
Abstract
Human rhinovirus (RV) is the most common cause of acute upper respiratory tract infections and has recently been shown to play a significant role in exacerbations of asthma and chronic obstructive pulmonary disease (COPD). There is a significant unmet medical need for agents for the prevention and/or treatment of exacerbations triggered by human RV infection. Phenotypic drug discovery programs using different perturbation modalities, for example, siRNA, small-molecule compounds, and CRISPR, hold significant value for identifying novel drug targets. We have previously reported the identification of lanosterol synthase as a novel regulator of RV2 replication through a phenotypic screen of a library of siRNAs against druggable genes in normal human bronchial epithelial (NHBE) cells. Here, we describe a follow-up phenotypic screen of small-molecule compounds that are annotated to be pharmacological regulators of target genes that were identified to significantly affect RV2 replication in the siRNA primary screen of 10,500 druggable genes. Two hundred seventy small-molecule compounds selected for interacting with 122 target gene hits were screened in the primary RV2 assay in NHBE cells by quantifying viral replication via in situ hybridization followed by secondary quantitative PCR-based assays for RV2, RV14, and RV16. The described follow-up phenotypic screening allowed us to identify Fms-related tyrosine kinase 4 (FLT4) as a novel target regulating RV replication. We demonstrate that a combination of siRNA and small-molecule compound screening models is a useful phenotypic drug discovery approach for the identification of novel drug targets.
Introduction
Human rhinovirus (RV) is responsible for more than 50% of cold-like illnesses and is the most common cause of acute upper respiratory tract infection.1,2 Recent studies have demonstrated that RV is also a lower respiratory pathogen and plays a significant role in asthma exacerbations and chronic obstructive pulmonary disease (COPD) exacerbations.3–5 Approximately two-thirds of respiratory virus infections associated with asthma or COPD exacerbations are due to RV,4,5 and exacerbations have been viewed as one major cause of morbidity and mortality in asthma and COPD. 6 Despite the recognized importance of RV infection in asthma and COPD exacerbations, the mechanism by which RV causes asthma or COPD exacerbations is not well understood, and there are currently no approved anti-RV agents. Therefore, identifying novel targets regulating RV replication and infection could offer new approaches for the treatment and/or prevention of asthma and COPD exacerbations.
RV is a single positive-strand RNA virus belonging to the family of Picornaviridae. There are more than 160 different RV serotypes grouped into three species (RV-A, RV-B, or RV-C) based on phylogenetic sequence identity. RV-A and RV-B can also be classified into a major and a minor group based on their binding to intercellular adhesion molecule (ICAM)-1 or to members of the low-density lipoprotein (LDL) receptor family, respectively. 2 RV-C was recently reported to bind to cadherin-related family member (CDHR)-3 as a potential entry receptor. 7 The large number of RV serotypes makes it challenging to develop anti-RV agents with broad-spectrum activity against the many RV serotypes.
Phenotypic screening approaches using different perturbation modalities, for example, small-molecule compounds, siRNA, and CRISPR, have been shown to hold significant value for identifying novel targets for drug discovery.8–11 Each perturbation modality has its own advantages and drawbacks. For siRNA screening, both pooled and arrayed siRNA libraries have been used since 2000 for functional genomics screens to elucidate novel gene functions and pathways.12–14 We have recently reported the identification of lanosterol synthase (LSS) as a novel regulator of RV2 replication through a phenotypic screening of a library of arrayed siRNAs against 10,500 druggable genes in normal human bronchial epithelial (NHBE) cells. 15 One major challenge in the siRNA screening field is that this approach is abundant in false positives mediated mainly by off-target effects,9,16–19 necessitating thorough validation of first-pass hits through follow-up experiments. In order to investigate for the possibility to identify more potential targets regulating RV2 replication from the siRNA screen, we performed a follow-up phenotypic screen using small-molecule compounds that are annotated to be pharmacological regulators of the target genes that were identified to significantly affect RV2 replication in the previously described siRNA screen. 15
Here, we describe the combined siRNA and small-molecule screening approach, including the compound selection strategy. We screened 270 small-molecule compounds selected for interacting with 122 target gene hits using the same high-content imaging RV2 assay applied in the previous siRNA screen, followed by the secondary quantitative PCR (qPCR) assays for RV2, RV14, and RV16. This follow-up screen allowed us to identify an additional novel target, Fms-related tyrosine kinase 4 (FLT4), that inhibits RV replication in human primary NHBE cells. We demonstrate that a combination of siRNA and small-molecule compound screening is a useful phenotypic drug discovery approach for the identification of novel drug targets.
Materials and Methods
Preparation of NHBE Cells
Cryopreserved NHBE cells were from Lonza (CC2540, Lot 7F4363; Lonza, Basel, Switzerland). Before screening, NHBE cells were thawed and expanded in complete bronchial epithelial growth medium (BEGM) (CC3171, CC4175; Lonza) according to Lonza’s instructions, and frozen at passage 4 with 0.45 million cells/vial and stored at −150 °C until use. Cryopreserved expanded NHBE cells were thawed, seeded in 175 cm2 cell culture flasks, and precultured for 7 days in complete BEGM. Cells were detached with TrypLe Express (12604-013; ThermoFisher, Waltham, MA) and used in the screen as described in the following sections.
RV2, RV14, and RV16 Viral Stocks
RV serotypes 2, 14, and 16 were obtained from ATCC (Manassas, VA; VR-482, VR-284, VR-283). Bulk virus stocks were prepared by inoculation of HeLa OHIO cells in cell culture flasks and incubating at 37 °C/5% CO2 until 100% cell death could be visually observed. The flask was then transferred to a −80 °C freezer overnight. The contents of the flask were then thawed and centrifuged at 600g for 10 min. The supernatant was then aliquoted and stored at −80 °C. To determine stock concentration, RV stocks were titrated on HeLa OHIO cells to determine the TCID50/mL using the Spearman Karber method.20,21 The concentrations of RV stocks are RV2 stock (4 × 108 plaque-forming units [PFU]/mL), RV14 stock (1.4 × 105 PFU/mL), and RV16 (3.3 × 107 PFU/mL).
siRNA Library Screening Using In Situ Hybridization
The siRNA library was the SilencerSelect Druggable Genome library (10,415 genes; Ambion, ThermoFisher, Waltham, MA), plus siRNA targeting 82 additional genes (Ambion). siRNA screening was performed as described previously. 15 Briefly, NHBE cells were seeded at 900 cells/well in 384-well plates (356663; BD Bioscience, San Jose, CA) and incubated for 24 h at 37 °C/5% CO2. siRNA transfections were then performed using 30 nM siRNA with 0.03 µL/well RNAiMax (13778; Invitrogen/ThermoFisher). After 72 h of siRNA transfection, cells were infected with 63,000 PFU/well of RV2 (reaching a multiplicity of infection [MOI] of 70 with respect to seeded cells) for 24 h at 33 °C/5% CO2. Cells were then fixed and stained with MitoTracker Deep Red (M22426; ThermoFisher), Hoechst 33342 (H3570; ThermoFisher), and a ViewRNA RV2-specific probe (VF1-10249-03; Panomics, ThermoFisher, Waltham, MA). The plates were sealed and imaged on an ImageXpress Micro automated microscope system (Molecular Devices, Berkshire, UK) using 20× objectives. Image analysis was performed using automated eCognition (Definiens) software, using algorithms for segmentation and classification of nuclei customized for NHBE cells.
Small-Molecule Compound Follow-Up Screening Using In Situ Hybridization
NHBE cells were seeded at a density of 5000 cells/well in 30 µL of complete BEGM in 384-well plates (356663; BD Bioscience, San Jose, CA) and incubated at 37 °C/5% CO2. After 24 h of incubation, DMSO-negative control, positive control ZD9720 (a LSS inhibitor), 22 and compounds dissolved in DMSO were prediluted in growth medium and then dosed in 5 µL/well to the appropriate wells using CyBio pipetting and incubated at 37 °C/5% CO2. The final DMSO concentration was 0.1% and the compounds were screened at eight concentrations ranging from 0.01 to 30 µM in triplicate. Twenty-four hours post-compound addition, cells were infected with 150,000 PFU/well of RV2 (MOI of 30). Wells for noninfected control received 5 µL of growth medium. Cells were infected at 33 °C/5% CO2 for 30 h until fixation. Cells were then stained, imaged, and analyzed as described under “siRNA Library Screening Using In Situ Hybridization.”
Small-Molecule Compound Secondary Screening Using Quantitative RT-PCR
NHBE cells were seeded at a density of 2500 cells/well in 30 µL of complete BEGM in 384-well plates (781098; Greiner, Kremsmünster, Austria) and incubated at 37 °C/5% CO2. After 24 h of incubation, DMSO-negative control, positive control pirodavir (an RV VP1 inhibitor), 23 and compounds dissolved in DMSO were directly dosed in 90 nL/well using an Echo liquid handler (Labcyte, Sunnyvale, CA). The final DMSO concentration was 0.3% and the compounds were screened at 10 concentrations ranging from 0.002 to 30 µM in duplicate. Twenty-four hours post-compound addition, cells were infected with 8745 PFU/well of RV2 (MOI of 3.5), 1.0 PFU/well of RV14 (MOI of 0.0004), or 15,000 PFU/well of RV16 (MOI of 6) for 24–26 h at 33 °C/5% CO2. Cells were then rinsed with cold DPBS (14190250; ThermoFisher), lysed with cell lysis buffer (5943523001; Roche Diagnostics, Basel, Switzerland), sealed, and stored at 4 °C for a maximum of 2 days, and then subjected to quantitative real-time reverse transcription polymerase chain reaction (RT-PCR). RT-PCR was conducted with One-Step RT-PCR assay kits (4991885001/4692136001/05189268001; Roche Diagnostics) to assess the RNA level of RV2 (forward primer 5′-3′ AATGTTGGTTACAACTACACACACC, reverse primer 5′-3′ GGATTCAATCTCGTCTCAGCTT; Sigma), RV14 (forward primer 5′-3′ CCTGAATGCGGCTAACCTT, reverse primer 5′-3′ GACCTTCAACCACTGGATCG; Sigma), and RV16 (forward primer 5′-3′ AGCCCAATGTGTGCTGAAT, reverse primer 5′-3′ ACTGGTAGACCTTGCACAATG; Sigma). Peptidylprolyl isomerase A (PPIA) (human PPIA gene assay, 05189268001; Roche Diagnostics) was used as an endogenous control. For performing RT-PCR analysis, 10× concentrated RT-PCR mix containing LightCycler 480 RNA Master Hydrolysis probes, RV2, RV14, or RV16 specific primers, and UPL probes (probe 96 for RV2, probe 1 for RV14, probe 50 for RV16; Roche Diagnostics) was prepared on ice according to the manufacturer’s instructions (4991885001; Roche Diagnostics). Concentrated RT-PCR mix and cell lysate were added to qPCR plates (4729749001; Roche Diagnostics) with 9 and 1 µL/well, respectively. Fifty cycles of reaction were performed on a LightCycler480 instrument (Roche Diagnostics). Only samples reaching threshold values before cycle 36 were included. The relative expression levels of specific genes were calculated using delta delta Cp. The compound inhibition effect for the tested compounds was calculated using GeneData Screener software using the following calculation method: compound % activity = 100*[(0% control – X)/(0% control – 100% control)], where X represents the normalized value for the compound based on the DMSO (100% control) and positive control pirodavir (0% control).
Small-Molecule Compounds Selected for Follow-Up Screening
All compounds and biological data were selected using ChemistryConnect, and filtered and analyzed with Pipeline Pilot24–26 (https://www.3dsbiovia.com/products/collaborative-science/biovia-pipeline-pilot). Initially, 270 compounds were selected to cover 122 targets identified by the RV siRNA experiment. These are annotated as antagonists and retrieved from internal and external sources. Clustering was performed within Pipeline Pilot using substructure definitions. The nearest-neighbor search was performed in the multifingerprint tool selecting from the available screening compound collection.24,25
Statistical Analyses
Redundant siRNA activity (RSA) analysis was used to aid the selection of hit mRNA targets from the siRNA library screen. 27 Data are presented as means ± SEM unless otherwise indicated in the figure legends. Compound concentration response (CR) data analysis using nonlinear regression fitted to a sigmoidal four-parameter equation, and statistical analysis were performed in GraphPad Prism 7 (La Jolla, CA). Statistical tests applied are indicated in the figure and table legends.
Results
Combined siRNA and Small-Molecule Compound Screening Approach for Target Identification
To identify putative targets that affect RV replication, we performed a combined siRNA and small-molecule compound phenotypic screening approach to first screen a library of siRNAs and, subsequently, screen a set of small-molecule compounds that are annotated to be pharmacological regulators of target genes that were identified to significantly affect RV2 replication in the siRNA primary screening. The siRNA library consists of 10,500 druggable genes with three siRNAs per gene, and the small-molecule compound set consists of 270 compounds. This set of 270 small-molecule compounds was selected using literature and AstraZeneca internal biological activity data via ChemistryConnect based on the criteria shown in
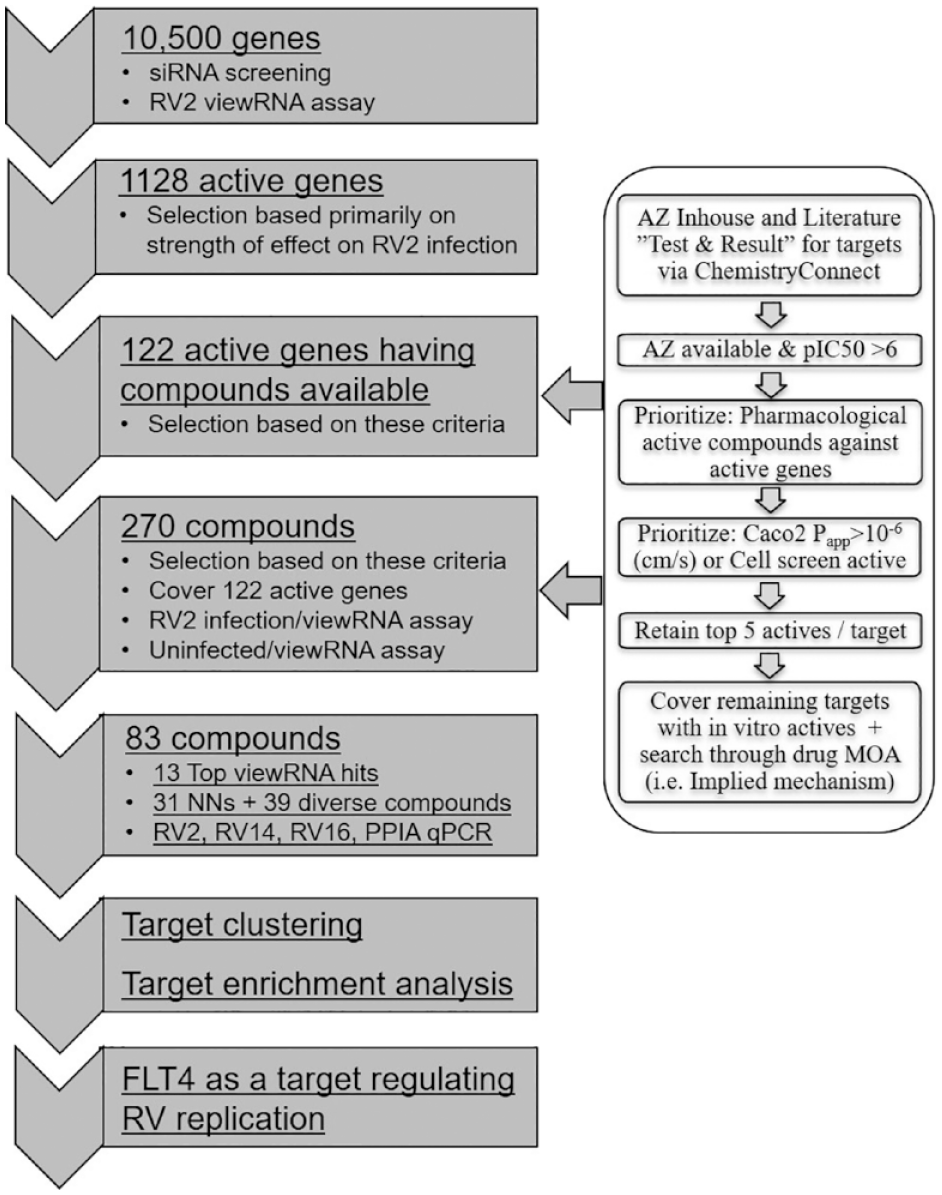
Overview of a combined siRNA and small-molecule compound screening cascade for identifying novel targets that inhibit RV replication in NHBE cells, number of siRNA and small molecules screened at each step, and criteria used for selecting compounds for follow-up screening.
Primary Screening of siRNA Library for RV2 Replication Regulators
A primary phenotypic screening of a library of siRNAs against druggable genes (three siRNAs per gene) in NHBE cells was performed to identify siRNAs that affect RV2 replication. RV2 replication was quantified via in situ hybridization with RV2 area index (the proportion of the cell cytoplasm positive for RV2 RNA) as an indication of viral replication, and number of nuclei (absolute number of nuclei per image field) as an indication of cell proliferative activity (
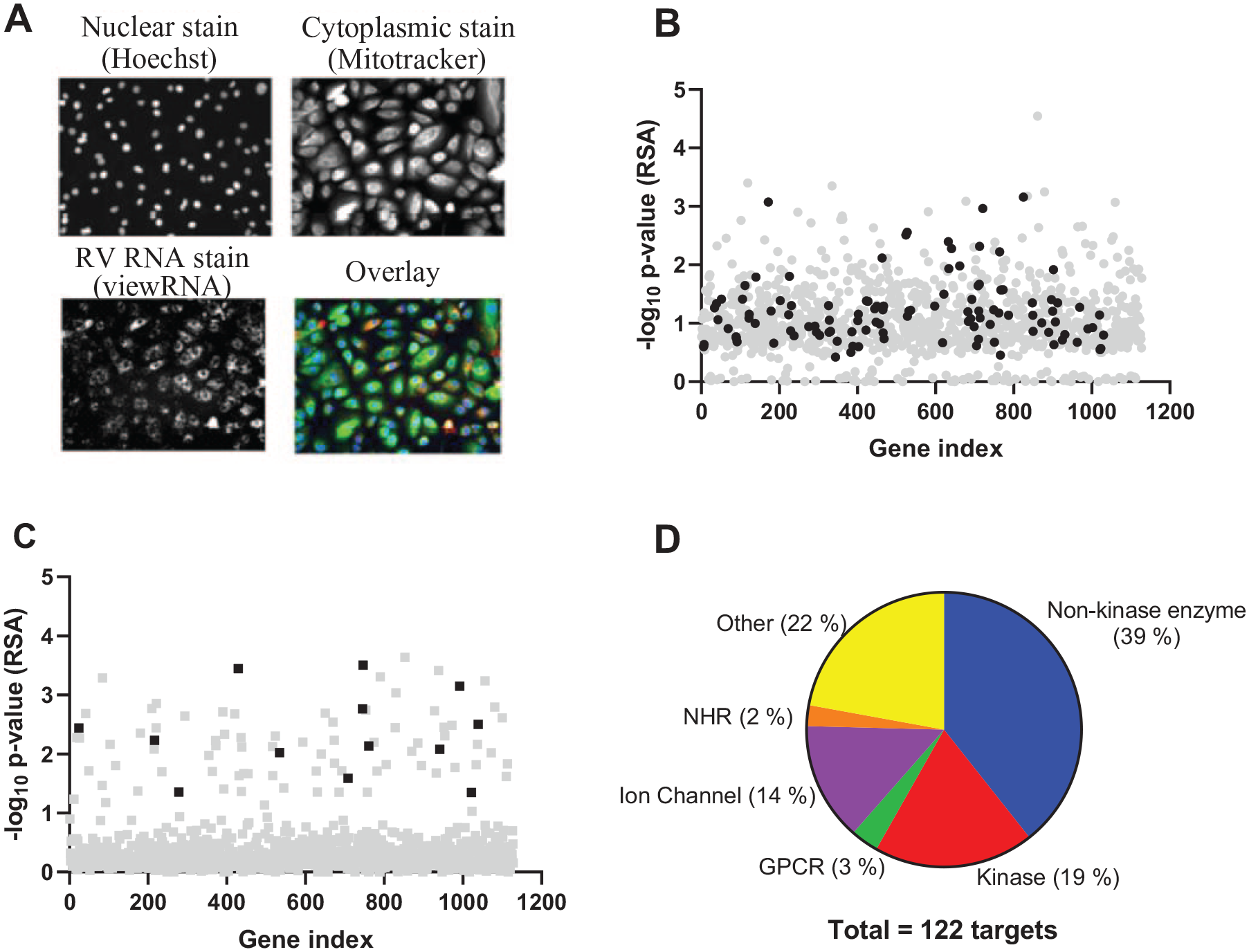
Overall results of RV2 siRNA screening using in situ hybridization in NHBE cells. NHBE cells were transfected with 30 nM siRNAs for 72 h, followed by infection with RV2 at an MOI of 70 for 24 h. Cells were then fixed and stained with MitoTracker Deep Red, Hoechst 33342, and a ViewRNA RV2-specific probe, and imaged with 20× objectives. (
Follow-Up Screening of Small-Molecule Compounds for RV Replication Regulators
Searching within the AstraZeneca available compound collection for compounds that are annotated to be pharmacological regulators of the 1128 target genes revealed that only 122 targets have available compounds in the compound collection.24,25 Among the 122 targets, siRNA knockdown of 109 targets inhibited RV2 replication in NHBE cells (
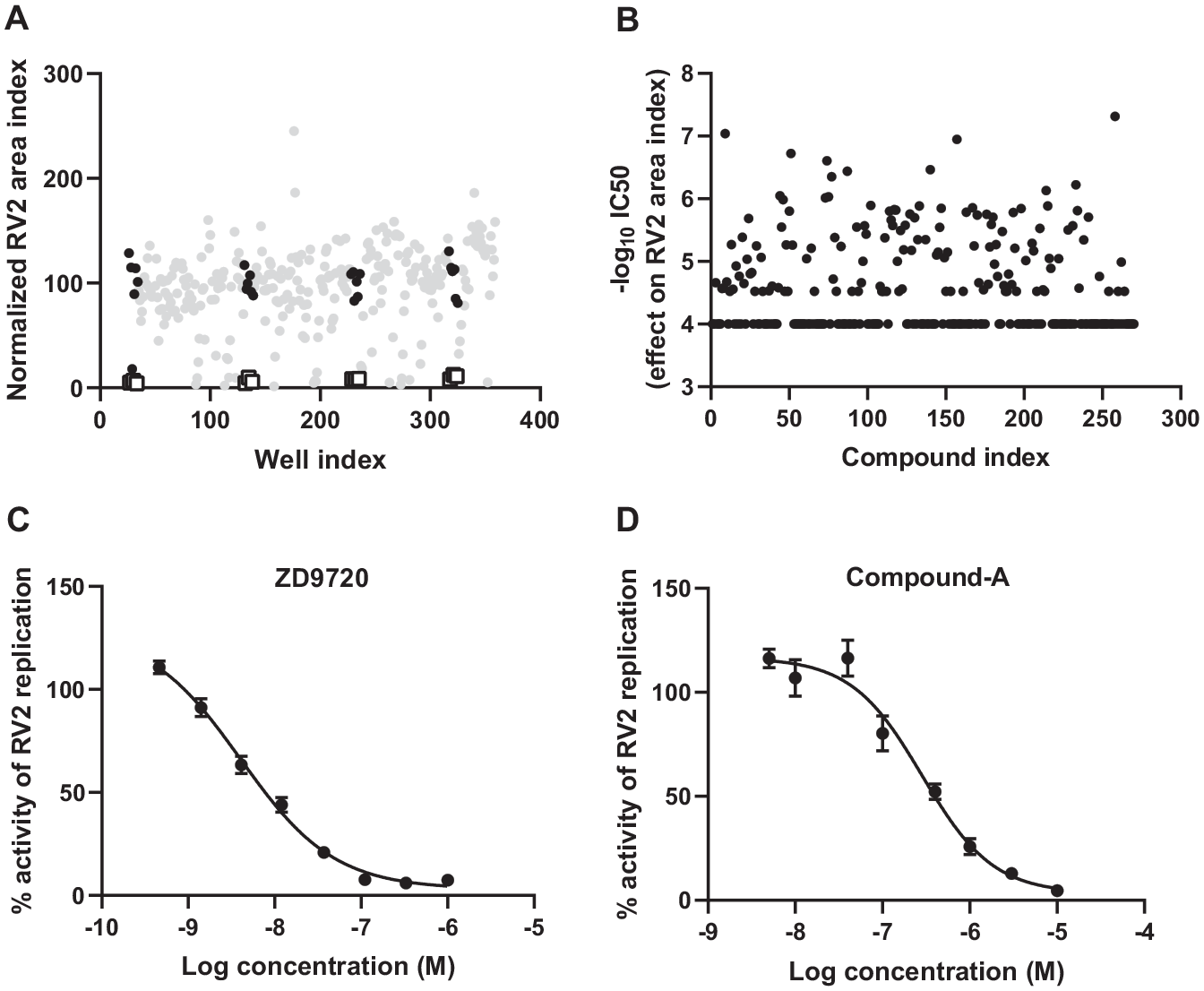
Overall results of the RV2 small-molecule follow-up screening using in situ hybridization in NHBE cells. NHBE cells were treated with small molecules for 24 h, followed by infection with RV2 at an MOI of 30 for 30 h. Cells were then fixed and stained with MitoTracker Deep Red, Hoechst 33342, and a ViewRNA RV2-specific probe, and imaged with 20× objectives. RV2 replication was quantified with RV2 area index and data were normalized as percent activity based on the on-plate DMSO (100% control) and positive control ZD9720 (0% control). (
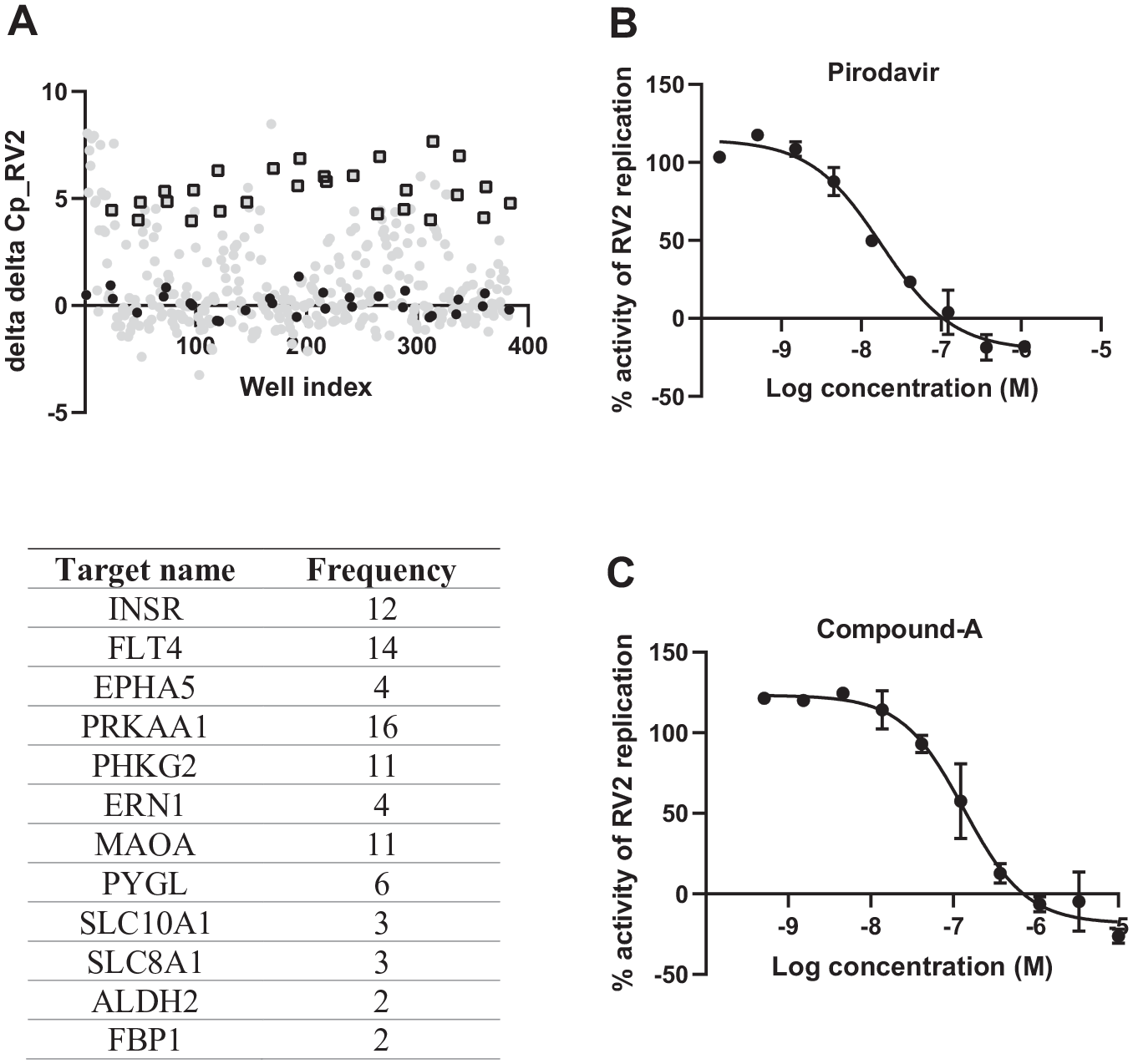
Overall results of the RV2 secondary screening using qRT-PCR in NHBE cells. NHBE cells were treated with small molecules for 24 h, followed by infection with RV2 at an MOI of 3.5 for 24–26 h. Cells were then lysed and cellular viral RNA analyzed by qRT-PCR for RV2 and a housekeeping gene PPIA. RV2 replication was quantified with Cq of RV2 and data were normalized to delta Cq of RV2, delta delta Cq, and then as percent activity based on the on-plate DMSO control (100% control) and positive control pirodavir (0% control). (
Identification of FLT4 as a Regulator of RV2 Replication in NHBE Cells
Biochemical validation of the potential targets was performed by correlating the potency of each compound in the RV2 assay with the potency of that compound in biochemical assays of the target retrieved from literature. From this analysis, FLT4 was found to be the most promising target. Overall, 14 compounds (CHEMBL347086, CHEMBL29197, CHEMBL3544937, CHEMBL357815, CHEMBL150187, CHEMBL443962, CHEMBL329201, CHEMBL276711, CHEMBL535, CHEMBL226471, CHEMBL21156, CHEMBL2105708, CHEMBL1230541, and CHEMBL1336) (see
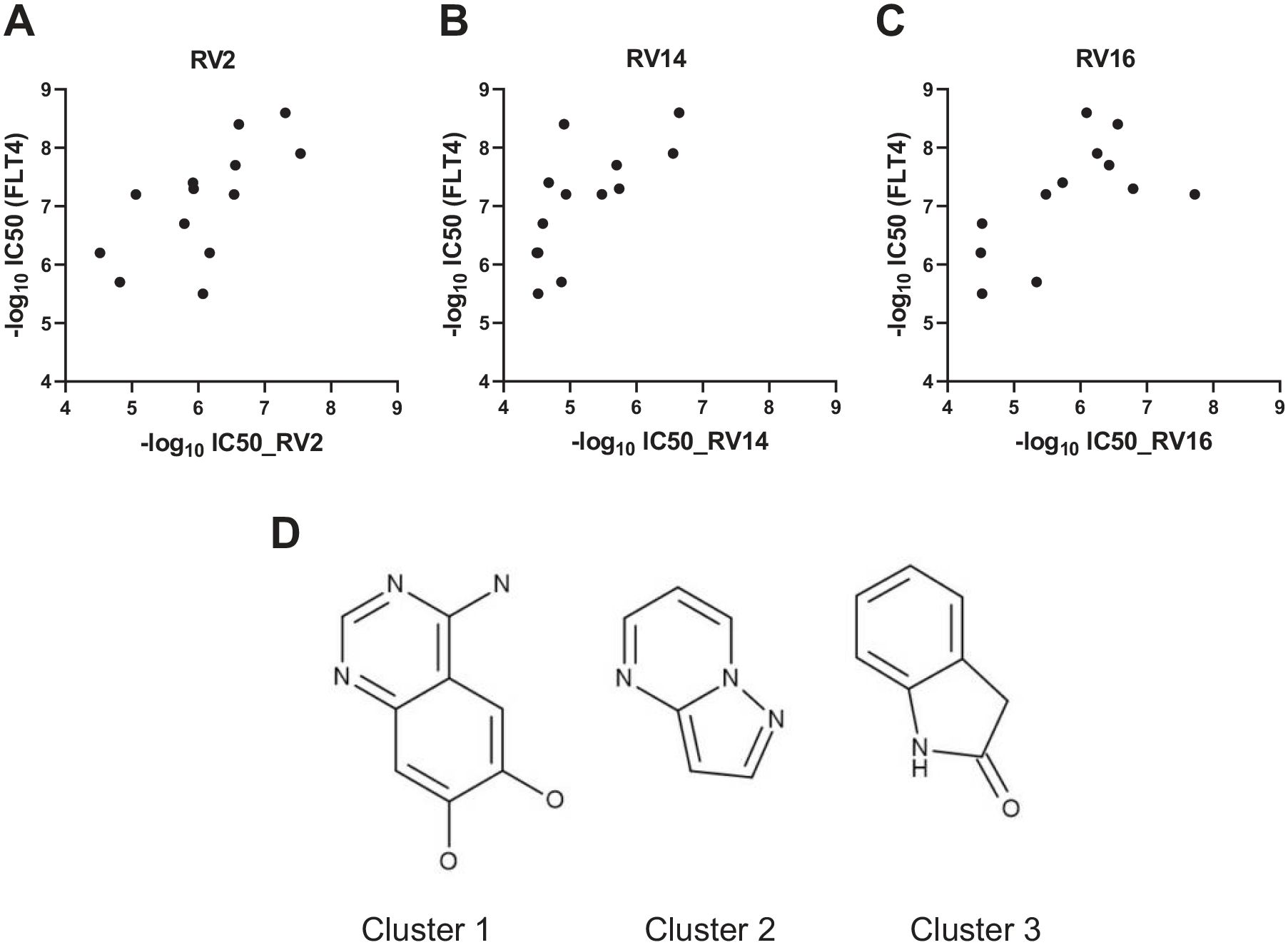
Potency correlation between compound effect on RV2, RV14, or RV16 and effect on FLT4. NHBE cells were treated with small molecules for 24 h, followed by infection with RV2 at an MOI of 3.5, RV14 at an MOI of 0.0004, or RV16 at an MOI of 6, respectively, for 24–26 h. Cells were then lysed and cellular viral RNA analyzed by qRT-PCR for RV2, RV14, or RV16 and the housekeeping gene PPIA. Anti-RV2 (
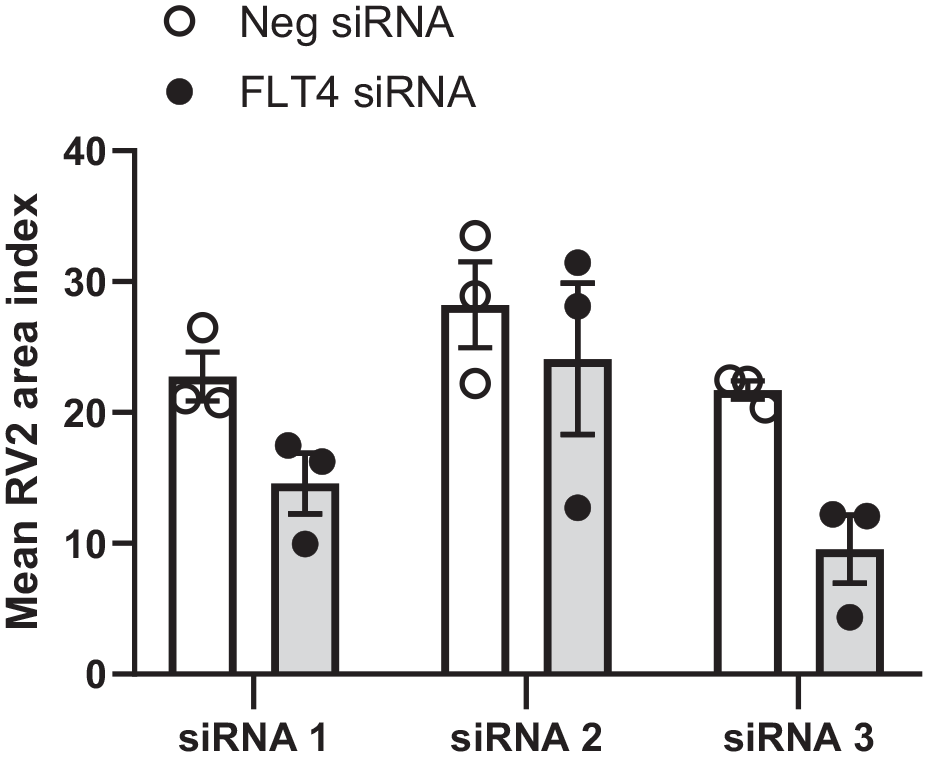
FLT4 results from the primary RV2 siRNA screening in NHBE cells. NHBE cells were transfected with three independent FLT4-specific siRNA variants (30 nM) for 72 h, followed by infection with RV2 at 70 MOI for 24 h. The proportion of the cytoplasmic area staining positive for RV2 RNA, “RV area index,” was calculated as described in Materials and Methods and normalized to the median of the wells treated with the negative control siRNA. FLT4 and negative control gene knockdown results are presented as mean ± SEM from three independent experiments.
Discussion
In this study, we report a combined siRNA and small-molecule phenotypic screening approach to identify targets regulating RV replication in human bronchial epithelial cells. This combination screening approach uses an in situ hybridization assay to first screen a library of siRNAs consisting of 10,500 druggable genes with 1128 active siRNA hits identified to either inhibit or enhance RV2 replication and, subsequently, using the same RV2 in situ hybridization assay, screen 270 small-molecule compounds that are annotated to be pharmacological regulators of 122 target genes that were identified in the siRNA primary screen. The active small-molecule compounds identified from the screen of 270 compounds, and chemically similar compounds, were further tested in the secondary qPCR assays for RV2, RV14, and RV16. With this combined siRNA and small-molecule screening approach, we have successfully identified FLT4 as a novel target regulating RV replication.
Phenotypic screens where physiologically relevant cells and biologically relevant in vitro assays are used to study perturbance-induced phenotypic changes have been shown to be very useful for identifying novel targets for treating human disease.10,28,29 Various genome-wide siRNA phenotypic screens studying the effects of siRNA-mediated gene knockdown on biological phenotypes, including viral replication, apoptosis, cell adhesion, and cell proliferation, have been reported.30–33 Lin and coworkers reported that the top-scoring siRNA hits, in a screen to identify novel regulators of the HIF-α transcription pathway, did not act through specific knockdown of the intended targets, but rather by targeting the HIF1-α mRNA through a microRNA-like mechanism. 34 Recent studies by Lin et al. and Smith et al. demonstrated that RNAi is more susceptible to off-target effects than CRISPR.19,35 One approach to reduce the number of false positives is to use a higher cutoff. However, at the same time as minimizing the false-positive rate, this approach may also increase the number of false negatives.
Small-molecule compound libraries, especially pharmacologically annotated tool compound collections, have been successfully used in various phenotypic screens for the identification of novel targets for biology of interest. Phenotypic effects induced by small-molecule inhibition of a target in a cell-based assay should in many cases be similar to the gene knockdown-induced effect of the same target. Using siRNA to knock down the gene of interest is a commonly used approach for the confirmation of any possible targets generated from small-molecule phenotypic screens as well as for the validation of cellular assays in the case that tool compounds for regulating the target of interest are not available.36,37 Following a rigorous process of technical and biological validation, our primary siRNA screen identified eight “high-confidence” hit mRNA targets, including LSS, which significantly reduced RV replication. 15 To investigate whether there is any potential for identifying more drug targets regulating RV replication from the siRNA screen, we applied a low threshold in the siRNA screen in order to reduce the number of false negatives, and this produced 1128 hits, a number that is not practical for detailed target validation studies. Instead, we integrated a follow-up small-molecule screen using the same high-content imaging RV2 assay applied in the primary siRNA screen. We did this by screening small-molecule compounds that are annotated to be pharmacological regulators of target genes that were identified in the siRNA primary screen. Our rationale was that a siRNA screen in combination with an annotated small-molecule compound screen might help to reduce false negatives by using a low threshold and facilitate the identification of additional drug targets that are not picked up as top-scoring primary siRNA hits. A similar approach has recently been reported in the search for host targets against influenza virus infection, whereby experimental or clinically approved drugs were selected based on the hits from an siRNA screen, enabling the identification of a host signaling pathway for targeting influenza virus. 38 Based on this hypothesis, we searched within the AstraZeneca compound collection for compounds that are annotated to be pharmacological regulators of the 1128 target genes. Only 122 targets had available compounds in the AstraZeneca compound collection. Two hundred seventy compounds were initially selected to cover these 122 targets. We used the same high-content imaging RV2 assay by quantifying viral replication via in situ hybridization as in the siRNA screen, to ensure comparability between the two screens. The confirmed small-molecule RV2 hits, and NNs of the confirmed hits, were further studied in a secondary RV2 assay as well as in RV14 and RV16 assays in which viral replication was quantified by qPCR. This combination screening approach allowed us to identify a novel target, FLT4, which had not been originally identified as a strong hit in the primary siRNA screen, that inhibits RV2 replication as well as shows a potential role in RV14 and RV16 inhibition. Thus, we were able to identify an additional target that would have been overlooked, had we not performed the follow-up small-molecule screen. This approach of combining genome-wide siRNA and small-molecule screens for the identification of novel drug targets could have wide applications. However, in our study, among the 1128 primary siRNA hits, only 122 siRNA hits had available compounds in the AstraZeneca compound collection, which limits our opportunity of identifying more additional targets. Interestingly, Lesch et al. identified available compounds for a similar proportion of the hits from their influenza virus siRNA screen (43 compounds covering 14 of the total 133 hit genes). 38 We might have been able to identify more targets using this combination screening approach if more compounds covering more primary siRNA hits had been available. Therefore, the success of this combination screening approach highly depends on the availability of small-molecule compounds.
FLT4 (VEGFR-3) is a receptor tyrosine kinase thta is primarily expressed on the lymphatic endothelium. The ligands for FLT4 are VEGF-C and VEGF-D, and receptor engagement and activation mediates lymph angiogenesis and maintenance of the lymphatic endothelium. Corresponding with this key function, mutations in the FLT4 gene cause hereditary lymphedema. FLT4 has also been linked to tumor metastasis. 39 Several drugs with activity against multiple receptor tyrosine kinases, including FLT4, have proven to be effective antitumor therapies, for example, erdafitinib, telatinib, sunitinib, and axitinib.40–43 However, to our knowledge, our finding of a link between FLT4 and RV infection is novel. In a study of natural RV infections in human subjects, VEGF levels were increased in nasal secretions. 44 However, it should be noted that the enzyme-linked immunosorbent assay (ELISA) used in this study detects VEGF-A, which does not bind to FLT4, and whether it can also detect VEGF-C or VEGF-D is unknown. Little is known about the role of FLT4 in viral infections in general. However, intriguingly, this is not the first time that FLT4 has emerged as a hit from an siRNA functional genomics screen for viral infection. For example, in the influenza virus siRNA screen performed by Lesch et al., not only was FLT4 one of the top 3 hit genes, but also 3 of the 14 selected compounds in their follow-up testing were active against FLT4. 38 FLT4 was also one of 5 genes that survived multiple rounds of testing and data analysis methods in an siRNA screen for hepatitis C infection. 45 In another study, siRNA-mediated knockdown of FLT4 inhibited chikungunya virus infection. 46 In a limited screen of siRNA against 65 genes against Ebola, virus-like particles showed that silencing of the FLT4 gene suppressed viral replication. 47 The authors went on to show by co-immunoprecipitation and Ch-IP that FLT4 protein interacts with both viral glycoprotein and viral RNA. The mechanism was suggested to be related to inhibition of cellular secretory functions.
The mechanism by which silencing of FLT4 suppresses viral infection is poorly understood. The FLT4 inhibitors tested by Lesch et al. in their influenza studies were shown to inhibit fusion of the viral and endosomal membranes. 38 FLT4 was shown to be induced via Ets 1 in Kaposi’s sarcoma virus-infected cells. 48 Ma et al. demonstrated that respiratory syncytial virus-, influenza-, or dsRNA-induced type I interferon (IFN) responses inhibit VEGF responses in human VEGF-transgenic mice, suggesting a potential link between VEGF and innate antiviral immunity. 49 However, it should be noted that the mechanism was shown to be via suppression of VEGFR-1 expression but not FLT4. A recent study showed a link between stimulation of VEGF-C expression and suppression of type I IFN responses. 50 Further studies would be required to investigate whether our findings could be explained by enhanced type I IFN responses. To our knowledge, there are no published studies investigating FLT4 in asthma. However, VEGF levels have been shown to be increased in the asthmatic airway, a finding that may relate to airway remodeling.51,52 As noted above, the ELISA used in these studies is most likely to detect VEGF-A, and it is not known whether these studies were also detecting VEGF-C or VEGF-D. In one study, FLT4 was shown to be expressed by cultured distal lung fibroblasts from COPD patients, and these cells produced VEGF in response to iloprost or TGF-β, implicating VEGF signaling in vascular remodeling in COPD. 53 However, to our knowledge, there are no published data linking FLT4 with susceptibility to viral exacerbations in COPD.
In summary, we performed a small-molecule phenotypic screen to follow up hits identified in a primary siRNA screen for modulators of RV replication. This combination screening approach allowed us to identify FLT4 as a novel target regulating RV replication. Our study demonstrates that a combination of siRNA and small-molecule compound screening models is a useful approach for the identification of novel drug targets.
Supplemental Material
Supplemental_Info_Combined_siRNA_and_SMs_HRV_screen_by_Ding_et_al – Supplemental material for Combined siRNA and Small-Molecule Phenotypic Screening Identifies Targets Regulating Rhinovirus Replication in Primary Human Bronchial Epithelial Cells
Supplemental material, Supplemental_Info_Combined_siRNA_and_SMs_HRV_screen_by_Ding_et_al for Combined siRNA and Small-Molecule Phenotypic Screening Identifies Targets Regulating Rhinovirus Replication in Primary Human Bronchial Epithelial Cells by Mei Ding, Christian Tyrchan, Elisabeth Bäck, Jörgen Östling, Steffen Schubert and Christopher McCrae in SLAS Discovery
Supplemental Material
Supplemental_Table_S1_Combined_siRNA_and_SMs_HRV_screen_by_Ding_et_al – Supplemental material for Combined siRNA and Small-Molecule Phenotypic Screening Identifies Targets Regulating Rhinovirus Replication in Primary Human Bronchial Epithelial Cells
Supplemental material, Supplemental_Table_S1_Combined_siRNA_and_SMs_HRV_screen_by_Ding_et_al for Combined siRNA and Small-Molecule Phenotypic Screening Identifies Targets Regulating Rhinovirus Replication in Primary Human Bronchial Epithelial Cells by Mei Ding, Christian Tyrchan, Elisabeth Bäck, Jörgen Östling, Steffen Schubert and Christopher McCrae in SLAS Discovery
Footnotes
Acknowledgements
The authors would like to thank Niklas Blomberg for initiating the work to select the small-molecule compounds for the follow-up screening, scientists at Cenix for supporting the in situ hybridization experiments, and Per-Erik Strömstedt and Ryan Hicks for valuable discussions.
Supplemental material is available online with this article.
Declaration of Conflicting Interests
The authors declared no potential conflicts of interest with respect to the research, authorship, and/or publication of this article.
Funding
The authors received no financial support for the research, authorship, and/or publication of this article.
References
Supplementary Material
Please find the following supplemental material available below.
For Open Access articles published under a Creative Commons License, all supplemental material carries the same license as the article it is associated with.
For non-Open Access articles published, all supplemental material carries a non-exclusive license, and permission requests for re-use of supplemental material or any part of supplemental material shall be sent directly to the copyright owner as specified in the copyright notice associated with the article.