion. The assay performance was robust (average RZ′ factor > 0.56) with the assay window and activity data for the standard control (TNF-α, LACT2B gRNA), neutral control (no TNF-α, NTC gRNA), and stimulator control (TNF-α, NTC gRNA) across all 384-well microwell plates used in the screen shown in
Figure 4A
. Data associated with 62 genes were removed from further analysis as a result of significant effects on cell number (toxicity index score < 0.3). In addition, there were individual replicate data points associated with a further 25 genes that also had toxicity index scores of <0.3. These replicates were excluded from further analysis as 0.3 was defined as the cutoff after which the cell number could artificially affect the response ratio. While there was cell loss among the other replicates for these 25 genes, it was at a level whereby the response ratio remained unaffected. gRNAs were ranked according to their percent inhibition, RZ′, SSMD, or T-statistic values. Ranking of gRNA effect by these parameters was found to be similar although not identical (
Arrayed CRISPR/Cas9 screen for mediators of TNF-α-mediated NF-κB signaling. (
Follow-Up of Arrayed CRISPR/Cas9 Screening Results
In an effort to better understand the performance (i.e., false discovery rate) of this CRISPR-based screening approach, we conducted further experiments for a subset of 152 genes. Eighty-three of the 102 genes identified as hits in the primary screen were selected for detailed follow-up. This included all those genes with some a priori association with TNF-α-mediated NF-κB signaling. Sixty-five nonhits were selected for further characterization on the basis of research in the literature that linked them to NF-κB signaling (e.g., identified in previous functional genomic screens related to TNF-α-mediated NF-κB signaling or linked to NF-κB). Three genes anticipated to play a role in TNF-α-mediated NF-κB signaling but toxic in the primary screen (PIM1, PDPK1, and MAP3K14) and an additional gene, IKBKG, not represented in the original studies but with a well-characterized role in canonical TNF-α-mediated NF-κB signaling, were also included. Genes were screened in quadruplicate across different plates within a single screening occasion using two to four individual gRNAs per gene (summarized in Suppl. Data). Data were analyzed as for the whole-kinome screens. Assay performance was robust (RZ′ > 0.44) with assay window and activity data for the standard control (TNF-α, LACT2B gRNA), neutral control (no TNF-α, NTC gRNA), and stimulator control (TNF-α, NTC gRNA) across all 384-well microwell plates used in the screen shown in
Figure 4D
. Data from one of four individual gRNAs were excluded from the analysis for each of the genes SMG1 and FASTKD5 as a result of toxicity. gRNAs to 45 genes were identified as hits using the consolidated and optimized aggregate ranking generated from the RZ′, SSMD, and T statistic using a median non-optimization-based method from Borda’s collection as described for the kinome screen (
Fig. 4E
). Hits resulted in activities of >27.7% inhibition and >2.5 SSMD (
Fig. 4F
) for at least one gRNA for any given gene target. In addition to measurement of phenotypic effect, two of the four replicate plates from the follow-up screen were subjected to a next-generation sequencing-based method to assess cutting efficiency. These data confirmed >70% cutting efficiency at the expected INDEL site for at least one gRNA per gene identified as a hit in 27 of 45 genes identified as hits in the focused screen, and in fact, in general, across the entire sample of 152 genes, >83% genes had at least one gRNA with >70% INDEL efficiency at the target site. As a result of the method used to assess cutting efficiency, optimized for throughput rather than ability to measure efficacy accurately across any gene, this is likely to be an underestimate. gRNA-associated gene targets related to canonical TNFR1-induced NF-κB signaling were found to be overrepresented in the dataset (p = 0.000012) and included RIPK1, CHUK, and PRKCZ. The activities (percent inhibition) of gRNAs to 18 genes (selected for their known roles in TNF-α-mediated NF-κB signaling28,48,49 or their known interactions with RelA or NFKBIA), along with their measured cutting efficiency and basal gene expression in the NF-κB-bla ME180-Cas9 cell line, are listed in rank order in
Table 1
. gRNAs to 13 of 18 of these genes were classified as active in contrast to the results from the primary screen (3/18 actives; IKBKG not screened in primary screen) (
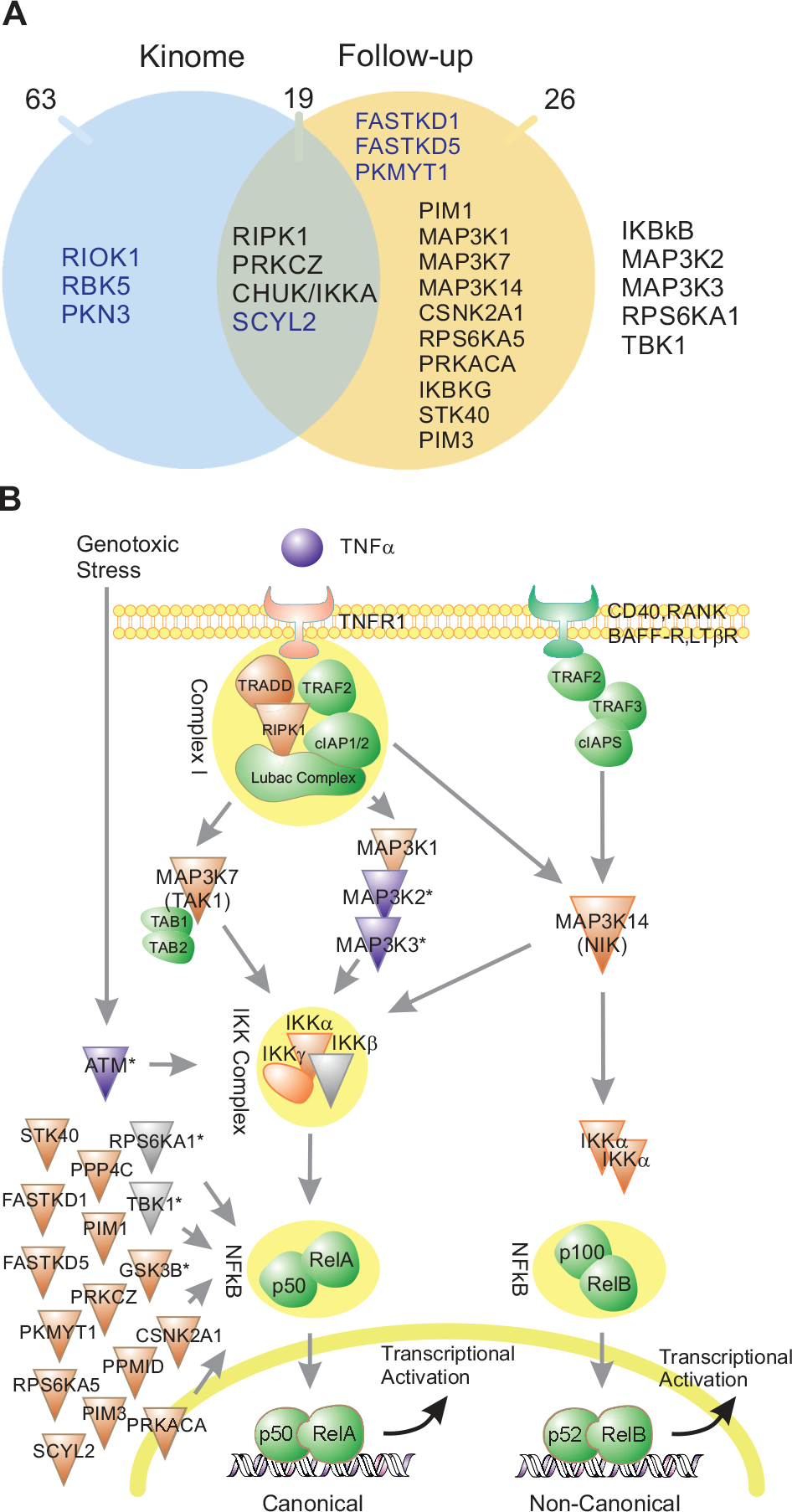
Global pathway analysis of gRNA hits. (
Activity of gRNAs Targeting Genes with Previously Characterized Roles in TNF-α-Mediated NF-κB Signaling.
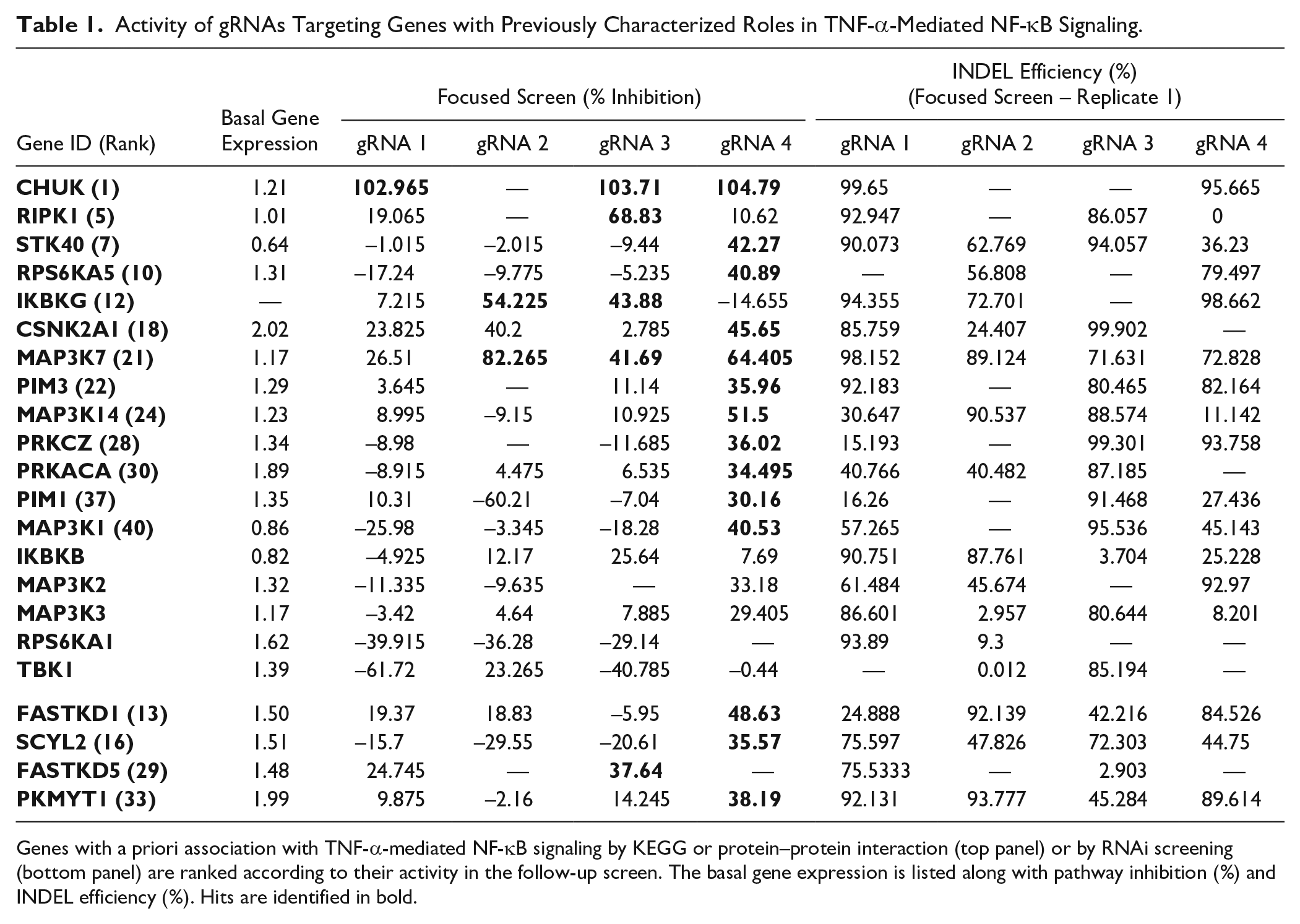
Genes with a priori association with TNF-α-mediated NF-κB signaling by KEGG or protein–protein interaction (top panel) or by RNAi screening (bottom panel) are ranked according to their activity in the follow-up screen. The basal gene expression is listed along with pathway inhibition (%) and INDEL efficiency (%). Hits are identified in bold.
In order to better understand the potential for false positives in the screen, an examination of all follow-up hits, not just those with previously characterized roles in signaling, was undertaken. This revealed 9 of 45 that were classified as hits in both screens but did not appear to be expressed at the mRNA level (<0.2 relative expression). For the genes not expressed at the RNA level, only one gRNA was identified as a hit for each, with the exception of two for MAST1. The probability of a coding region off-target for these gRNAs was calculated using an algorithm based on predicting the number of off-targets with up to three mismatches for either NGG or NAG PAM sites.50,51 There were a range of probabilities, for example, 0.089, 0.136, 0.258, 0.413, and 0.498 for the gRNAs targeting RIPK3, MAST1, MYO3A, STK33, and MYO3A, respectively. The gRNAs were unique in the genome (0 bp mismatch) but had the potential for off-targets with either a 2 bp (between 4 and 25) or a 3 bp (between 82 and 397) mismatch. The off-target cutting potential of these gRNAs could explain the false positives observed. Notably, RIPK3 expression loss is a relatively common phenomenon across many cancer cell lines and is thought to correlate with tumor cell escape from necroptosis, 50 so although it might be expected to play a role in canonical signaling, it is reasonable that it is not expressed within this cell type.
Meta-Analysis of TNF-α-Mediated NF-κB Signaling Functional Genomic Data
We went on to complete a systematic overlap analysis of these data with previously published RNAi and CRISPR-based functional genomic screens relevant to TNF-α-mediated NF-κB signaling (Suppl. Data). The RNAi studies were reflective of genome-wide, 34 druggable, 35 kinome, 33 and focused scale 37 screens using chemically synthesized siRNA libraries across different cell types (HEK293T and A549) and using different reporter systems (NF-κB luciferase, NF-κB GFP, and IL8 luciferase). A total of 44 kinases were identified as having putative roles on TNFα/NF-κB signaling from across these RNAi-based screens. Of these, only the kinase, IKKB, was identified as a hit in more than a single study despite significant overlap of the genes targeted by the respective libraries. Also notable is that only 1–3 of 18 genes with known roles in the pathway were identified as hits in each of the three larger RNAi screens, in contrast with our arrayed CRISPR approach (13/18). Eleven of the 44 kinase hits from the RNAi studies were identified as hits using our singlicate arrayed CRISPR approach. These represented all but one of the hits identified by Gewurz et al.54 (CHUK, IKBKG, STK40, PIM3, FASTKD1, FASTKD5, and PKMYT1) and 4 of 34 hits from Choudhary et al. (MAP3K14, MAP3K7, IKBKG, and SCYL2). 33 Four of these genes (FASTKD1, FASTKD5, PKMYT1, and SCYL2) had not been associated with NF-κB prior to the completion of the RNAi screens. It is therefore notable that in our study, which utilized independent technology, the same genes were identified as hits ( Table 1 ). The recently published pooled CRISPR approach comprised the screening of a 963-gene pooled CRISPR library across three cell backgrounds (HeLa, A549, and HCT-116) using either a fluorescent reporter or immunocytochemical method of assessing p65 translocation as a measure of TNF-α-mediated NF-κB signaling. 55 Fifty-five genes (52 kinases and IKBKG, TNFRS1A, and TRADD) were overlapping between this pooled CRISPR study and our arrayed approach. Hit/nonhit classification correlated between the two CRISPR studies with the exception of four genes. IKBKB was not identified as active in our screen but was identified as weakly active in the pooled CRISPR screens conducted in HeLa or A549 cell lines. IKBKB was not active in the pooled CRISPR screen conducted in the HCT116 colorectal cancer cell line, raising the possibility that the lack of activity of IKBKB in our assay was perhaps not due to lack of sensitivity but specific cell line differences. MAP3K14 (NIK), PIM3, and PRKD2 were identified as active in our screen but inactive via the pooled CRISPR approach. MAP3K14 is thought to play a key role in the noncanonical activation of NF-κB via p52:RelB, in addition to activation by translocation of p65/RelA:p50 dimers. 52 A consequence of the use of a p65/RelA translocation phenotypic endpoint in the pooled CRISPR screen is an inability to detect molecules signaling either exclusively or partially through alternative NF-κB dimers such as p52:RelB, and in this case suggests a reason for the disparate hit calling compared with our study.
Discussion
Functional genomic screening enables unbiased identification of proteins important for biological processes. In particular, arrayed vector-based CRISPR/Cas9-based screening offers selective targeting of genes across a range of cellular assays of greater diversity than is possible by other CRISPR/Cas9 screening approaches. We demonstrated the utility of large-scale lentiviral-based arrayed CRISPR/Cas9 libraries to the systematic interrogation of cellular signaling. In particular, singlicate arrayed CRISPR screening was successful at identifying the majority of kinase genes with known regulatory roles in TNF-α-mediated NF-κB and corroborated novel findings from earlier RNAi screens. For the gRNA library and assay system under study here, we found arrayed screening using singlicate gRNAs to be more robust than when mixtures of gRNAs targeted to a single gene were used. We highlight the library composition, screening parameters, and quality control procedures important to the generation of robust arrayed vector-based screening data.
In previous arrayed CRISPR studies, either viral vector-based libraries of up to 200 genes,13,17 –19 chemically synthesized gRNA libraries of up to 640 genes,14,20 –22 or plasmid-based libraries of up to 1514 genes15,16 were used for loss-of-function screens. Our study was the first to use a lentiviral library at the whole-kinome 743-gene library scale. The use of high-throughput lentiviral generation and quality control methods alongside use of automated liquid dispensing and a Cas9-expressing reporter cell line resulted in screening data of high quality. To identify actives and rank gRNAs, we used multiple statistics and rank-order analysis rather than just selection of a single statistic. The multiple hit selection methods produced largely similar results, but the main advantage was that the consolidated and amalgamated rank-order analysis identified those genes most consistently identified as hits. A secondary advantage was that an objective data-driven approach was enabled by which to identify hits. Through the selection of a well-studied pathway, TNF-α-mediated NF-κB signaling, 28 we were able to confirm the utility of the lentiviral library in identifying key kinase regulators of the pathway. To further evaluate the library and screening approach, we selected a subset of actives and inactives from the kinome-scale screen for target gene expression analysis, assessment of INDEL formation, and evaluation of the effect of gRNA sequence and targeting position. This follow-up screen, in which gRNAs were screened as singlicates, detected the majority of known kinase regulators of the pathway and thus appeared more sensitive to detection of a priori associated canonical genes than the kinome-wide screen in which gRNAs were evaluated at three or four gRNAs per target per well. Both MAP3K7 and MAP3K14 were flagged as toxic in the primary screen but identified as active in the focused screen when individual gRNAs were deployed. One possibility is that the presence of multiple active gRNAs targeted to distinct genomic loci in the same cell resulted in large deletions that mobilize DNA/cellular repair/damage mechanisms that result in apoptosis. A second possibility is that somehow multiplexed gRNAs resulted in interference as a result of either competition for Cas9 or some other mechanism that prevented the active gRNA exhibiting its phenotypic effect. Our use of a high-throughput next-generation amplicon sequencing technique to estimate INDEL formation efficiency allowed us to look at the overall cutting efficiency of the library through extrapolation of testing on 152 genes. More than 83% of the genes had at least one gRNA with a specific cleavage efficiency of >70%, and 75% genes had at least one gRNA with >90% editing efficiency. It is of note, though, that while efficient editing was a prerequisite for a phenotypic effect, this was not predictive of an effect. Therefore, while it was of use to confirm appropriate INDEL formation for those genes identified as hits, utilization of INDEL formation efficiency as a sole measure to guide future library design evolution appears to have limited utility. There could be a number of reasons for this lack of correlation; INDEL formation could have resulted in in-frame deletions but not loss of product, delayed degradation of gene product beyond the length of the assay, or other not yet understood parameters with effects on gRNA function (e.g., DNA structure). 53
The use of lentivirus-based arrayed CRISPR screening to annotate TNF-α-mediated NF-κB signaling proved to be superior at the detection of known regulators versus previous kinome and larger-scale siRNA-based approaches. A significant number of genes identified as hits in previous RNAi-based screens were corroborated by our CRISPR-based screen, in particular all those kinases identified by Gewurz et al. 54 There were fewer overlapping hits with other previous RNAi screens.33 –35 The reasons for this disparity could be a result of superior gene targeting by CRISPR, the extended assay timeline (11 days) in our study, cell line-specific differences or differences in reporter technology (luciferase33 –35 vs ratiometric FRET), and associated handling of perturbants that resulted in toxicity. It is of note that good concordance, including for known reference genes, was obtained by comparison of our data with those generated using a recently published pooled CRISPR approach in cell lines containing a fluorescent reporter for p65 translocation. 38 There are some examples of genes, identified by our study, that would have been expected to be identified as active but were not. These included TBK1, RPS6KA1, MAP3K2, MAP3K3, GSK3B, and ATM. gRNA knockout of ATM and TBK1 resulted in cellular toxicity. Therefore, in at least these cases it appears that modulation of kinase activity rather than knockout is advantageous for screening. Where toxicity as a result of gene knockout is not a factor, it is possible that gRNA design or delivery was not optimal or that pathway redundancy in the ME-180 cell line precluded detection. As a consequence of using study of the TNF-α-mediated NF-κB signaling pathway to develop and evaluate the new screening approach, a number of genes that have not been previously linked to TNF-α-mediated NF-κB signaling were identified. Of those identified with highest confidence is FGFR1, which was identified as a hit in both gRNA screens. There is evidence that in prostate cancer cells, FGF signaling via FGFR1 promotes NF-κB signaling via TAK1/MAP3K7. 55 It is therefore also possible that our data indicate a role for FGFR1 signaling in NF-κB activation in ME180 cells and thus potentially also in inflammation of the tumor microenvironment in cervical cancer. It is clear that as with any screen, the validation of hits to fully understand roles will be important. The availability of diverse functional genomic data (small molecule, RNAi, and CRISPR) is now a possibility to aid in the convergence of these downstream efforts.
Much is still being learned about the potential and limitations of CRISPR screening. We are confident that this developed arrayed CRISPR platform could be extended across a range of pathway reporter systems, functional assays, and cell types to bring greater molecular understanding of cell signaling across different cell backgrounds. For example, comparative profiling in different cancer backgrounds could identify differences in NF-κB activation, providing insight to pathway dysregulation associated with specific cancers. In our study, the use of a Cas9-expressing cell line was a key determinant in acceptable screening data quality. Future screens could be enhanced through the use of technology that inserts Cas9 to a safe harbor insertion site, such as the adeno-associated virus site 1 (AAVS1), a naturally occurring site of integration of AAV on chromosome 19, rather than random integration of Cas9, which could result in altered characteristics to the cell line under study. Furthermore, methods that enable direct in-assay delivery of Cas9 to cells at a high-throughput scale could enable screening in primary cell systems given the gRNA library is vector based. New generations of gRNA libraries will also be made available that build on the learnings and data of this and other studies to include highly validated functional gRNAs to the entire human genome, and potentially the ability to generate kinase-dead rather than kinase-knockout phenotypes. Ultimately, continued investment in library design will lead to improved fidelity of screening data. While the high-throughput next-generation sequencing-based method we adopted to measure INDEL formation was adequate for validation of cutting at the desired loci at the hit follow-up scale, cost would preclude a larger study aimed at gRNA library optimization. Our data also indicate this to be an insufficient measure in the prediction of gRNA efficacy in generating a functional knockout. The emergence of single-cell transcriptomics could prove to be a valuable tool in the ultimate optimization of gRNA libraries of the future. It is also the case that it will ultimately be desirable to consider the effects of gene upregulation and the phenotypic effect of gene combination knockout studies driving the development of further novel gRNA libraries.
We demonstrate one of the first applications of arrayed CRISPR screening at scale. Using a pathway screen for TNF-α-mediated NF-κB signaling, we demonstrate the requisite screening parameters and workflows in order to obtain meaningful data from the use of arrayed virally delivered CRISPR libraries. We highlight the use of a known list of genes as a reference set to explore the specificity and sensitivity of any emergent functional genomic screening approach. In so doing, we share a screening approach that was successful at identifying the majority of kinase genes with known regulatory roles in TNF-α-mediated NF-κB and corroborating novel findings from previous RNAi screens. Our data demonstrate a unique approach for investigating NF-κB regulation and a new resource for wider cellular biology research and target identification for drug discovery.
Research Data
R2_Supplemental_Data_for_Arrayed_CRISPRCas_Screen_by_OShea_et_al – Supplemental material for A Novel Screening Approach for the Dissection of Cellular Regulatory Networks of NF-κB Using Arrayed CRISPR gRNA Libraries
Supplemental material, R2_Supplemental_Data_for_Arrayed_CRISPRCas_Screen_by_OShea_et_al for A Novel Screening Approach for the Dissection of Cellular Regulatory Networks of NF-κB Using Arrayed CRISPR gRNA Libraries by Patrick O’Shea, Jan Wildenhain, Mathew Leveridge, Chetana Revankar, Jian-Ping Yang, Jenna Bradley, Mike Firth, James Pilling, David Piper, Jonathan Chesnut and Beverley Isherwood in SLAS Discovery
Supplemental Material
R2_Supplemental_Material_for_Arrayed_CRISPRCas_Screen_by_OShea_et_al – Supplemental material for A Novel Screening Approach for the Dissection of Cellular Regulatory Networks of NF-κB Using Arrayed CRISPR gRNA Libraries
Supplemental material, R2_Supplemental_Material_for_Arrayed_CRISPRCas_Screen_by_OShea_et_al for A Novel Screening Approach for the Dissection of Cellular Regulatory Networks of NF-κB Using Arrayed CRISPR gRNA Libraries by Patrick O’Shea, Jan Wildenhain, Mathew Leveridge, Chetana Revankar, Jian-Ping Yang, Jenna Bradley, Mike Firth, James Pilling, David Piper, Jonathan Chesnut and Beverley Isherwood in SLAS Discovery
Footnotes
Acknowledgements
The authors would like to thank Damla Etal and Babak Alaeimahabadi (AstraZeneca) for completion of the next-generation sequencing and RNASeq data analysis, respectively. This work was jointly funded by AstraZeneca and Thermo Fisher Scientific.
Declaration of Conflicting Interests
The authors declared the following potential conflicts of interest with respect to the research, authorship, and/or publication of this article: P.O., M.L., J.B., M.F., J.P., and B.I. are or were employed by AstraZeneca; J.W. is employed by Syngenta; and C.R., J.P.Y., D.P., and J.C. are or were employed by Thermo Fisher Scientific during the completion of these studies.
Funding
The authors received no financial support for the research, authorship, and/or publication of this article.
Supplemental material is available online with this article.
References
Supplementary Material
Please find the following supplemental material available below.
For Open Access articles published under a Creative Commons License, all supplemental material carries the same license as the article it is associated with.
For non-Open Access articles published, all supplemental material carries a non-exclusive license, and permission requests for re-use of supplemental material or any part of supplemental material shall be sent directly to the copyright owner as specified in the copyright notice associated with the article.