
Abstract
The cellular thermal shift assay (CETSA) was introduced in 2013 to investigate drug–target engagement inside live cells and tissues. As with all thermal shift assays, the response measured by CETSA is not simply governed by ligand affinity to the investigated target protein, but the thermodynamics and kinetics of ligand binding and protein unfolding also contribute to the observed protein stabilization. This limitation is commonly neglected in current applications of the method to validate the target of small-molecule probes. Instead, there is an eagerness to make direct comparisons of CETSA measurements with functional and phenotypic readouts from cells at 37 °C. Here, we present a perspective of the early CETSA literature and put the accumulated data into a quantitative context. The analysis includes annotation of ~270 peer-reviewed papers, the majority of which do not consider the underlying biophysical basis of CETSA. We also detail what future technology developments are needed to enable CETSA-based optimization of structure–activity relationships and more appropriate comparisons of these data with functional or phenotypic responses. Finally, we describe ongoing developments in assay formats that allow for CETSA measurements at single-cell resolution, with the aspiration to allow differentiation in cellular target engagement between cells in co-cultures and more complex models, such as organoids and potentially even tissue.
Introduction
An understanding of cellular target engagement is critically important to validate the underlying mechanisms of action of chemical probes and drugs. 1 Such measurements not only shed light on compound availability in live cells and tissues, but also link evidence of physical interaction with a target protein to intended, and sometimes unintended, functional and phenotypic cellular responses. A series of retrospective analyses of compound failure in clinical studies2–5 from leading pharmaceutical companies emphasized the need to verify compound exposure and physical target engagement in relevant tissue to understand whether the clinical hypothesis was in fact tested. Following these reports, recent years have seen a boost of new technologies to quantify physical binding events in cells and tissues.6,7
One of these approaches is the cellular thermal shift assay (CETSA).8,9 It relies on the established thermodynamic principle of ligand-induced stabilization, in which physical interaction of the ligand shifts the equilibrium between denatured and native forms of the target protein by preferentially binding to either of these states.10–12 In short, a ligand that binds exclusively to the native protein, such as a specific active site binder, will increase the total concentration of folded protein when the ligand concentration is titrated toward and beyond the dissociation constant. Thermal shift assays exploit this coupled equilibrium by monitoring how these events change with temperature, reported as a shift in melting temperature (ΔTm) for the studied protein. If the specific interaction of the ligand with the native protein persists at elevated temperatures, stabilization of the target protein is observed (i.e., ΔTm > 0), while a temperature-dependent loss of the protein–ligand interaction can mask an interaction occurring at physiological temperature, such that ΔTm = 0. This limitation, or bias, is well described, and application of Tm shift assays for screening activities comes with a risk of missing true binders unless adequately compensated for by increased compound concentrations or clever adjustments of solvent conditions. 13
Since Tm shift assays report on binding equilibria at elevated temperatures, CETSA data must be appropriately extrapolated to physiological temperatures before quantitative comparisons are made with other cellular readouts.14,15 As highlighted below, this consideration is poorly recognized in the literature, and abundant direct comparisons between CETSA measurements and cell-based readouts are reported without consideration of the temperature dependence of the investigated interaction. This comes with the risk of under- and/or overestimation of the relevance of observed cellular target engagement and can in the worst case lead to incorrect conclusions on the importance of the studied pharmacology. Here, we use literature data to exemplify the need to understand the underlying driving forces behind the binding event to support such extrapolations and comparisons.
These considerations are important also in the context of recent developments using mass spectrometry (MS)-based CETSA, also referred to as thermal proteome profiling (TPP).16–18 It is becoming increasingly clear that live cell MS CETSA offers the possibility to broadly capture changes in the cellular protein interactome, including ligand-induced changes to protein-protein complexes and impact on the metabolome through “metabolite sensing” proteins.19–24 Such measurements offer the possibility to combine primary CETSA responses with secondary alterations of protein stability that can be reliably derived from the primary pharmacology. In analogy with primary Tm shift responses, such functional biomarkers can be either stabilizing, destabilizing, or invisible, because secondary interactions are also expected to respond differently to temperature changes. An additional critical parameter toward a more complete mechanistic insight is offered by treatment duration, because the relatively rapid establishment of the primary interaction will differ in temporal appearance from long-term changes to the protein interactome (coined as “horizontal cell biology” in a recent review). 25 As discussed below, critical considerations in temperature, time, and CETSA amenability space will be required to define a suitably combined biomarker of treatment.
Literature Annotation
The first description of CETSA, in 2013, already demonstrated applicability to several different model systems, with a focus on intracellular kinases and enzymes involved in metabolic activation of antimetabolites.
8
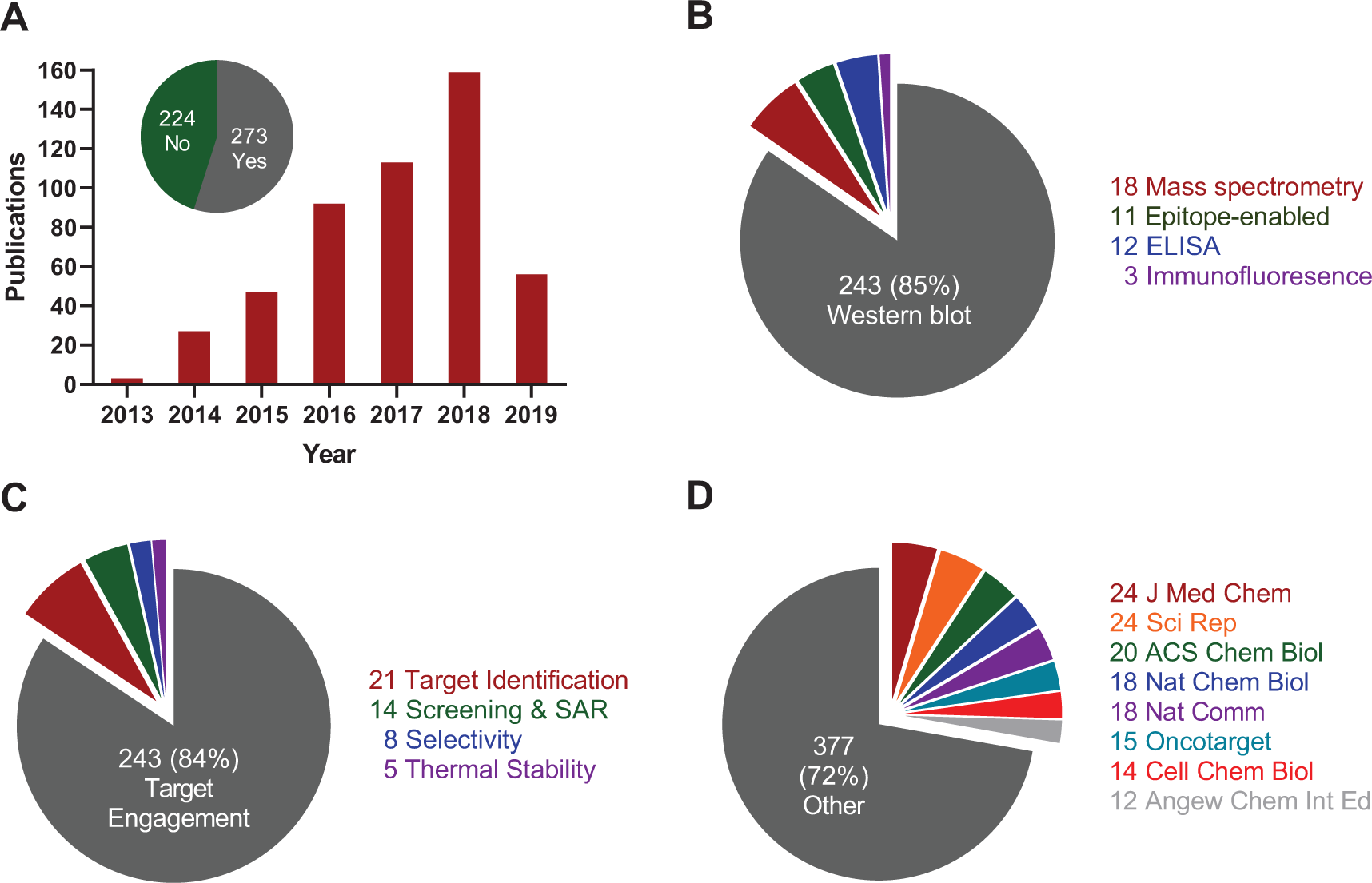
As recently reviewed, CETSA has subsequently been applied to a broad range of different protein classes25,26 and now goes beyond soluble cytoplasmic proteins to also include membrane-bound proteins.27–29 A survey of the literature using Web of Science (Clarivate) alongside Google Scholar and PubMed identifies close to 500 peer-reviewed articles citing the original study
8
and/or the follow-up methods paper
9
(
(
Not surprisingly, contributions are especially abundant in journals that cover aspects of drug discovery and chemical biology (
Heat Pulse Design
While applications of CETSA have broadened to new types of model systems and readouts, most studies adhere to the same treatment regime and transient heat pulse (3 min) as applied in the original study (
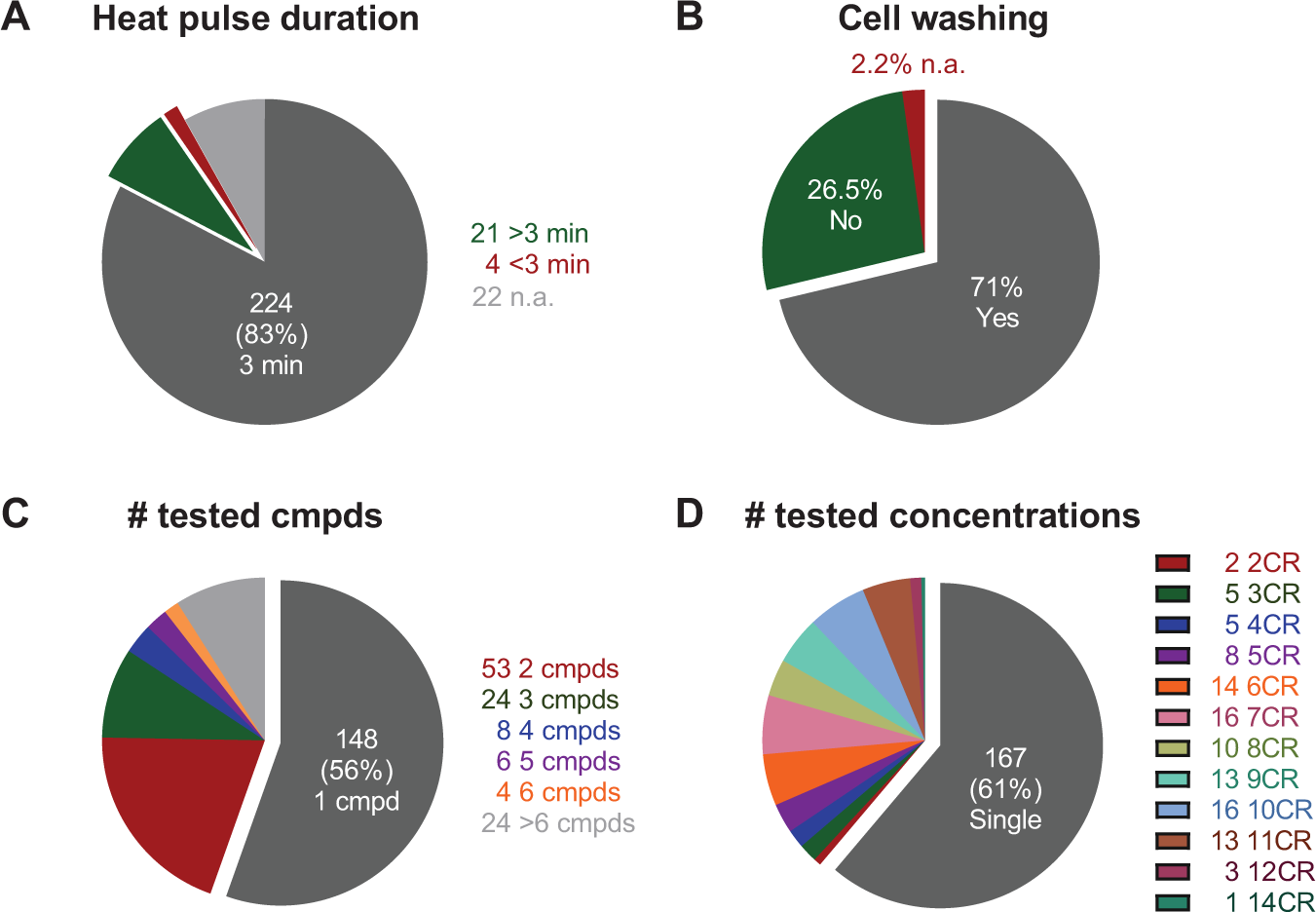
(
The continuous drift from equilibrium conditions during heating results in progressive loss of agreement with other cellular readouts, prompting the need for more effective heat transfer in shorter heat pulses. 15 Such technological developments come with the potential of increasing the space of CETSA-amenable target proteins, but this area is hitherto left relatively unexplored. Instead, recent efforts toward high-throughput screening–compatible formats have seen a development toward longer heating times to increase assay windows, 34 although this comes with a compromise in terms of lost assay sensitivity. Based on learnings we have made since the original CETSA descriptions,8,9 we instead recommend that users invest time in characterizing each model system (protein) with regard to the minimal time required to establish a thermal aggregation curve. If further experimentation is limited to a single heating time, this represents the best compromise to avoid unnecessary overheating, while additional experimentation at several heating times is highly encouraged for improved mechanistic insight.
To Treat or Not to Treat?
Examination of the literature on live cell CETSA experiments shows that, in general, four different experimental protocols are applied when considering the full treatment and heat sequence (i.e., including all steps from initial preincubation of cells with the compound to cell lysis after the transient heating and cooling of samples). Most studies (71%) follow the original 2013 protocol, in which cells are first exposed to compound and then harvested and washed in phosphate-buffered saline (PBS) to remove excessive compound prior to heat pulse. The advantage of this procedure is that it avoids the unwanted appearance of poorly permeable binders (i.e., compounds that become permeable and result in target engagement only because of the heat pulse). The extent of this problem is not well understood from the current literature, although recent cell membrane integrity experiments have shown that cells become permeable to some compounds when heated higher than 55–58 °C for 3 min (temperatures vary somewhat depending on which cells and which dye or substrate are used for the cell integrity assessments).8,9,32–34 These considerations are critically important, since the intent of CETSA experiments is often to make quantitative comparisons of target engagement with other functional cellular readouts to draw conclusions on the pharmacological consequence of the observed interaction. This suggests the inclusion of complementary measures of cell permeability to validate the observed data or, alternatively, examination of whether responses change as a function of time after washing.
There are also obvious drawbacks with the removal of treatment prior to application of the heat pulse, especially if this includes harvesting cells through detachment and extensive wash procedures in the absence of compound. Equilibria over the cell membrane are known to reestablish on the timescale of minutes,
36
especially if the cellular efflux is mediated by active transport mechanisms, such that true binders may be washed away unless interactions are long-lived or the compounds are retained intracellularly through specific mechanisms (e.g., metabolized to cell-impermeable derivatives). We were more concerned with such appearance of false negatives when first formatting CETSA for screen purposes
9
and instead opted to leave compound present throughout the heat step. In addition, this makes the protocol more compatible with large-scale profiling or screening, because it removes the cumbersome wash steps. This alternative practice has now appeared in about 26% of follow-up studies (
Our survey identified two additional practices besides these major approaches. First, there are a few studies in which cells are first treated with compound and then lysed (with or without inclusion of wash steps) prior to the heat step37–40 (
One Compound, One Condition—Appropriate Interpretations?
A key limitation of Western blot–based CETSA is that it significantly limits the number of experimental permutations that can be tested. This becomes immediately apparent in published studies, which to a large extent involve testing a single compound or a few compounds (1 compound in 56% of studies, and 2–3 compounds in 28% of studies;
Frameworks for quantitative analysis including temperature extrapolations are well worked out for traditional Tm shift assays on isolated proteins,43,44 including an understanding of the natural bias toward interactions that are increasingly favored at elevated temperatures (entropy driven). Enthalpically driven interactions, in contrast, are at risk of being lost, especially for proteins with high Tm’s, because these require extrapolations further away from 37 °C. There are, however, encouraging examples of such interactions for which the CETSA response persists to extreme temperatures, as exemplified by a chemical probe toward NUDT5. 45 The NUDT5 study represents the first use of CETSA as the main SAR-driving cellular assay alongside a biochemical assay, because it was deliberately chosen to avoid undesirable influence from nonselective cell viability assays in probe optimization. CETSA profiling for this thermostable protein was conducted at 83 °C, in which cell permeability is severely affected, such that what starts as a live cell assay becomes a permeabilized cell assay or even lysate during the heat pulse.
CETSA is expected to be governed by the same principles as conventional Tm shift assays with regard to temperature extrapolation, although with two major differences. First, in equilibrium Tm shift experiments, the samples are heated continuously (~1 °C/min), thus allowing enough time for sample reequilibration at all temperatures, whereas CETSA is based on the application of a transient heat pulse (commonly 3 min;
Several interactions have been shown to rearrange substantially on the timescale of the heat challenge, such as the interaction between lactate dehydrogenase A and a selection of 15 inhibitors, 34 resulting in a 20-fold shift in apparent potencies when varying the heat pulse duration between 5 and 50 min. Similarly, variation of the heat pulse between 0.5 and 20 min showed a several orders of magnitude shift of apparent potency for a selection of mature p38α inhibitors. 15 The impact of temperature-induced reequilibration can also be deduced from changes to the heat pulse magnitude (as exemplified for, e.g., p38α15,32 and SMYD3 33 ). Similar changes are apparent in the emerging two-dimensional MS CETSA literature, in which both heat pulse temperature and compound concentration are varied in the same experiment, as exemplified by the interactions between panobinostat and HDAC1, HDAC2, and phenylalanine hydroxylase. 46 Collectively, these data demonstrate the need to account for reequilibration in both experimental setup and analysis. Failure to adequately compensate for this (by, e.g., raising compound concentrations) comes with the risk of missing true interactions, potentially explaining why CETSA is commonly tested at high µM concentrations. Given the promiscuity of small molecules, especially early in drug development, this creates somewhat of a catch-22 situation, because the elevated concentrations can also promote target engagement that is not necessarily responsible for the observed functional responses.
A critical question then becomes: What can be regarded as good practice for interpretation of CETSA responses when there is a significant potency offset between assays? Systematic measurements of heat pulse duration and magnitude dependence require access to high-throughput amenable readouts, explaining the present lack of more quantitative CETSA studies. While the development of immunoassays for measurements of nonmodified endogenous protein requires the selection of affinity reagents and extensive assay development efforts,9,30,31,35 assays based on tagged proteins are more readily available.32–34 Pending improved availability of these tools in additional labs, there are encouraging data emerging on the correlation between CETSA data and functional data,33–35 including retained ranking of compounds even when there are significant potency offsets between assays. 34 It is important to remember, though, that this may not hold true when the tested compounds come from different structural clusters or target different binding sites, because the molecular driving forces behind the interactions may differ significantly. Since many studies still rely on Western blot for readouts, we appreciate there are significant practical limitations in capacity and throughput. Our advice in these situations is to conduct the CETSA experiments with the shortest possible heat duration and as close to the thermal aggregation temperature as possible. 15
Collectively, these data illustrate the need to study multiple cell-active compounds over a sufficiently broad potency range to probe the correlation between observed cellular target engagement through CETSA and functional responses, experiments that are feasible also with Western blot–based readouts. When building such mechanistic understanding, it is important to also properly consider the “vulnerability” of the investigated pharmacology; 47 that is, potency offsets between assays can also result from the need to modulate target activity to different extents (biological redundancy controls what target occupancy is required to achieve pharmacological effects). While a low vulnerability can be expected to negate some of the potency drop-off in CETSA (although serendipitous), cases with high vulnerability may see justifiably large differences between assays without jeopardizing the link between these events.
Further Miniaturization and Single-Cell-Resolution CETSA
A particularly attractive feature of CETSA is its potential application for discovery of new biomarkers and testing in patient-derived cells to support translational studies. While most published studies are based on in vitro treatment of lysates or isolated cells (
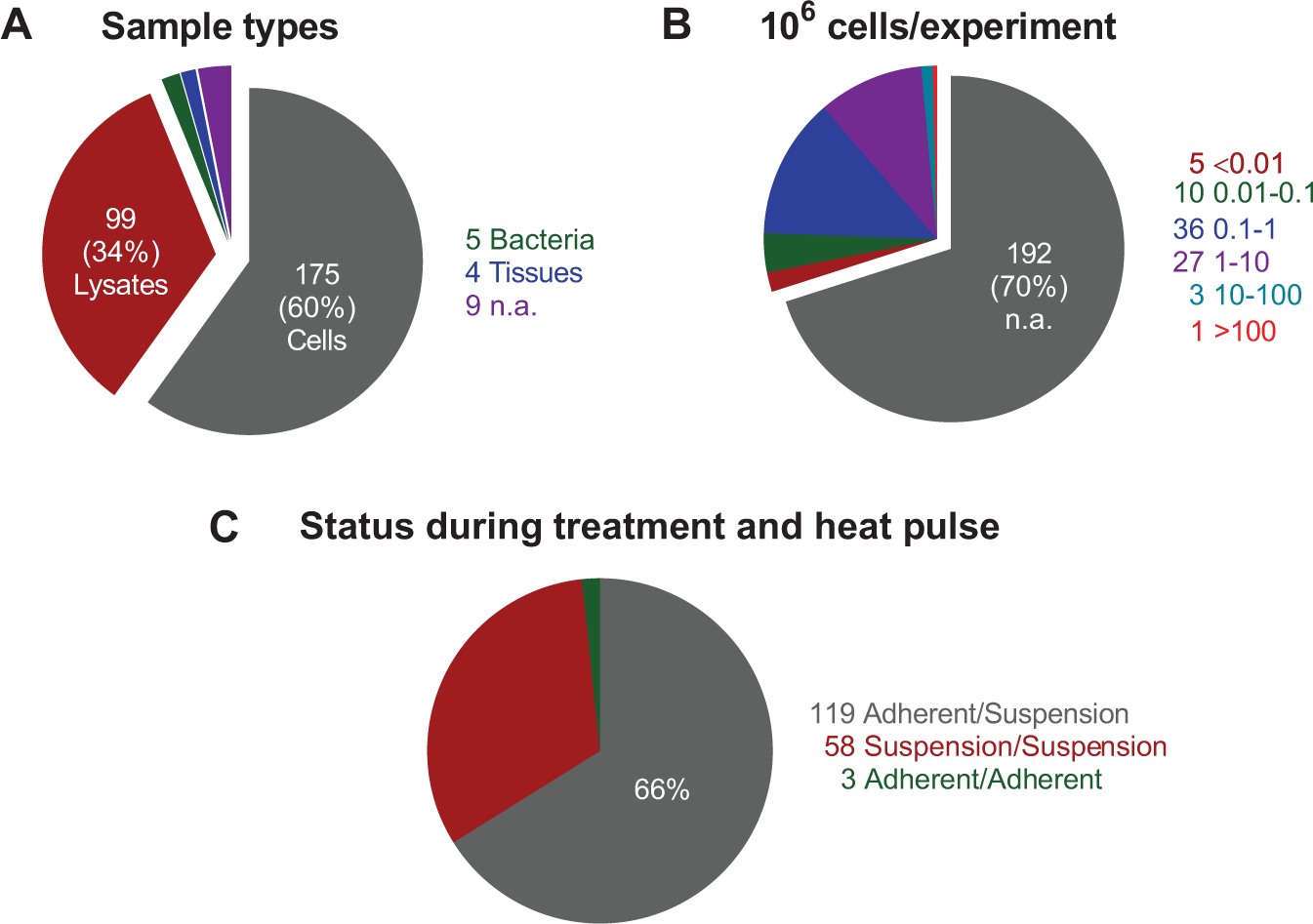
(
Such application requires demonstration that signatures (in, e.g., whole blood) are reflective of pharmacological response in the relevant physiological tissue, or, alternatively, that primary cultures of patient-derived cells can be shown to reflect individual patient responses toward the investigated pharmacology. One important aspect currently limiting such latter application to a few compounds is the requirements for large cell numbers in CETSA measurements, commonly on the order of 104–107 cells per individual sample (
These sample requirements currently restrict experimentation to a few selected compounds or, worse, may prevent testing in patient-derived cells because of limited cell availability. This is a key argument for development of more sensitive assay formats, ideally beyond those that are currently applied for screen purposes based on, for example, AlphaLISA (~104 cells per well in published assays). Of potential interest in this space are imaging-based readouts, because they in principle can be applied to populations of a few hundred cells. These assays also offer the possibility to better understand heterogeneity in response, although at present it is not known whether responses in relevant cell populations can be masked when using bulk readouts in tissue samples. A critical limitation at present is that imaging CETSA has been practically demonstrated for only two model systems,53–55 one of which is the highly amenable p38α system, and its broader application remains unclear at present.
Additional arguments for alternative CETSA formats come from the present literature’s bias toward testing of cells in suspension (
Summary and Outlook
CETSA has received much attention since its introduction in 2013, to the extent that it is now commonly requested by reviewers for demonstration of physical target engagement in live cells. While most published studies focus on assessment of intracellular drug binding to a primary target protein of interest, more recent studies using MS-based CETSA also highlight the potential to also pursue alternative responses within the cellular protein interactome as biomarkers of treatment. These include changes to protein stability resulting from posttranslational modification, coordinated thermal unfolding of members of multiprotein complexes, 22 and alterations in protein stability due to changes in endogenous metabolite levels. 57
This opens the possibility that secondary CETSA events could represent more robust measures of response, especially in cases in which the primary pharmacology demonstrates poor CETSA amenability, and when considered collectively could signify a more complete fingerprint of treatment. A prominent example of an apparently nonresponsive interaction can be found in the first publication on thermal proteome profiling, 16 in which the well-established binding of dasatinib to the fusion protein BCR–ABL appeared blind to CETSA at first glance. Closer examination of the MS CETSA data showed co-stabilization of several interaction partners, however, thus providing secondary evidence of the interaction. A more integrated view that considers both primary and secondary responses was recently described as “horizontal cell biology,” 25 including also knowledge about the temporal appearance of each of these events. To fully exploit these opportunities, the analysis should incorporate also the CETSA amenability (i.e., the extent to which primary pharmacologies and downstream events respond to different heat pulses). It will be interesting to follow how such developments are integrated with, for example, blood–CETSA 51 and other innovative approaches that can bring us closer to measuring individual patient responses.
Supplemental Material
Seashore-Ludlow_Supplemental_Table_1 – Supplemental material for Perspective on CETSA Literature: Toward More Quantitative Data Interpretation
Supplemental material, Seashore-Ludlow_Supplemental_Table_1 for Perspective on CETSA Literature: Toward More Quantitative Data Interpretation by Brinton Seashore-Ludlow, Hanna Axelsson and Thomas Lundbäck in SLAS Discovery
Footnotes
Acknowledgements
We acknowledge Helena Almqvist for early assistance with the literature annotation and Daniel Martinez Molina at Pelago Bioscience for reviewing the manuscript.
Supplemental material is available online with this article.
Declaration of Conflicting Interests
The authors declared no potential conflicts of interest with respect to the research, authorship, and/or publication of this article.
Funding
The authors disclosed receipt of the following financial support for the research, authorship, and/or publication of this article: Chemical Biology Consortium Sweden (HA and TL) is financially supported by SciLifeLab and Karolinska Institutet. BS-L and HA also acknowledge SciLifeLab Technology Development Grants.
References
Supplementary Material
Please find the following supplemental material available below.
For Open Access articles published under a Creative Commons License, all supplemental material carries the same license as the article it is associated with.
For non-Open Access articles published, all supplemental material carries a non-exclusive license, and permission requests for re-use of supplemental material or any part of supplemental material shall be sent directly to the copyright owner as specified in the copyright notice associated with the article.