
Abstract
The rapid rise in the emergence of multidrug-resistant (MDR) and extensively drug-resistant (XDR) strains of Mycobacterium tuberculosis (Mtb) mandates the discovery of novel tuberculosis (TB) drugs. Mur enzymes, which are identified as essential proteins in Mtb and catalyze the cytoplasmic steps in the peptidoglycan biosynthetic pathway, are considered potential drug targets. However, none of the clinical drugs have yet been developed against these enzymes. Hence, the aim of this study was to identify novel inhibitors of Mur enzymes in Mycobacterium tuberculosis. We screened an antitubercular compound library of 684 compounds, using MurB and MurE enzymes of the Mtb Mur pathway as drug targets. For experimental validation, the top hits obtained on in silico screening were screened in vitro, using Mtb Mur enzyme-specific assays. In all, seven compounds were found to show greater than 50% inhibition, with the highest inhibition observed at 77%, and the IC50 for these compounds was found to be in the range of 28–50 μM. Compound 5175112 showed the lowest IC50 (28.69 ± 1.17 μM), and on the basis of (1) the binding affinity, (2) the stability of interaction noted on molecular dynamics simulation, and (3) an in vitro assay, MurE appeared to be its target enzyme. We believe that the overall strategy followed in this study and the results obtained are a good starting point for developing Mur enzyme-specific Mtb inhibitors.
Introduction
The increase in the number of multidrug-resistant (MDR) and extensively drug-resistant (XDR) strains of Mycobacterium tuberculosis (Mtb) is a major cause of concern and highlights the need to discover new and more effective drugs. One-third of the world’s population is infected with Mtb, and approximately 10.4 million people are affected with tuberculosis (TB). 1 Despite the availability of vaccines and effective drug therapy, Mtb continues to claim more lives than any other single infectious agent. Therefore, there is an urgent requirement to find and develop novel drug candidates against untapped target enzymes in the pathogen.
Currently available TB drugs inhibit enzymes of various metabolic pathways in Mtb.
2
Isoniazid and ethambutol inhibit the mycolic acid synthetic pathway and pyrazinamide inhibits the fatty acid synthetic pathway in Mtb. Expanding the information on enzymes that catalyze metabolic pathways that are essential for the survival of the bacterium but have not yet been explored in the drug discovery process has its advantages.
3
Peptidoglycan (PG) is the key component of the cell wall, which is present outside the cytoplasmic membrane in almost all bacteria,
4
and so far, has not been targeted by the clinically approved TB drugs. PG’s primary function is to preserve cell integrity by withstanding osmotic pressure; additionally, it also contributes to the maintenance of a defined cell shape and serves as a platform for anchoring other cell envelope components.
5
Its biosynthesis is a complex process and takes place in the cytoplasm as well as in the cell membrane. The cytoplasmic steps in the biosynthetic pathway of PG involve the synthesis of uridine diphosphate-N-acetylmuramyl pentapeptide (UDP-MurNAc-pentapeptide) catalyzed by the Mur enzymes. Mur enzymes are highly conserved among bacteria and have no counterparts in eukaryotes. They catalyze the formation of UDP-MurNAc-pentapeptide in two stages: The first step in the Mur pathway is catalyzed by MurA, where the enolypyruvate moiety from phosphoenolpyruvate (PEP) is transferred to UDP-N-acetylglucosamine (UDP-GlcNAc). Subsequently, MurB catalyzes the reduction of the enolypyruvate group to a lactoyl moiety using β-nicotinamide adenine dinucleotide phosphate (NADPH) as the cofactor and forms UDP-N-acetylmuramic acid (UDP-MurNAc). In the next four steps, Mur ligases (MurC-MurF) catalyze the sequential addition of amino acids
Hitherto, most of the available drugs were discovered by screening assays that used one enzyme as the target protein in a pathogen. With this approach, the odds of gain of resistance to such drugs by the bacterium are very high.9,10 In contrast, a microorganism would take longer to develop resistance against a molecule that can inhibit more than one of its essential proteins. Hence, finding a drug molecule that has multiple targets within the pathogen would not only require robust computational screening but also mandate the development of an assay method, where multiple enzymes (potential targets) can function optimally in a single reaction. To test this hypothesis and in order to establish the potential of Mur enzymes as drug targets, a one-pot assay that reconstructs the Mtb Mur pathway in vitro has been developed by us. 11 The assay offers the advantage of carrying out the activity of six enzymes (Mtb MurA-MurF) in a single reaction and hence is conducive for the screening and identification of molecules that could possibly disrupt more than one of the Mur enzymes. Using this assay, we found furan-based benzene mono- and dicarboxylic acid derivatives and benzene-1,3-dicarboxylic acid 2,5-dimethylpyrrole derivatives (previously tested for their inhibitory effects on Mur ligases from Escherichia coli11–14) to inhibit two Mur ligases (MurE and MurF) in Mtb. Since the crystal structures of Mur ligases (MurC-MurF) from E. coli15–18 and other bacteria19,20 were solved, we can find several reports on inhibitors against them.12,13,21–28 In Mtb, however, most of the reported inhibitors are against the MurE enzyme since the crystal structure of only this Mur ligase is available in Mtb.28–33 Only recently has the structure of Mtb MurB been solved, 34 and therefore reports on its inhibitors are few. 35
In the present study, a set of 684 antitubercular compounds, obtained from the Council of Scientific and Industrial Research—Indian Institute of Integrative Medicine (CSIR-IIIM) repository, was screened to identify Mur inhibitors in Mtb. We first screened the compounds in silico by docking them against MurB and MurE as the target proteins and then further analyzed their effect in vitro, using the one-pot assay for Mtb Mur enzymes. The IC50 value was determined for compounds showing an inhibitory effect greater than 50% on the assay. The inhibitor showing the lowest IC50 value was then selected for molecular dynamics (MD) simulation studies.
Materials and Methods
Chemicals and Reagents
Uridine 5′-diphospho-N-acetylglucosamine (UDP-GlcNAc), PEP, adenosine triphosphate (ATP), NADPH, Bis-Tris propane, and amino acid substrates (
Screening of Antitubercular Compounds
A set of 684 antitubercular compounds was used in this study. These compounds were obtained on whole-cell (Mtb H37Rv) screening (
Preparation of Protein Structure
Structures of Mtb MurB 36 and MurE 37 were retrieved from the Protein Data Bank (PDB) (https://www.rcsb.org/pdb/home/home.do) and used for docking studies. Structures were prepared using MGLTools software (The Scripps Research Institute) by (1) adding hydrogen and Gasteiger charges, (2) removing water, and (3) minimizing the energy using the UCSF Chimera program.
Grid Generation and In Silico Screening
All 684 compounds were independently screened against Mtb MurB (PDB ID: 5JZX) and MurE (PDB ID: 2WTZ) enzymes using the AutoDock Vina script for multiple compounds. Rigid docking was performed with the 40, 40, 40 dimension of the grid, with a spacing of 0.375 Å, centered on the active site residues of each Mur enzyme. Vina predicted the binding mode of the ligands and binding affinity was observed in kilocalories per mole. The threshold cutoff score for docking was set on the basis of the binding affinity scores of Mur enzymes with their respective UDP sugar nucleotide substrates. The binding affinity of UDP-GlcNAc enolpyruvate for MurB and of UDP-N-acetylmuramoyl-
In Vitro Screening by One-Pot Assay
The top hits from in silico screening were screened for their in vitro effect using the one-pot assay developed for Mtb Mur enzymes, 11 where each compound was tested at a fixed concentration of 50 μM. Each reaction was carried out in triplicate in microtiter plates. Briefly, Mur enzymes (A-F) were mixed together and were preincubated with each inhibitor for 15 min at room temperature, followed by the addition of other reaction components to initiate the reaction. Assays were carried out for 30 min at 37 °C and the absorbance was read at 630 nm to estimate the release of inorganic phosphate (Pi) using the Pi ColourLock Kit. Positive control reactions were carried out without the inhibitors.
After this preliminary screening, active compounds (showing ≥50% inhibition at 50 μM concentration) were then evaluated for determining IC50 values. Each compound at concentrations in the range of 10–50 μM was preincubated with enzymes for 15 min, followed by the addition of the remaining constituents of the assay. The final reaction mixture was incubated for 30 min at 37 °C and the absorbance was read at 630 nm to estimate the release of Pi using the Pi ColourLock Kit. Assays with the inhibitors were done in triplicate and the percentage inhibition at each concentration was calculated. IC50 values were determined by nonlinear regression analysis using GraphPad Prism software (GraphPad Software, La Jolla, CA).
For studying the inhibitory activity of compound 5175112 against MurE, a sequential coupled assay was carried out as described previously. 11 Briefly, after the completion of MurA to MurD coupled reactions, to the reaction mix, MurE preincubated with compound 5175112 (at a concentration range of 10–50 μM) and 1 mM meso-diaminopimelic acid were added and the final reaction was incubated for 30 min at 37 °C. The control reaction contained all the components except the MurE enzyme. In the “no inhibitor” control reaction, the organic solvent (DMSO) was added in place of the inhibitor at 5% concentration. The net Pi released was calculated as described earlier. 11
MD Simulations of the Protein–Inhibitor Complexes
MD simulation incorporates Newton’s laws of motion to understand the time-dependent behavior of the molecular system. The best fitted conformations were subjected to MD simulation using GROMACS 4.5.3. Ligand topology files were generated using PRODRG. MD simulation was carried out for the protein and protein–ligand complex. MD simulation was performed up to 10, 20, and 40 ns using GROMOS53a6.ff all atom force field. After neutralizing the system, energy minimization was performed using the steepest descent method for the relaxation of the initial solvent as well as elimination of any residual strain. Temperature and pressure coupling were performed using a b\Berendsen thermostat and the Parrinello–Rahman method, respectively. For understanding the ligand stability in the binding pocket of respective proteins, we plotted RMSD values of backbone atoms for the protein and protein–ligand complex.
Results
The screening methodology adopted in this study for the identification of hit molecules was a combination of a phenotypic (whole-cell-based) screen and target (Mur enzyme)-based approach (in silico and in vitro), as depicted in Figure 1 .
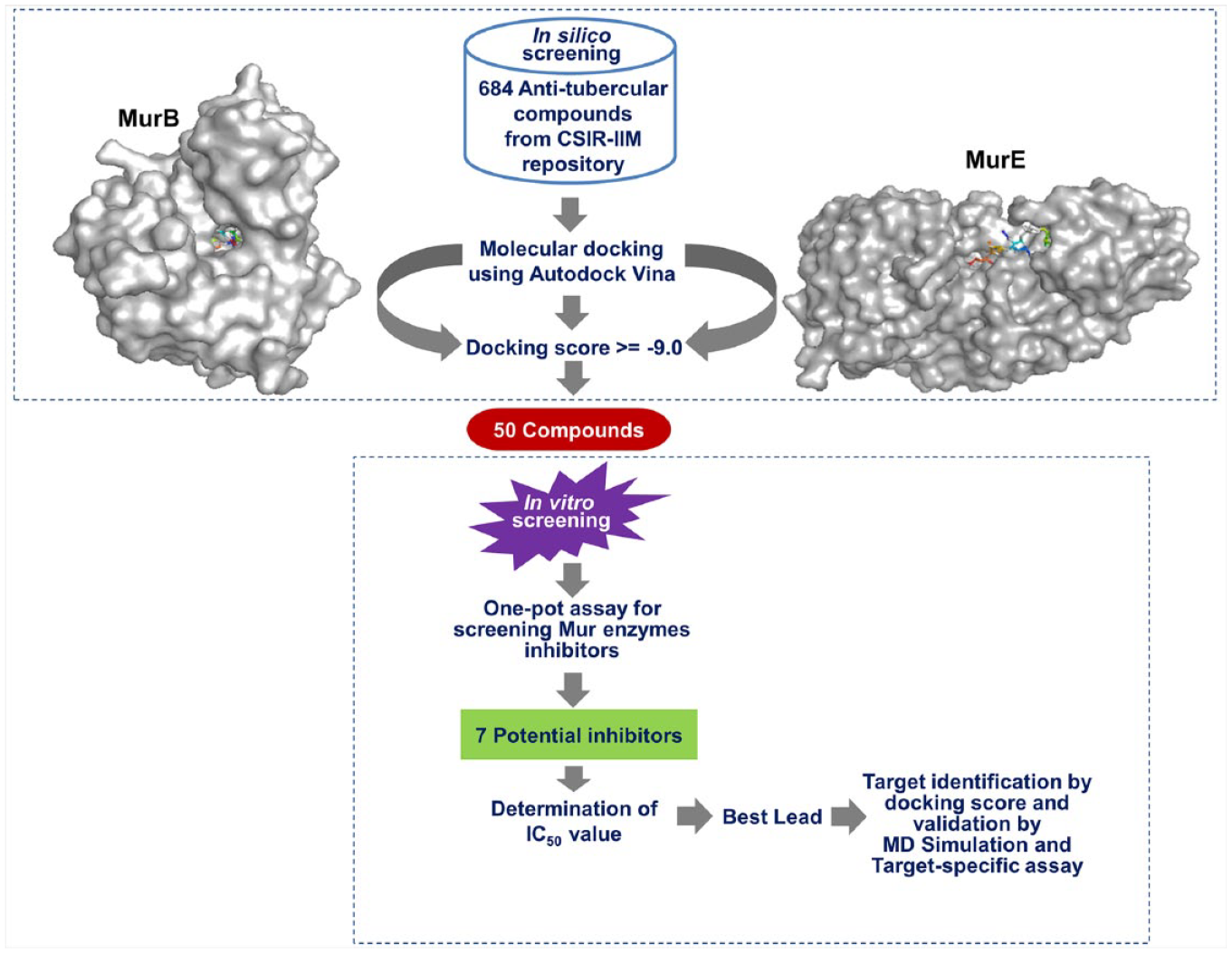
Schematic representation of the steps adopted in the study for the identification of potential inhibitors of Mur enzymes of Mtb.
Molecular Docking
Molecular docking for the set of 684 antitubercular compounds was performed for both MurB and MurE enzymes. On in silico screening, 50 compounds (
In Vitro Screening
The in vitro evaluation of 50 inhibitors (at 50 μM concentration each) by the one-pot assay resulted in the identification of seven compounds that showed more than 50% inhibition of the assay (the remaining compounds showed a 20%–40% inhibitory effect [data not shown]). Out of these seven compounds, five compounds with IDs 5174183, 5175112, 5212357, 5213336, and 5238312 were found to show 63.26%, 76.99%, 55.89%, 70.15%, and 54.59% inhibition, respectively ( Table 1 ). Significantly, we found these hits to be novel compounds since none of them have previously been reported against a Mur enzyme of any pathogen.
List of Compounds Showing More than 50% Inhibition of the One-Pot Assay.
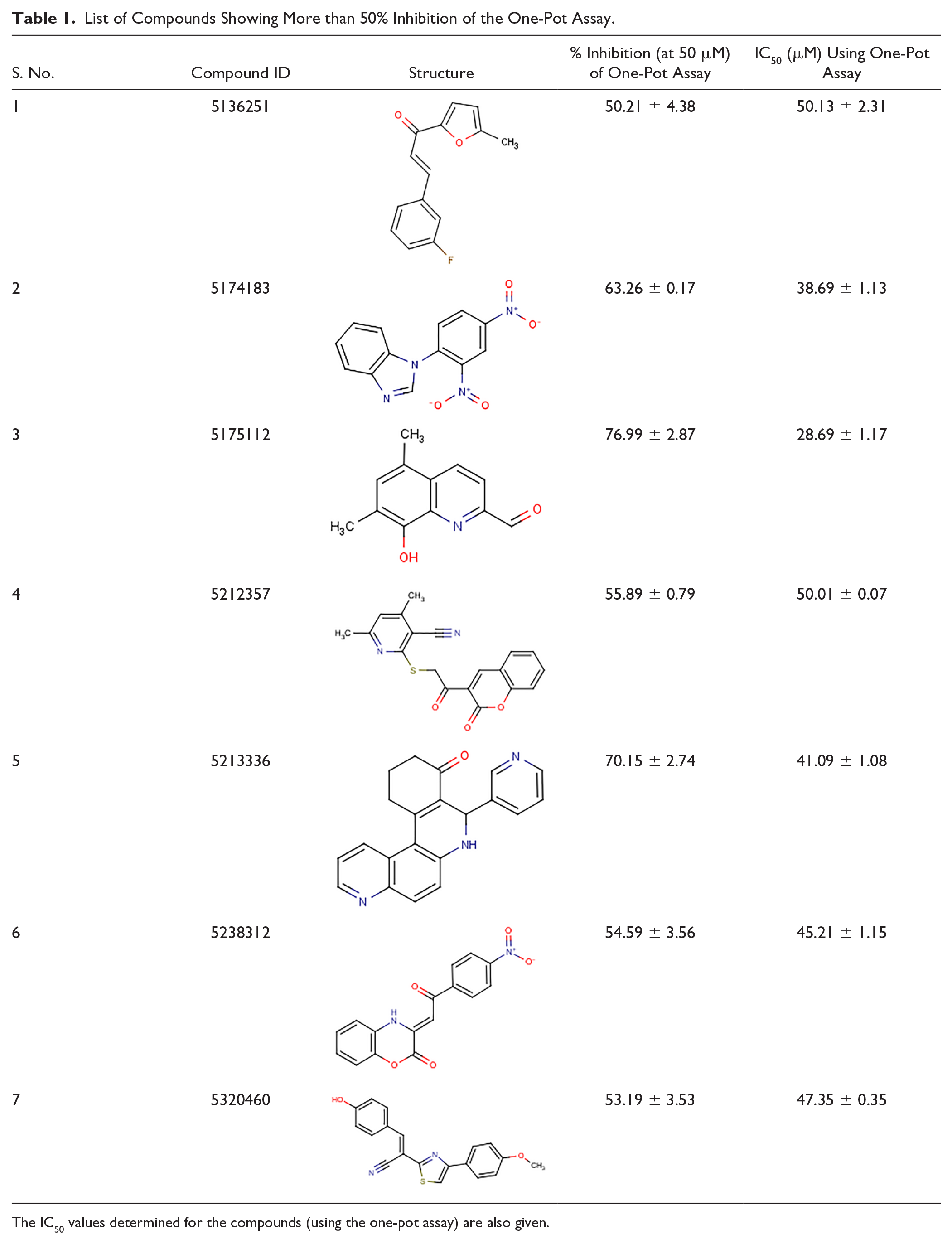
The IC50 values determined for the compounds (using the one-pot assay) are also given.
IC50 Determination
IC50 values of the compounds were found to be in the range of 28–50 μM (
Table 1
), with the lowest value shown by the compound with ID 5175112 (28.69 ± 1.17 μM). On the basis of the docking score (–9.6 kcal/mol), MurE was identified as the target enzyme for this compound (
MD Simulation
MD simulation was carried out up to 40 ns and the stability of the protein–ligand complex, as well as per-residue fluctuations in protein, was examined by evaluating the root mean square deviation (RMSD) and root mean square fluctuation (RMSF), respectively. RMSD values of the protein–inhibitor complex and those of the native protein were compared to check whether the ligand remains bound to the active site or drifts away during simulation. Our analysis, as depicted in
Figure 2A
, illustrates lower RMSD values, thus indicating compound 5175112 to have occupied the active site pocket of MurE during simulation. On observing the RMSF of terminal regions of the complex (
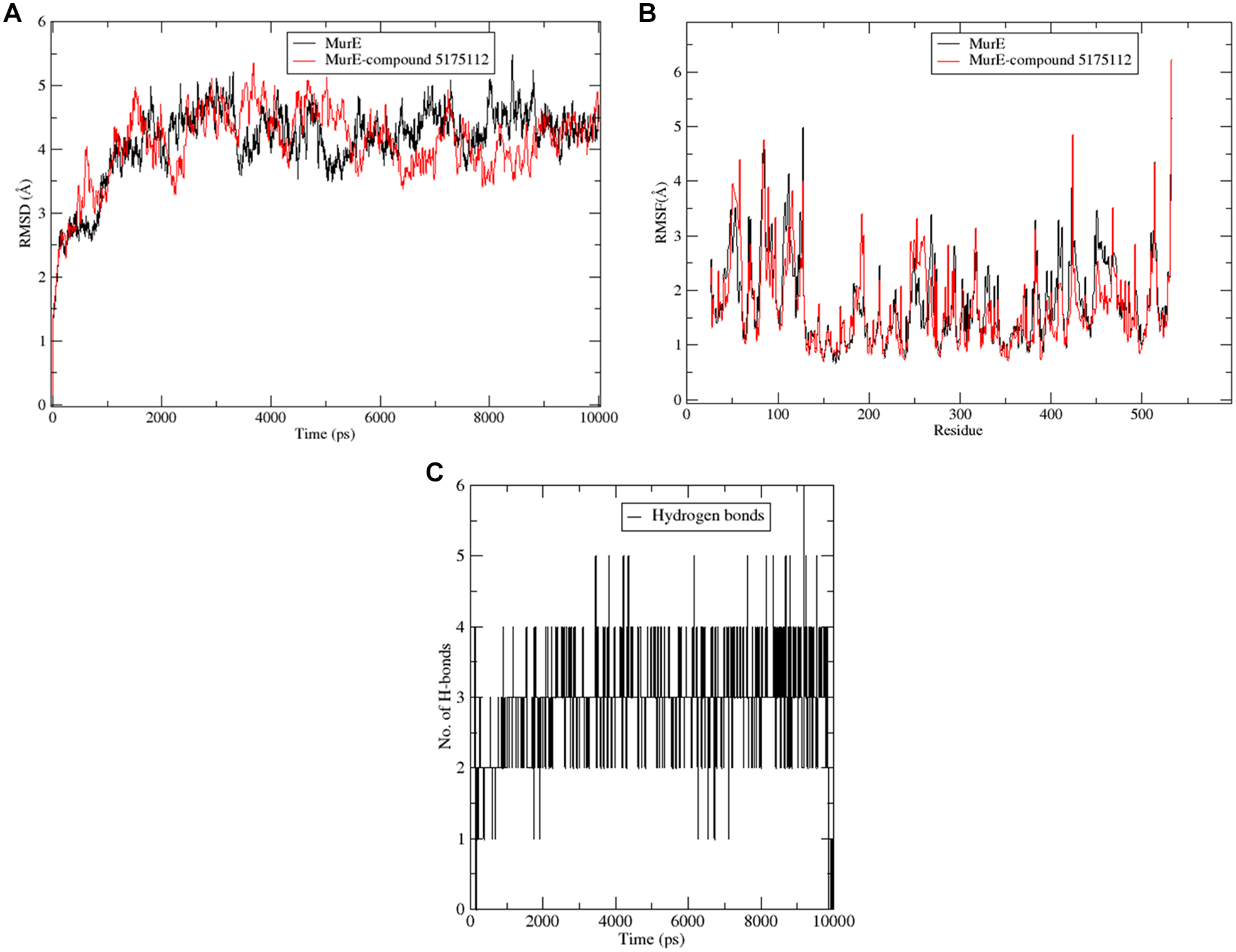
Exploration of the MD simulation results of the protein complex with compound 5175112. (
We further studied binding of 5175112 with MurE at different time frames (
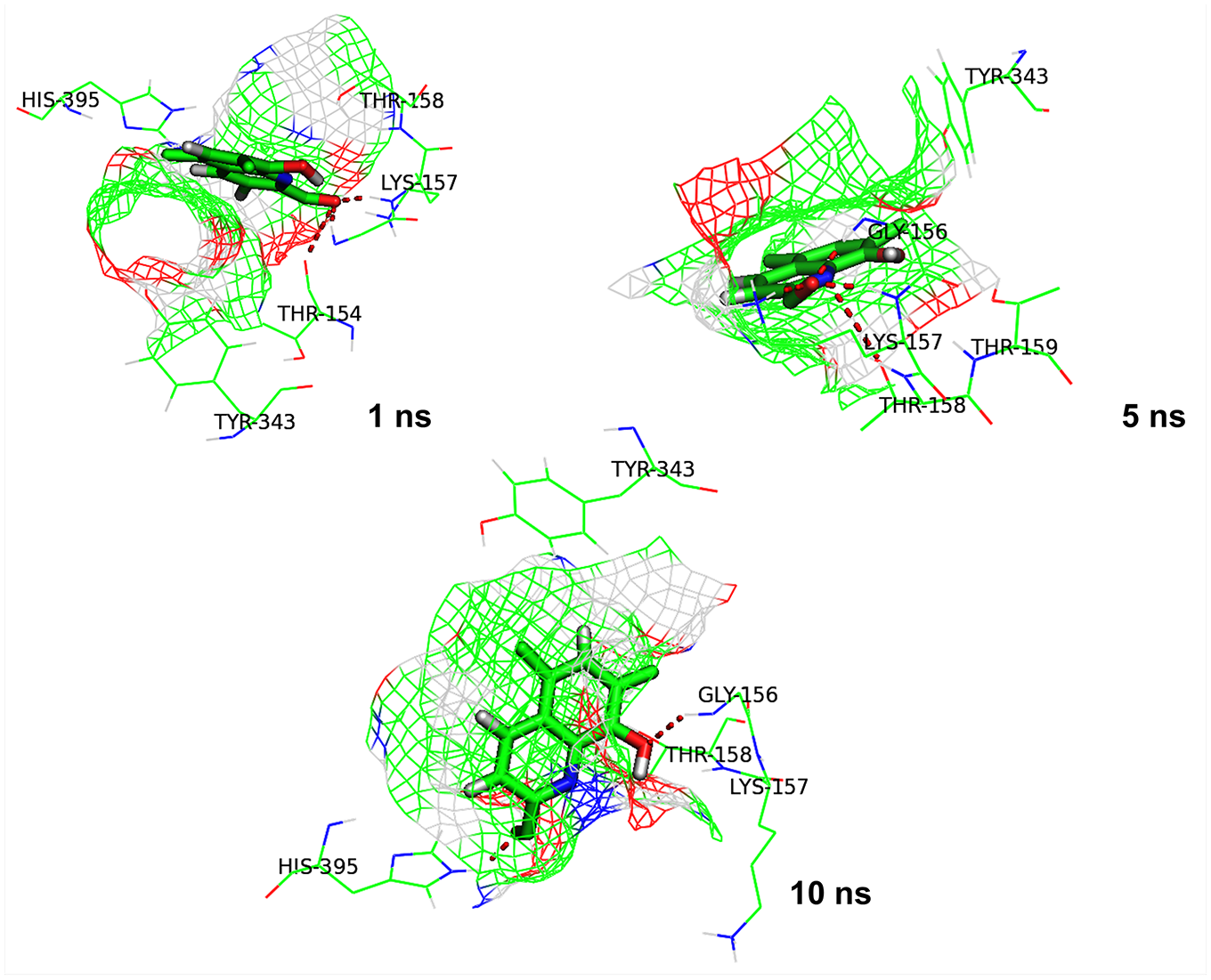
Interaction between MurE and compound 5175112 at different time frames (1, 5, and 10 ns) during simulation. The red dotted lines depict hydrogen bonds.
Discussion
Novel scaffolds with potent antimycobacterial activity are urgently needed to treat MDR and XDR strains of Mtb. The development of new molecules of any class involves several processes, including the identification of potential inhibitors, discovery of new molecules with novel mechanisms of action, and the chemical alteration of existing drugs. Methods such as whole-cell-based screening, combinatorial synthetic chemistry, and target-based high-throughput screening (HTS) have been followed widely for the identification of potential inhibitors. These methods have led to the identification of several Mur enzyme inhibitors from other organisms24,39,40 and many antitubercular scaffolds that are currently being evaluated in different stages of clinical trial. 41 Bedaquiline is one such example of HTS-based identification of an antitubercular drug, which works by inhibiting the ATP synthase enzyme in Mtb.42–44 With the availability of small-molecule libraries and considering the advancements made in computational methods, opportunities for the identification of novel chemical scaffolds for a target protein have increased manyfold. However, in the absence of experimental validation of in silico hits and/or due to a lack of translation of in vitro activity into mycobactericidal activity and vice versa, 42 a large body of research work toward TB drug discovery is unfortunately rendered ineffective.
In recent years, Mur enzymes in Mtb have received attention as promising drug targets due to their essential requirement in the survival of the pathogen.9,10,28,45,46 To identify Mur enzyme-specific inhibitors, an attempt was made in this study to screen compounds with previously demonstrated anti-Mtb activity.45,47 MurB and MurE enzymes were selected because in Mtb, crystal structures of only these two Mur enzymes have been solved.
We performed structure-based screening of 684 antitubercular compounds having low MIC value and other druglike properties against MurB and MurE enzymes. After docking, the top hits (50) obtained were further tested in vitro using the one-pot assay for Mtb Mur enzymes and the best chemical scaffolds were tested for IC50 determination. A compound with ID 5175112 showing the lowest IC50 value (28.69 ± 1.17 μM) emerged to be the most promising among the screened compounds and hence was selected for further investigations. On analyzing the binding affinity (docking score of −9.6 kcal/mol) and stability of interaction (as observed by MD simulation) of the ligand, MurE appeared to be the possible target of 5175112. Further, a target-specific in vitro assay showed inhibition (43%) of the MurE enzyme, hence strongly indicating MurE to be the prime target enzyme of 5175112. In addition, MD simulation also identified GLY156, LYS157, THR154, THR158, and HIS395 as the key interacting residues. Importantly, Lys157 residue, which has been reported earlier as the most essential residue in the active site of Mtb MurE, 38 was found to form multiple hydrogen bonds with compound 5175112. In fact, both LYS157 and THR158 residues have been reported to be present as the conserved residues in the ATP binding site of all four Mur ligase enzymes.10,38 Taken together, compound 5175112 shows promising potential as a MurE inhibitor in Mtb, with the possibility of a novel mechanism of action. Through medicinal chemistry, the compound could serve as a starting point for chemical modification and be developed as a higher-affinity scaffold with enhanced inhibitory activity.
In conclusion, this study describes an efficient strategy that employs a combination of structure-based screening followed by in vitro assay to test antitubercular compounds. We believe that this methodology could be used for HTS of larger compound libraries, while ensuring that computational results are corroborated by experimental analysis.
Supplemental Material
Supplemental_Material_for_Screening_of_anti-tubercular_compound_Mycobacterium_tuberculosis_by_K_Eniyan,_et_al.Final – Supplemental material for Screening of Antitubercular Compound Library Identifies Inhibitors of Mur Enzymes in Mycobacterium tuberculosis
Supplemental material, Supplemental_Material_for_Screening_of_anti-tubercular_compound_Mycobacterium_tuberculosis_by_K_Eniyan,_et_al.Final for Screening of Antitubercular Compound Library Identifies Inhibitors of Mur Enzymes in Mycobacterium tuberculosis by Kandasamy Eniyan, Jyoti Rani, Srinivasan Ramachandran, Rahul Bhat, Inshad Ali Khan and Urmi Bajpai in SLAS Discovery
Footnotes
Acknowledgements
We thank the Council of Scientific and Industrial Research (CSIR), India, for funding the project through Open Source Drug Discovery (OSDD). Jyoti Rani thanks the Indian Council of Medical Research (ICMR) for fellowship support. We thank the Principal of Acharya Narendra Dev College; the Director of CSIR—Institute of Genomics and Integrative Biology (IGIB), New Delhi; and the Director of CSIR—Indian Institute of Integrative Medicine (IIM), Jammu, for providing the infrastructural support and other facilities. We give special thanks to Dr. Vasanthanathan Poongavanam, Uppsala University, Sweden, for his helpful input on the computational experiments.
Supplemental material is available online with this article.
Declaration of Conflicting Interests
The authors declared no potential conflicts of interest with respect to the research, authorship, and/or publication of this article.
Funding
The authors disclosed receipt of the following financial support for the research, authorship, and/or publication of this article: This work was financially supported by the Council of Scientific and Industrial Research—Open Source Drug Discovery (OSDD/HCP0001/12FYP/2012-13/Fin/2416).
References
Supplementary Material
Please find the following supplemental material available below.
For Open Access articles published under a Creative Commons License, all supplemental material carries the same license as the article it is associated with.
For non-Open Access articles published, all supplemental material carries a non-exclusive license, and permission requests for re-use of supplemental material or any part of supplemental material shall be sent directly to the copyright owner as specified in the copyright notice associated with the article.