
Abstract
Mono(ADP-ribosylation) (MARylation) and poly(ADP-ribosylation) (PARylation) are posttranslational modifications found on multiple amino acids. There are 12 enzymatically active mono(ADP-ribose) polymerase (monoPARP) enzymes and 4 enzymatically active poly(ADP-ribose) polymerase (polyPARP) enzymes that use nicotinamide adenine dinucleotide (NAD+) as the ADP-ribose donating substrate to generate these modifications. While there are approved drugs and clinical trials ongoing for the enzymes that perform PARylation, MARylation is gaining recognition for its role in immune function, inflammation, and cancer. However, there is a lack of chemical probes to study the function of monoPARPs in cells and in vivo. An important first step to generating chemical probes for monoPARPs is to develop biochemical assays to enable hit finding, and determination of the potency and selectivity of inhibitors. Complicating the development of enzymatic assays is that it is poorly understood how monoPARPs engage their substrates. To overcome this, we have developed a family-wide approach to developing robust high-throughput monoPARP assays where the enzymes are immobilized and forced to self-modify using biotinylated-NAD+, which is detected using a dissociation-enhanced lanthanide fluorescence immunoassay (DELFIA) readout. Herein we describe the development of assays for 12 monoPARPs and 3 polyPARPs and apply them to understand the potency and selectivity of a focused library of inhibitors across this family.
Keywords
Introduction
ADP-ribosylation has evolved as a posttranslational protein modification heavily involved in cellular stress response pathways and regulating host–pathogen interactions. The writers of the modification, poly(ADP-ribose) polymerase (PARP) enzymes are expressed throughout evolution. Human PARPs are responsible for the mono(ADP-ribosylation) (MARylation) and poly(ADP-ribosylation) (PARylation) of serine, aspartic acid, glutamic acid, lysine, tyrosine, and cysteine.
1
The protein family is divided into two groups such that there are 12 mono(ADP-ribose) polymerases (monoPARPs) and 4 poly(ADP-ribose) polymerases (polyPARPs) and 1 inactive member (
The polyPARPs have been extensively studied using small-molecule inhibitors; however, there is much less known about the monoPARPs, as evidenced by the number of publications for each PARP subfamily (
Typically, a family-wide approach to the generation of potent and selective chemical probes begins with the development of high-throughput enzyme activity or biophysical assays to enable rapid, high-fidelity rank-ordering of inhibitors. A major barrier to the development of enzyme activity assays for the monoPARPs is the gap in knowledge regarding what their substrates are and how they engage them, and at the time this work was initiated, there were no antibodies available that recognized MARylation. There have been limited reports describing inhibitor screening using enzyme assays, and in many cases high concentrations of enzyme have been required to generate sufficient signal to background to make the assays robust.8,16–19 Also, previous reports have used thermal shift assays (TSAs), 20 surface plasmon resonance (SPR), 20 DNA-encoded libraries (DELs) 21 and small-molecule microarrays (SMMs) 7 to identify and characterize monoPARP ligands; however, these techniques are either not quantitative (i.e., TSAs) or low-throughput (i.e., SPR), or require the development of expertise in complex chemistry (i.e., DELs and SMMs). Additionally, there has been a report of an AlphaScreen assay to identify inhibitors of the ADP-ribose binding macrodomains of PARP9, PARP14, and PARP15; however, this strategy was not shown to identify inhibitors of the catalytic activity of these proteins. 22 While there have been efforts to identify substrates for monoPARPs,23–27 none of the substrates identified have been used to develop high-throughput inhibitor screening assays, and many of our attempts to develop assays using several of these substrates have not been successful. Additionally, there are no x-ray structures of monoPARPs bound to their substrates in the Protein Data Bank (PDB).
To enable the discovery of potent and selective monoPARP inhibitors, we have developed self-modification assays for nearly all of the PARPs. The enzymes are immobilized on Ni-NTA plates to bring them into close proximity and overcome the weak KM for self-modification, generating a low, yet detectable level of enzymatic activity in the absence of a true physiologically relevant substrate. The incorporation of a biotinylated ADP-ribose moiety derived from biotin-NAD+ is detected using a highly sensitive dissociation-enhanced lanthanide fluorescence immunoassay (DELFIA) readout capable of monitoring less than 1% enzymatic turnover. To calibrate the potency values generated in self-modification enzyme assays, we developed SPR assays for nearly all of the PARPs, and found the Kd values generated were similar to the potencies measured in the enzyme assays. Additionally, we employed a variation of this DELFIA assay format to validate a published report that ubiquitin is an ADP-ribosylation substrate of the PARP9/DTX3L complex. 28 We used these assays to screen a focused library of PARP inhibitors against all PARP enzymes, generating a comprehensive dataset of potency and selectivity across the enzyme family that can be further used to develop chemical probes for understanding the biology of MARylation and its role in human disease.
Materials and Methods
Reagents
β-Nicotinamide-N 6 -(2-(6-(6-[biotinyl]aminohexanoyl)aminohexanoyl)aminoethyl)-adenine dinucleotide (Biotinylated-NAD+) was purchased from Biolog (Bremen, Germany). DELFIA Eu-N1 streptavidin, DELFIA assay buffer, diethylenetriamine pentaacetic acid (DTPA)-purified bovine serum albumin (BSA), DELFIA Enhancement Solution, and biotinylated hexahistidine peptide were purchased from PerkinElmer (Waltham, MA). Olaparib and PJ-34 were purchased from AdooQ Biosciences (Irvine, CA); rucaparib, niraparib, and veliparib were purchased from Selleckchem (Houston, TX); and RBN010860 and AZ12629495 29 were synthesized. All other buffer reagents were purchased from Millipore-Sigma (Burlington, MA) at the highest level of purity possible. White polystyrene 384-well nickel-nitrilotriacetic acid (Ni-NTA)-coated microplates were custom-made by Thermo Fisher (Waltham, MA) and clear 384-well polypropylene microplates and white polystyrene high-binding 384-well microplates were purchased from Greiner (Monroe, NC). DNA oligomers were synthesized by Integrated DNA Technologies (Skokie, IL). 32P-NAD+ was purchased from American Radiolabeled Chemicals (St. Louis, MO). Histone H1, histone H2A, histone H2B, histone H3.1, and histone H4 were purchased from New England Biolabs (Ipswich, MA).
Protein Purification
PARP enzymes were purified to greater than 80% purity using an N-terminal hexahistidine (His6) tag. Construct, expression, and purification details can be found in
PARP9 and DTX3L were co-expressed in Sf9 insect cells on two separate pFastBacI plasmids. PARP9 (NM_001146102.1) was fused to an MBP tag with a TEV cleavage site between the MBP tag and PARP9. DTX3L (NM_138287.3) was fused to a His6 tag with a thrombin cleavage site between the His6 tag and DTX3L. The complex was purified on a nickel affinity column followed by an MBP column. The two proteins remained as a complex throughout the purification. Full-length proteins, UBE1 (NM_003334.3), UBE2D1 (NM_003338.4), and ubiquitin (NM_021009), were purified by affinity chromatography for their respective tags, followed by size exclusion chromatography. UBE2D1 and ubiquitin were both fused to a His6 tag, while UBE2D1 also had a TEV cleavage site between the tag and the gene sequence. UBE1 was fused to a Flag tag. UBE2D1 was treated with TEV protease and subjected to an additional round of purification on a nickel column to remove the His-tagged protease. Ubiquitin required one additional passage over a monoQ column following the size exclusion column to achieve high purity.
Equipment
Compounds were serially diluted on a Fluent (Tecan, Mannedorf, Switzerland) and spotted into white 384-well polystyrene Ni-NTA-coated microplates using a Mosquito (TTP Labtech, Melbourn, UK). During assay development, reagents were added to the microplates with multichannel pipets for some assay development steps; otherwise, they were added by Multidrop Combi (Thermo Fisher). During screening assays, all reagents were added by Multidrop Combi. Microplates were washed using an Elx-406 (Biotek, Winooski, VT) and read on an Envision plate reader (PerkinElmer) using a LANCE/DELFIA top mirror and a 340 nm TRF filter for excitation and 615 nm TRF filter for emission. SPR assays were developed on Biacore T200, Biacore 4000, and Biacore 8K systems (GE Healthcare Life Sciences, Marlborough, MA).
General Self-Modification Enzymatic Activity Assay Procedure
Reactions were performed in a 25 µL volume in 384-well white polystyrene Ni-NTA-coated microplates at 25 °C. Enzyme assay buffer was 20 mM HEPES (pH = 7.5), 100 mM NaCl, 2 mM DTT, 0.1% DTPA-purified BSA, and 0.002% Tween 20. Compounds were stored in 100% DMSO and 0.5 µL was dry-spotted into the microplates. Uninhibited control wells contained DMSO (final concentration [f.c.] = 2%) and fully inhibited control wells contained rucaparib, RBN010860, or AZ12629495 (f.c. = 200 µM), depending on the PARP being tested. His-tagged PARP enzymes were added in a 20 µL volume to the microplates and incubated for 30 min before the addition of 5 µL of biotinylated-NAD+ to initiate the reaction. The assays were ended while in the linear range of product versus time formation by the addition of 5 µL of NAD+ (f.c. = 2 mM) to outcompete the incorporation of biotinylated-NAD+. PARP1, PARP2, and PARP3 are activated by DNA;30,31 therefore, DNA oligomers were included in the reactions by addition to the biotinylated-NAD+ solution. The sequences of the DNA oligomers used for each PARP are listed in
PARP5a Enzymatic Activity Assay Procedure
White polystyrene high-binding 384-well microplates were coated with 25 µL of 0.5 mg/mL histone H1 for 16 h at 4 °C, and then washed three times with 100 µL of TBS-T, followed by blocking with 100 µL of TBS Superblock for 1 h, and washed three times with 100 µL of TBS-T. Reactions were performed in a 25 µL volume in 384-well white polystyrene Ni-NTA-coated microplates at 25 °C. Enzyme assay buffer was 20 mM HEPES (pH = 7.5), 100 mM NaCl, 2 mM DTT, 0.1% DTPA-purified BSA, and 0.002% Tween 20. Compounds were stored in 100% DMSO and 0.5 µL was dry-spotted into the microplates. Uninhibited control wells contained DMSO (f.c. = 2%) and fully inhibited control wells contained rucaparib (f.c. = 200 µM). PARP5a (f.c. = 10 nM) was added in a 20 µL volume to the microplates and incubated with compound for 1 h before the addition of 5 µL of a mixture of biotinylated-NAD+ (f.c. = 3 µM) and NAD+ (f.c. = 10 µM) to initiate the reaction. The assay was ended after 2 h by the addition of 5 µL of NAD+ (f.c. = 2 mM) to outcompete the incorporation of biotinylated-NAD+. Quenched reactions were washed five times using 100 µL of TBS-T, followed by the addition of 1:1000 DELFIA Eu-N1 streptavidin diluted in DELFIA assay buffer, and then incubated for 30 min at 25 °C to allow the streptavidin to bind to the incorporated biotin. Next, the reactions were washed five times with 100 µL of TBS-T, followed by the addition of 25 µL of DELFIA enhancement solution. Microplates were incubated 30 min; then the DELFIA signal was read on an Envision plate reader (excitation = 340 nm, emission = 615 nm).
PARP9 Enzymatic Activity Assay Procedure
Reactions were performed in a 25 µL volume in clear 384-well polypropylene microplates at 25 °C. Enzyme assay buffer was 20 mM HEPES (pH = 7.5), 100 mM NaCl, 0.1% DTPA-purified BSA, and 0.002% Tween 20. Compounds were stored in 100% DMSO and 0.5 µL was dry-spotted into the microplates. Uninhibited control wells contained DMSO (f.c. = 2%) and fully inhibited control wells contained NAD+ (f.c. = 10 mM). PARP9/DTX3L (f.c. = 8 nM), UBE1 (f.c. = 50 nM), UBE2D1 (f.c. = 400 nM), His-tagged ubiquitin (f.c. = 1 µM), and biotinylated-NAD+ (f.c. = 3 µM) were added in a 20 µL volume and incubated with test compounds for 1 h before the addition of 2.5 µL of biotin-NAD+. The reaction was initiated by adding 2.5 µL of ATP (f.c. = 20 µM), and the reaction proceeded at 25 °C for 3 h before adding 25 µL of NAD+ (f.c. = 10 mM) to stop the incorporation of biotinylated-NAD+. Next, the stopped reactions were transferred from the clear 384-well polypropylene microplate to a 384-well white polystyrene Ni-NTA-coated microplate and incubated for 30 min to allow the His-tagged ubiquitin to bind. The microplate with captured His-tagged ubiquitin was washed five times using 100 µL of TBS-T, followed by the addition of 1:1000 DELFIA Eu-N1 streptavidin diluted in DELFIA assay buffer, and then incubated for 30 min at 25 °C to allow the streptavidin to bind to the incorporated biotin. Next, the reactions were washed five times with 100 µL of TBS-T, followed by the addition of 25 µL of DELFIA enhancement solution. Microplates were incubated 30 min; then the DELFIA signal was read on an Envision plate reader (excitation = 340 nm, emission = 615 nm).
DELFIA Metal-Chelation Counterscreen Assay Procedure
Reactions were performed in a 25 µL volume in 384-well white polystyrene Ni-NTA-coated microplates at 25 °C. The counterscreen assay buffer was 20 mM HEPES (pH = 7.5), 100 mM NaCl, 2 mM DTT, 0.1% DTPA-purified BSA, and 0.002% Tween 20. Compounds were stored in 100% DMSO and 0.5 µL was dry-spotted into the microplates. All control wells contained DMSO (f.c. = 2%). Biotinylated hexahistidine peptide (f.c. = 5 nM) was incubated with test compound in the counterscreen assay buffer for 60 min. Counterscreen assay buffer without peptide was added to fully inhibited control wells. The microplates were washed five times using 100 µL of TBS-T, followed by the addition of 1:1000 DELFIA Eu-N1 streptavidin diluted in the counterscreen assay buffer, and then incubated for 30 min at 25 °C to allow the streptavidin to bind to the incorporated biotin. Next, the reactions were washed five times with 100 µL of TBS-T, followed by the addition of 25 µL of DELFIA enhancement solution. Microplates were incubated for 30 min, and then the DELFIA signal was read on an Envision plate reader (excitation = 340 nm, emission = 615 nm).
Enzyme Inhibition Assay and Counterscreen Assay Data Analysis
Enzyme kinetics and parameters such as KM and kcat were determined using Prism software (GraphPad Software Inc., San Diego, CA) to analyze Michaelis–Menten fits of steady-state enzyme velocities. Screening data were processed using Dotmatics (Bishop’s Stortford, UK) and IC50 values and Hill slopes were generated using four-parameter fits. The quality and robustness of the assay was determined by analysis of the Z′ factor 32 and performance of the 50% inhibition and IC50 control compounds.
General Surface Plasmon Resonance Procedure
The PARP SPR buffer was 50 mM HEPES (pH = 7.5), 100 mM NaCl, 1 mM TCEP, and 0.05% Tween 20, and assays were run at 25 °C. PARPs were captured either via N-terminal His6 tags on CM5 Ni-NTA sensor chips or via N-terminal biotinylated Avi tags on streptavidin SA sensor chips (GE Healthcare Life Sciences). The constructs used in the SPR assay are listed in Table 1 . Typically, 3000–6000 RU of protein was immobilized for each assay. Compounds were screened using a flow rate of 30 µL/min, using 60 s of association time, typically followed by observing dissociation for 90 s. For higher-affinity compounds, the dissociation window was increased, and in some instances compound binding was analyzed using single-cycle kinetics. Solvent correction was applied using a six-point curve from 1.5% to 2.75% DMSO injected before and after every 96 cycles. Data were normally fit to an equilibrium binding model to derive the binding constant KD, and when significant off-rates were observed, data were fit to a kinetic model to derive parameters for association and dissociation (ka and kd) and the binding constant Kd.
Results
Determining Conditions That Activate Enzymes to Perform ADP-Ribosylation
Several commercially available proteins and/or synthesized peptides corresponding to proteins reported to be substrates of PARP7,25,33,34 PARP14,35–37 and PARP16
38
were tested as substrates under a variety of conditions. We looked for the incorporation of 32P-NAD+ or biotinylated-NAD+ using gel-, far-Western blot-, scintillation proximity counting-, or microplate-based readouts, but did not observe any signal above background under multiple reaction conditions. In contrast, we observed robust activity of PARP1 and PARP2 on purified histones, which are reported to be substrates for those enzymes
39
(e.g., see
PARP enzymes are reported to self-ADP-ribosylate; however, in the literature high concentrations of proteins have been needed to observe this activity. In the work of Vyas et al.,
2
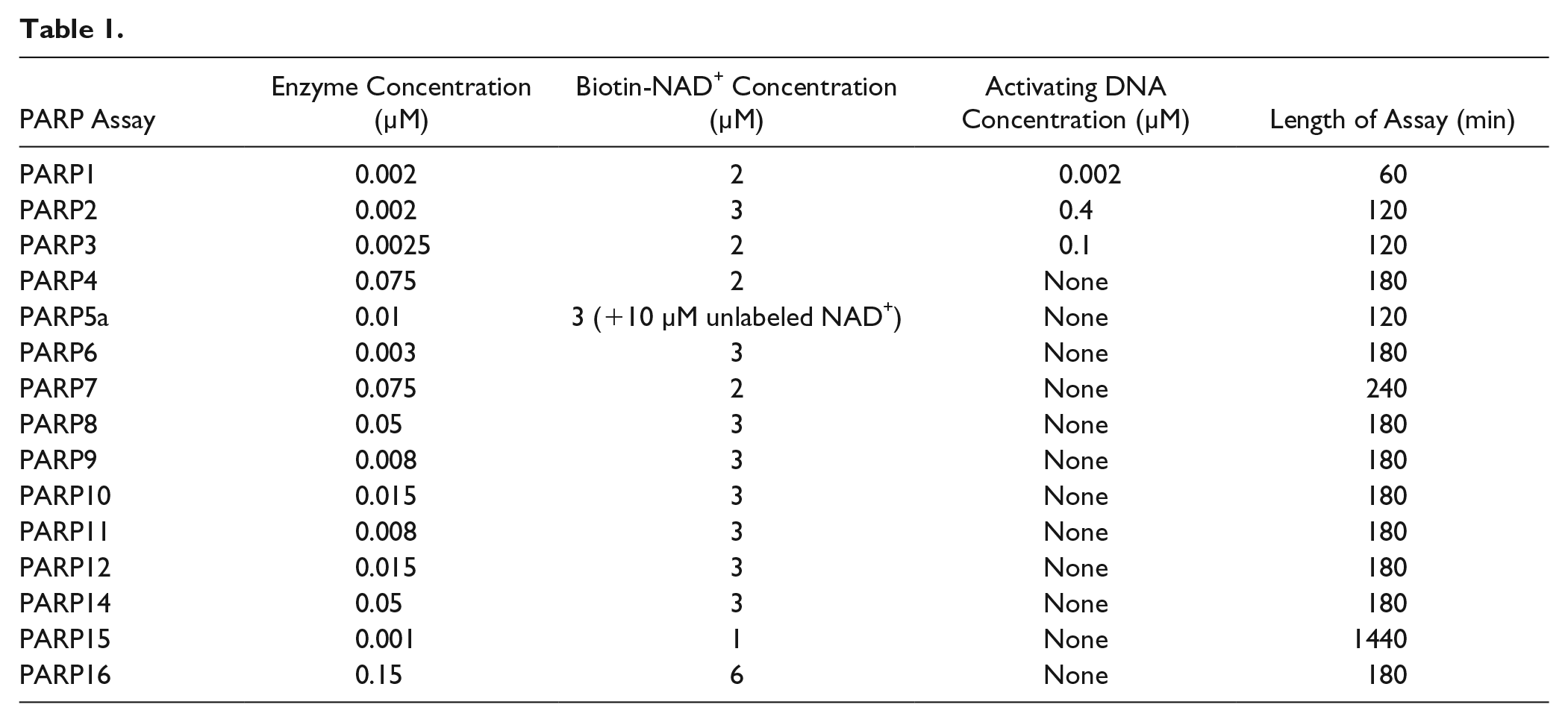
self-ADP-ribosylation of nearly all monoPARP and polyPARP enzymes was observed on immunoprecipitated enzymes that were immobilized on beads and incubated with 32P-NAD+. To recapitulate this condition, recombinant purified His6-tagged PARP7, PARP14, and PARP16 enzymes were immobilized on Ni-NTA microplates, incubated with biotinylated-NAD+, and then an enzyme-linked immunosorbent assay (ELISA) readout was used to detect incorporation of biotinylated-MAR. The plates were washed prior to detection to remove the unincorporated biotinylated-NAD+ to avoid interfering with the streptavidin-based detection. Parallel reactions were run where the enzymes were incubated with biotinylated-NAD+, followed by Ni-NTA capture and ELISA detection. We observed robust activity for the monoPARPs only in conditions where the enzyme was immobilized before addition of the biotinylated-NAD+; however, PARP1 was active in both scenarios (
Immobilization of monoPARPs forces a self-modification enzymatic reaction. (
Development of Self-Modification Assays for Screening Inhibitors of the PARP Family
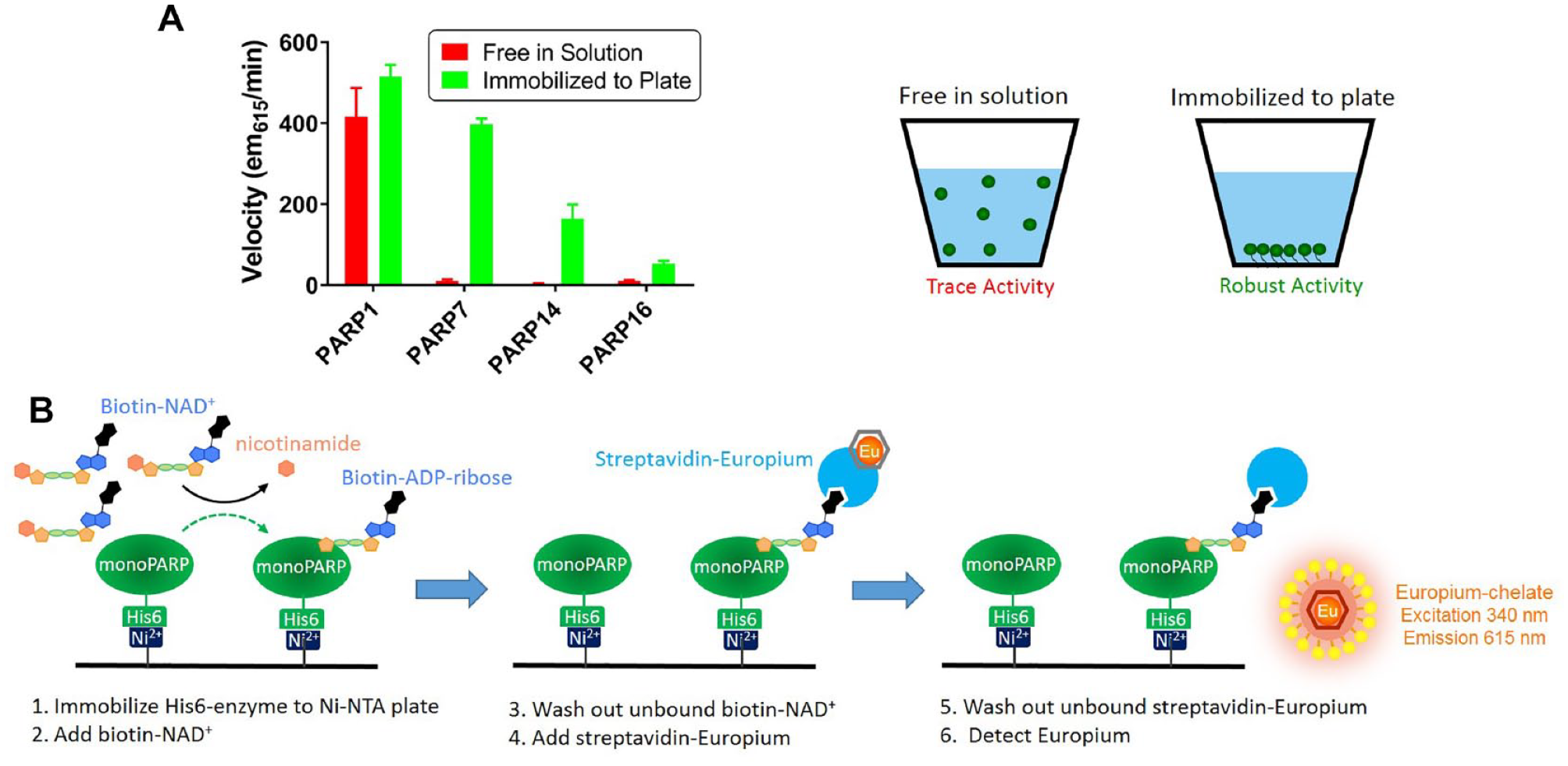
Based on the observation that immobilization led to forced self-ADP-ribosylation of PARP7, PARP14, and PARP16, we postulated that this strategy could be a family-wide solution to developing monoPARP enzyme assays without knowledge of their physiologically relevant substrates. We then subjected all reported enzymatically active monoPARP and polyPARP enzymes to assay development beginning with recombinant protein that was at least 80% pure (in most cases, purity exceeded 95%). Multiple assay development steps were performed to generate robust assays that would be sensitive to inhibitors of all mechanisms of inhibition. An example of this assay development process is demonstrated for PARP16 in
Figure 2
and for the remaining PARPs in
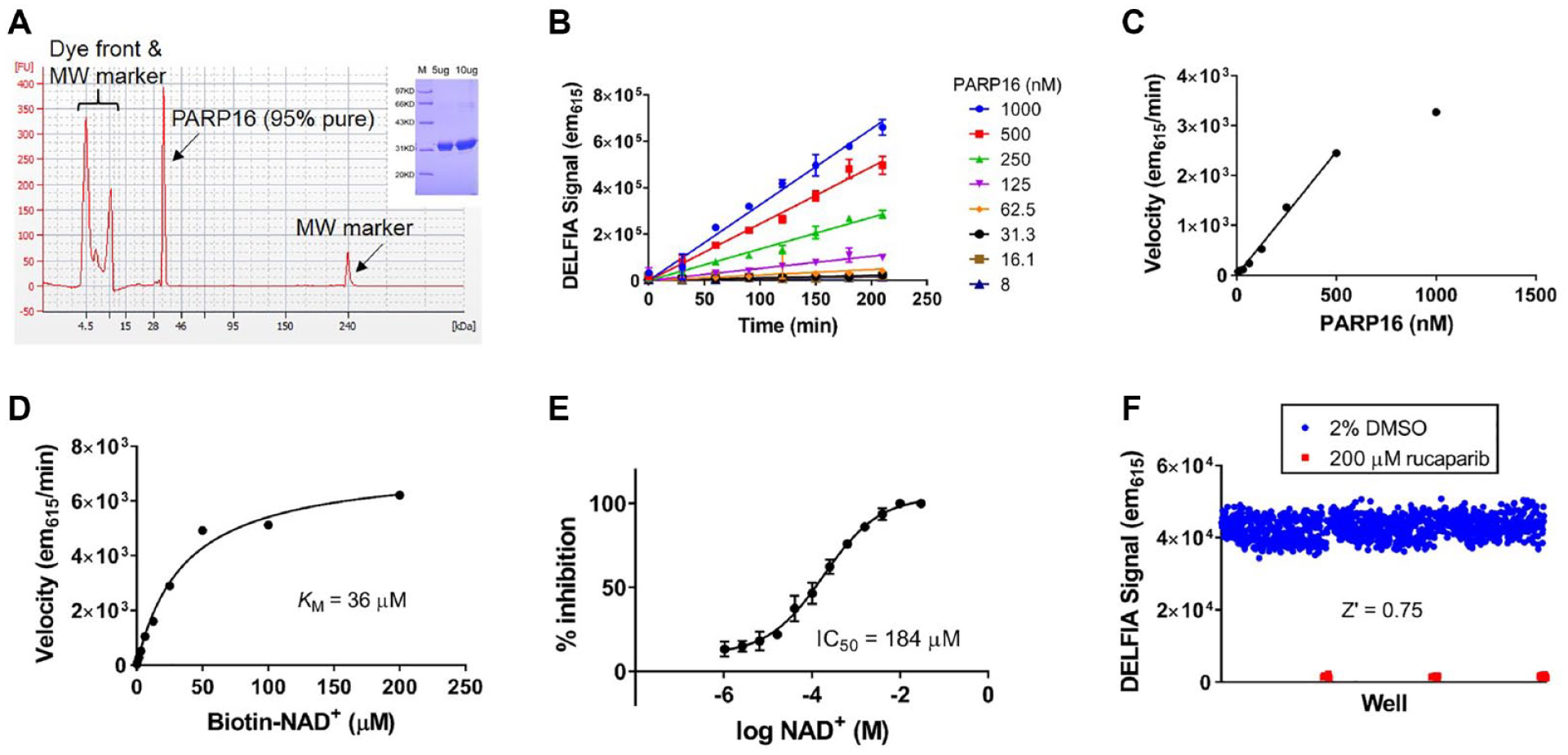
Assay development of PARP16 using the strategy of immobilizing enzyme to force self-modification. (
Enzyme assays were calibrated by determining the IC50 values of six key literature PARP1 inhibitors (structures shown in
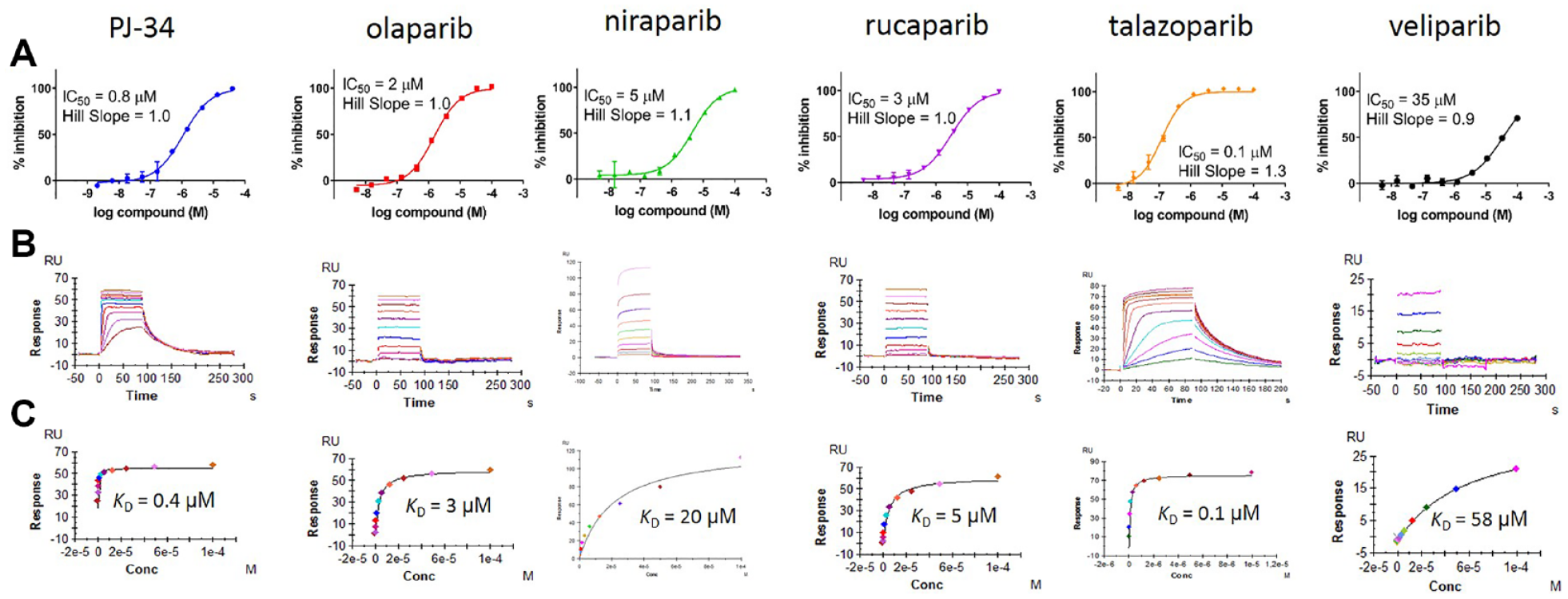
Testing PARP1 literature inhibitors versus PARP16. (
Development of an Assay Measuring the ADP-Ribosylation of Ubiquitin by the PARP9/DTX3L Complex
PARP9 is a macrodomain-containing PARP that heterodimerizes with DTX3L, an E3 ligase. Based on sequence alignments of its PARP domain, it has been predicted to be catalytically inactive.
40
Empirical testing of this prediction via self-modification reactions has not shown PARP9 to possess enzymatic activity,
2
and we have confirmed this ourselves (data not shown). However, a recent report was published showing that the PARP9/DTX3L complex MARylates the C-terminal carboxy terminus of ubiquitin when incubated with an E1 activating enzyme, an E2 conjugating enzyme, and ATP. We were able to confirm this activity using biotin-NAD+ to modify the ubiquitin and a far-Western blot readout to detect the incorporation of biotinylated-MAR (
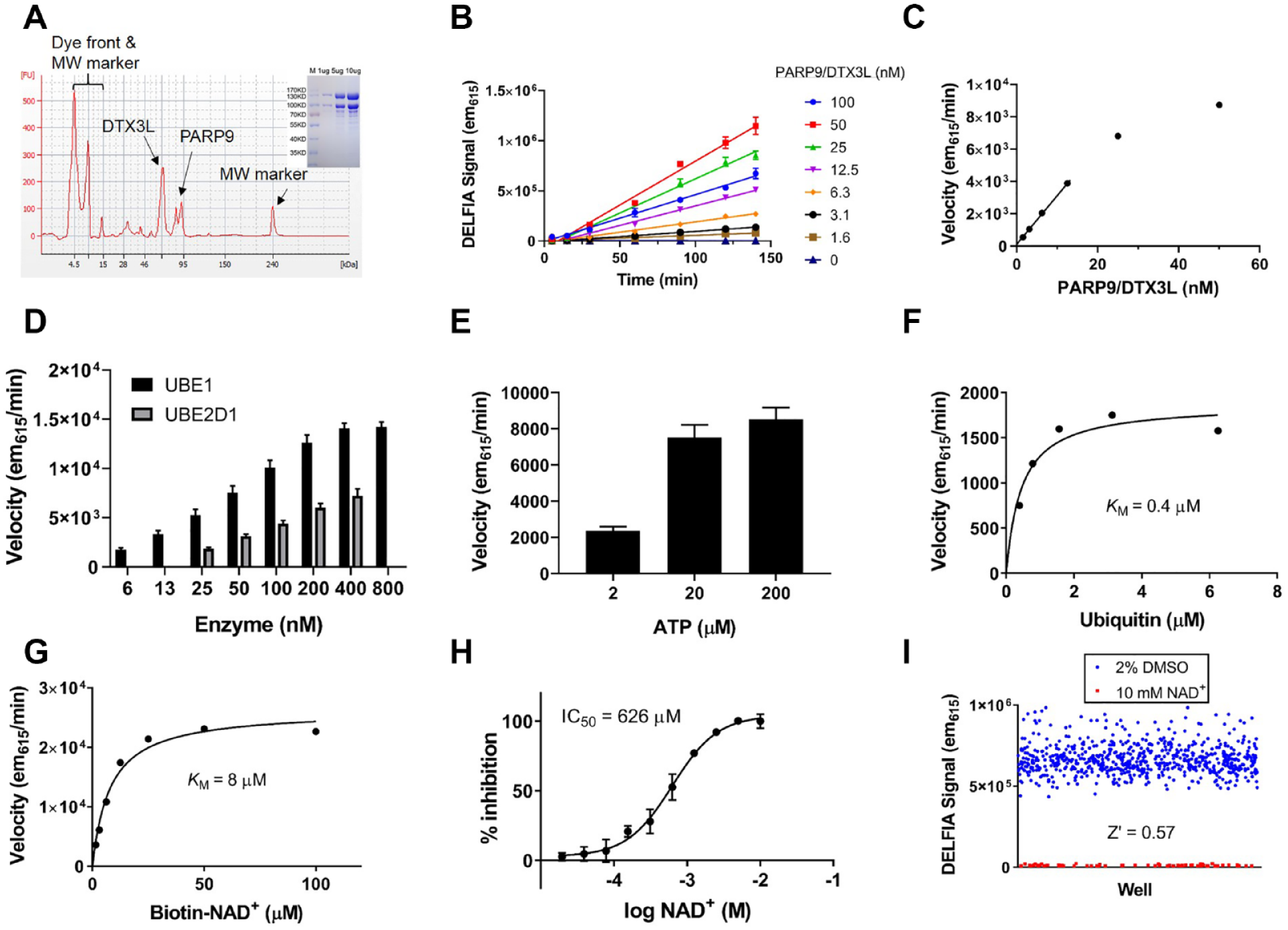
Assay development to follow ubiquitin MARylation by the PARP9/DTX3L complex. (
Counterscreen Assay to Identify Compounds That Bind Nickel
Since the capture of His6-tagged PARP enzymes on Ni-NTA microplates is critical to observing enzymatic activity, compounds containing pharmacophores capable of binding to the nickel on the Ni-NTA microplates can lead to undesirable inhibition of the DELFIA signal. To quickly counterscreen compounds for this behavior, we developed a counterscreen assay examining the displacement of a biotinylated His6 peptide from the Ni-NTA microplates; the overall scheme for this assay is shown in
Screening a Collection of PARP-Focused Compounds across All PARP Enzymes
The PARP assays were used to generate potency and selectivity information across the entire enzyme family for a selection of literature PARP1 inhibitors and Ribon’s internal collection of >4000 PARP-focused inhibitors. Compounds were screened in duplicate using a single concentration screening paradigm, and selected compounds were further followed up with dose–response titrations to generate IC50 values. A correlation plot of n = 1 and n = 2 screening data for PARP16 (
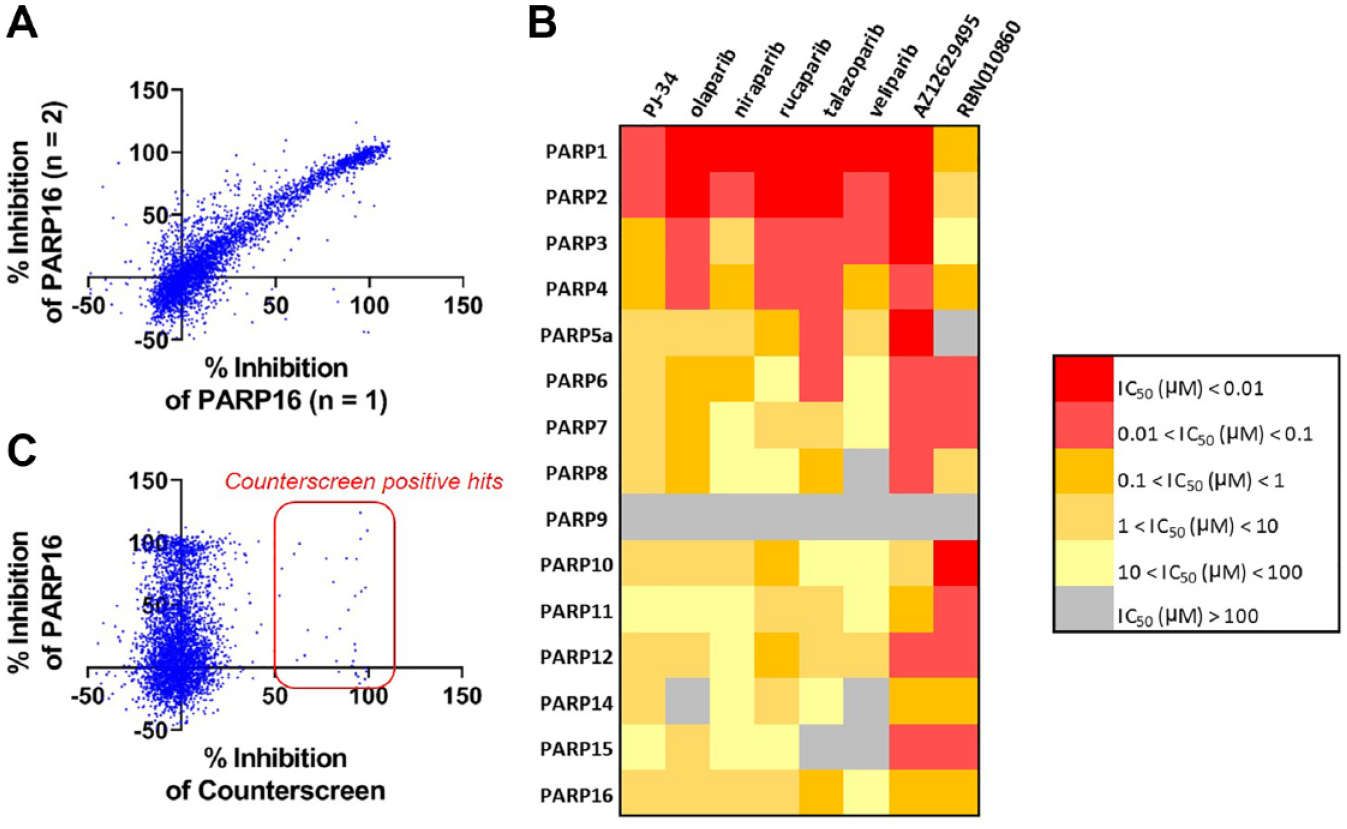
Screening a library of PARP inhibitors against PARP16. (
Discussion
ADP-ribosylation is increasingly being recognized for playing a key role in stress response pathways in the context of immune function, inflammation, and cancer.12–14 The four known polyPARP enzymes have been extensively studied, and in the case of PARP1 and PARP2, Food and Drug Administration (FDA)-approved cancer drugs targeting those enzymes exist; however, much less is known about the 12 monoPARP enzymes. The lack of a clear understanding of how the monoPARP enzymes interact with their substrates has been a barrier to developing robust high-throughput assays that would enable a family-wide approach to generating chemical probes to study the function of these enzymes. We tested the ability of multiple monoPARPs to MARylate their published substrates by incubating recombinant proteins or synthetic peptides in the presence of 32P-NAD+ or biotinylated-NAD+. In most cases, we did not observe any detectable incorporation of MAR and determined that another paradigm to screening PARP enzymes was needed for this protein family.
PARP enzymes are noted to modify themselves with MAR; however, when we attempted to perform self-modification reactions using radioactive and ELISA-based formats, many PARPs showed little to no measurable activity and assay development was not possible. We discovered that when PARPs are immobilized via His6 tags onto Ni-NTA-coated microplates, we were able to observe robust self-modification that was detectable using a DELFIA readout. We hypothesize that immobilization of the PARPs on the microplate surface induces molecular crowding, which overcomes weak KM values for these interactions to generate detectable levels of self-modification. DELFIA assays are a variation of an ELISA that relies on lanthanide fluorescence to detect incorporation of a biotinylated ADP-ribose moiety. We compared DELFIA to other ELISA formats such as colorimetric, fluorometric, and chemiluminescence, and DELFIA gave the most reproducible data with the highest Z′ factors. Using this approach, we were able to develop robust self-modification assays for nearly all monoPARPs and polyPARPs. Ultimately, immobilization as a means to force self-modification may be a useful approach in the development of assays for targets where substrate information is lacking.
Notable exceptions to the paradigm of using self-modification reactions to generate a screening assay were observed for PARP5a and the PARP9/DTX3L complex. In the case of PARP5a, we found that the enzyme was able to PARylate immobilized histones, and we developed DELFIA screening assays with a similar detection workflow to the self-MARylation assays on histone-coated microplates. PARP9 and DTX3L, an E3 ligase, are located on chromosome 3q21 in a head-to-head orientation and are regulated by an IFN-γ responsive bidirectional promoter.
41
Those proteins are always found in a heterodimer and we were not able to express and purify PARP9 without co-expressing DTX3L and purifying as a complex. Recently, it was shown that the PARP9/DTX3L complex MARylates the free C-terminal carboxylic acid on glycine 76 of ubiquitin only under conditions where the E2 enzyme is charged with ubiquitin.
28
Since glycine 76 is normally used to conjugate ubiquitin to proteins, this modification precludes this ubiquitination in an NAD+-dependent manner. We were able to confirm these findings using a far-Western blot to visualize the ADP-ribosylation of ubiquitin and subsequently developed a microplate-based assay where the enzymatic reaction is run with all components free in solution followed by capture and detection of the ADP-ribosylated products. The PARP9/DTX3L reaction specific activity is several orders of magnitude larger than any self-modification reaction using immobilized PARP enzymes and underscores the need to perform further studies to identify monoPARP substrates (
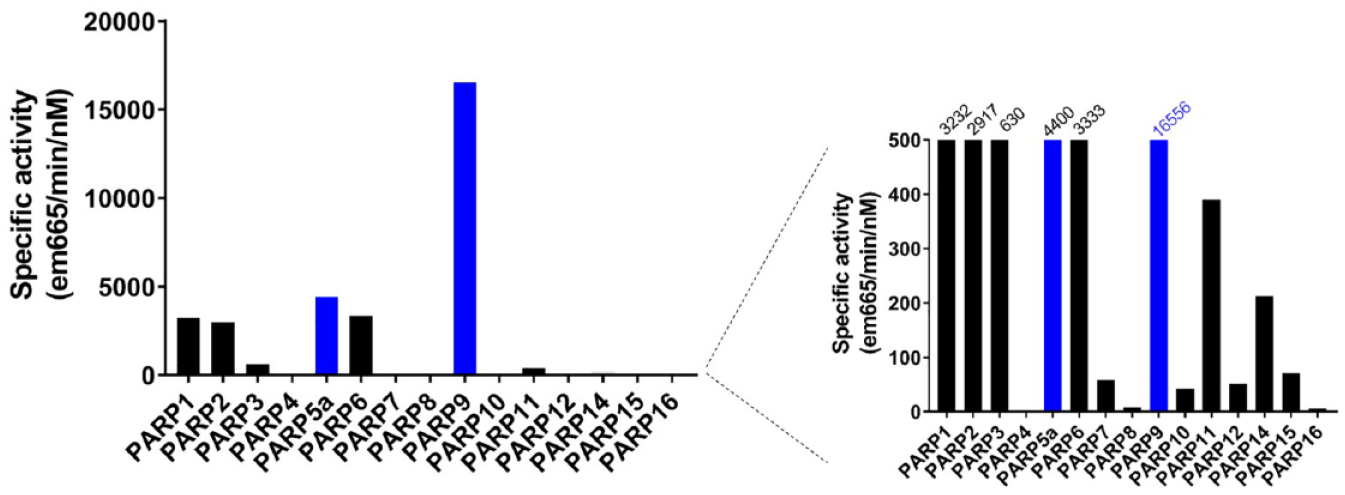
Comparing specific activities of PARP enzymes under final assay conditions. The specific activity for each enzyme assay was calculated. The forced self-modification of immobilized enzymes (black bars) overall showed lowered specific activity than assays where an enzyme and its substrate were allowed to react freely in solution (blue bars).
Using assays generated for nearly the entire PARP family, we are able to rapidly and efficiently screen inhibitors in parallel to identify hits for each PARP as well as to build structure–activity relationships within the family. The ability of the assays to accurately assess inhibitor potency was validated by generating SPR assays for all PARPs and comparing IC50 values from the enzyme assays generated at KMapp or as close to KMapp as possible. The fact that SPR assays could be generated for nearly all PARPs tested also indicates the amenability of these proteins to biophysical approaches for hit identification, such as fragment screening. The strategies presented here are being used to generate potent and selective chemical probes for each family member to enable interrogation of the role of their catalytic activity in disease biology, and ultimately develop targeted therapeutics.
Supplemental Material
Wigle_DELFIA_and_automodification_Supp_Info_Revised – Supplemental material for Forced Self-Modification Assays as a Strategy to Screen MonoPARP Enzymes
Supplemental material, Wigle_DELFIA_and_automodification_Supp_Info_Revised for Forced Self-Modification Assays as a Strategy to Screen MonoPARP Enzymes by Tim J. Wigle, W. David Church, Christina R. Majer, Kerren K. Swinger, Demet Aybar, Laurie B. Schenkel, Melissa M. Vasbinder, Arne Brendes, Claudia Beck, Martin Prahm, Dennis Wegener, Paul Chang and Kevin W. Kuntz in SLAS Discovery
Footnotes
Acknowledgements
We thank Mr. Phil Anwer for his efforts to obtain the custom Ni-NTA-coated microplates, Mr. Andrew Santospago for preparing compound plates, and Dr. Heike Keilhack and Dr. Victoria Richon for helpful discussions and critical review of this manuscript. Additionally, we are grateful to Ping Wei and her team at Viva Biotech and Hara Black and her team at Evotec SE for their contributions to protein production efforts.
Supplemental material is available online with this article.
Declaration of Conflicting Interests
The authors declared the following potential conflicts of interest with respect to the research, authorship, and/or publication of this article: T.J.W., W.D.C., C.R.M., K.K.S., D.A., L.B.S., M.M.V., P.C., and K.W.K. are current or former employees and stockholders of Ribon Therapeutics Inc.; A.B., C.B., M.P., and D.W. are employees of Evotec SE.
Funding
The authors received no financial support for the research, authorship, and/or publication of this article.
References
Supplementary Material
Please find the following supplemental material available below.
For Open Access articles published under a Creative Commons License, all supplemental material carries the same license as the article it is associated with.
For non-Open Access articles published, all supplemental material carries a non-exclusive license, and permission requests for re-use of supplemental material or any part of supplemental material shall be sent directly to the copyright owner as specified in the copyright notice associated with the article.