
Abstract
Thermal shift assays (TSAs) can reveal changes in protein structure, due to a resultant change in protein thermal stability. Since proteins are often stabilized upon binding of ligand molecules, these assays can provide a readout for protein target engagement. TSA has traditionally been applied using purified proteins and more recently has been extended to study target engagement in cellular environments with the emergence of cellular thermal shift assays (CETSAs). The utility of CETSA in confirming molecular interaction with targets in a more native context, and the desire to apply this technique more broadly, has fueled the emergence of higher-throughput techniques for CETSA (HT-CETSA). Recent studies have demonstrated that HT-CETSA can be performed in standard 96-, 384-, and 1536-well microtiter plate formats using methods such as beta-galactosidase and NanoLuciferase reporters and AlphaLISA assays. HT-CETSA methods can be used to select and characterize compounds from high-throughput screens and to prioritize compounds in lead optimization by facilitating dose–response experiments. In conjunction with cellular and biochemical activity assays for targets, HT-CETSA can be a valuable addition to the suite of assays available to characterize molecules of interest. Despite the successes in implementing HT-CETSA for a diverse set of targets, caveats and challenges must also be recognized to avoid overinterpretation of results. Here, we review the current landscape of HT-CETSA and discuss the methodologies, practical considerations, challenges, and applications of this approach in research and drug discovery. Additionally, a perspective on potential future directions for the technology is presented.
Introduction
Analyses of the drug discovery process have suggested that for successful translation of molecules from phase I to phases II and III clinically, ascertaining drug–target interaction at the site of action is crucial (AstraZeneca’s “5R framework” 1 and Pfizer’s “3 Pillars” 2 ). Developing methods to establish drug–target interaction in human tissues is a high hurdle, and can only be performed on a select few molecules, explaining why this task is often not completed before the clinical phases of a program. As a step toward the ultimate goal of working in human tissues, prior evidence of target engagement in cell lines informs the mechanism of action and can help drive the structure–activity relationship (SAR). Human cell lines can be easily grown and used as model systems, and therefore serve as an important transition into living specimens, especially if initial experiments with molecules of interest were carried out on a purified protein. Confirmation that a tool molecule binds to the target of interest in a model cell system provides valuable information that informs drug discovery and basic research.
Popular methods for determining cellular target engagement involve fluorescence (FRET) or bioluminescence (BRET) resonance energy transfer approaches to assess interactions, which require compound immobilization and binding of target proteins. 3 In these approaches, the small-molecule ligand must be modified, requiring chemistry efforts and knowledge around SARs for the molecule. Other methods rely on ligand-directed modification of target proteins, and again, the small-molecule ligand must be modified. These methods exploit binding events to capture protein or allow ligand-directed modification.
Other approaches involve sensing a change in protein structure after ligand binding, and while historically more broadly performed on purified proteins, some of the techniques have been applied to cellular systems. An example of this is protease sensitivity (reviewed in Chang et al. 4 ) where cells or pure proteins are treated with compounds, subjected to proteases, and then examined by sodium dodecyl sulfate–polyacrylamide gel electrophoresis (SDS-PAGE) and staining/Western blotting, or mass spectrometry methods to quantify resulting digestion products. Different structural forms of proteins have been shown to display different sensitivities to proteases because the protease cleavage sites become more or less exposed with the different protein conformations. Methionine oxidation is another method that relies on surface exposure; in this case the methionine residues become more oxidized when exposed on the outer surface of the protein.
Altered thermal stability of proteins is another method that has been widely used to detect changes in protein structure induced by ligand binding. In thermal shift, proteins are subjected to increasing temperatures that ultimately cause denaturation and aggregation. With treatment over a full range of temperatures, a melt or aggregation curve can be used to calculate the melting temperature (Tm or Tagg). Small-molecule binding that effects a change in the protein structure and/or dynamics can result in increased stability of the protein during thermal challenge, and this approach is described as the thermal shift assay (TSA). The TSA was traditionally applied to purified proteins, where thermally induced changes in structure can be detected using intrinsic tryptophan fluorescence, 5 fluorescent hydrophobic dyes (e.g., SYPRO Orange), 6 light scattering, 7 thiol reactive compounds that become fluorescent upon forming an adduct, 8 or sensitivity to a thermostable protease at elevated temperatures, 9 among others. Many of these approaches are compatible with high-throughput screening (HTS).
More recently, thermal shift was reported in a cellular context, giving rise to the cellular thermal shift assay (CETSA). 10 In these first CETSA studies in 2013, intact cells or cell lysates were heated to a range of temperatures, aggregated proteins were removed by centrifugation, and soluble protein was detected by immunoblot. Drug–target engagement was demonstrated for various clinical targets using lysates from cell lines or tissues of systemically treated mice. After this initial report, other experiments describing proteome profiling with CETSA11,12 and the application of the technique to membrane proteins 13 using mass spectrometry readouts were published. These approaches extended CETSA to the full detectable proteome, including membrane proteins, and described methods for experiments that informed on both on-target and off-target interactions.
As more reports on the use of CETSA were published, there was interest in extending CETSA capability to increase the assay throughput. The goal was to enable microtiter plate-based CETSAs in 384-well and higher-density plate formats, such that target engagement assays could be used to profile larger numbers of compounds, 14 as shown in Figure 1 . In a drug discovery setting, these assays could be used to select and characterize compounds of interest from high-throughput screens and characterize compounds generated in lead optimization chemistry efforts. To enable high-throughput CESTA (HT-CESTA), various groups have combined thermal shift with high-throughput protein detection methods ( Table 1 ). Reported methods and applications are described here in, and listed in Table 2 .

Adaptation of CETSA from the originally reported Western blot method to higher-throughput plate-based methods to increase assay throughput. Approaches discussed in this review enable high-throughput plate-based CETSA in 384- and 1536-well microtiter plates. The equivalent of hundreds of Western blots can be performed in a small number of microtiter plates.
HT-CETSA Approaches.
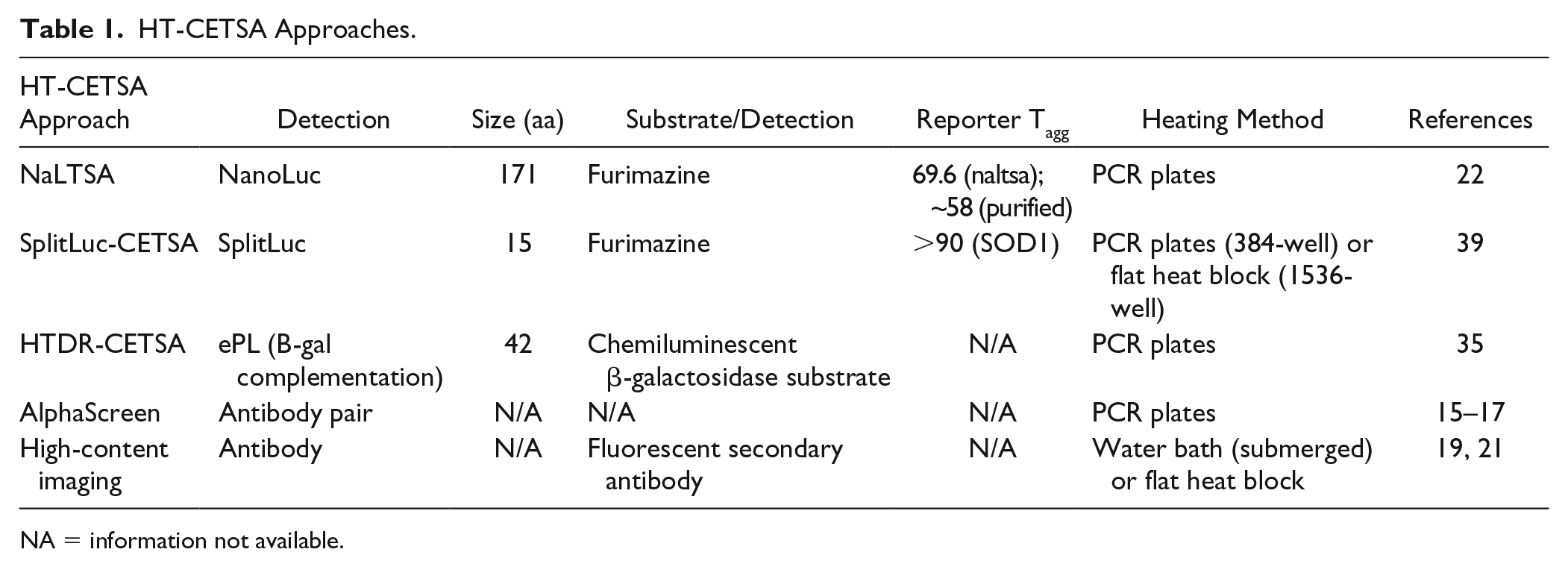
NA = information not available.
HT-CETSA Experimental Details.
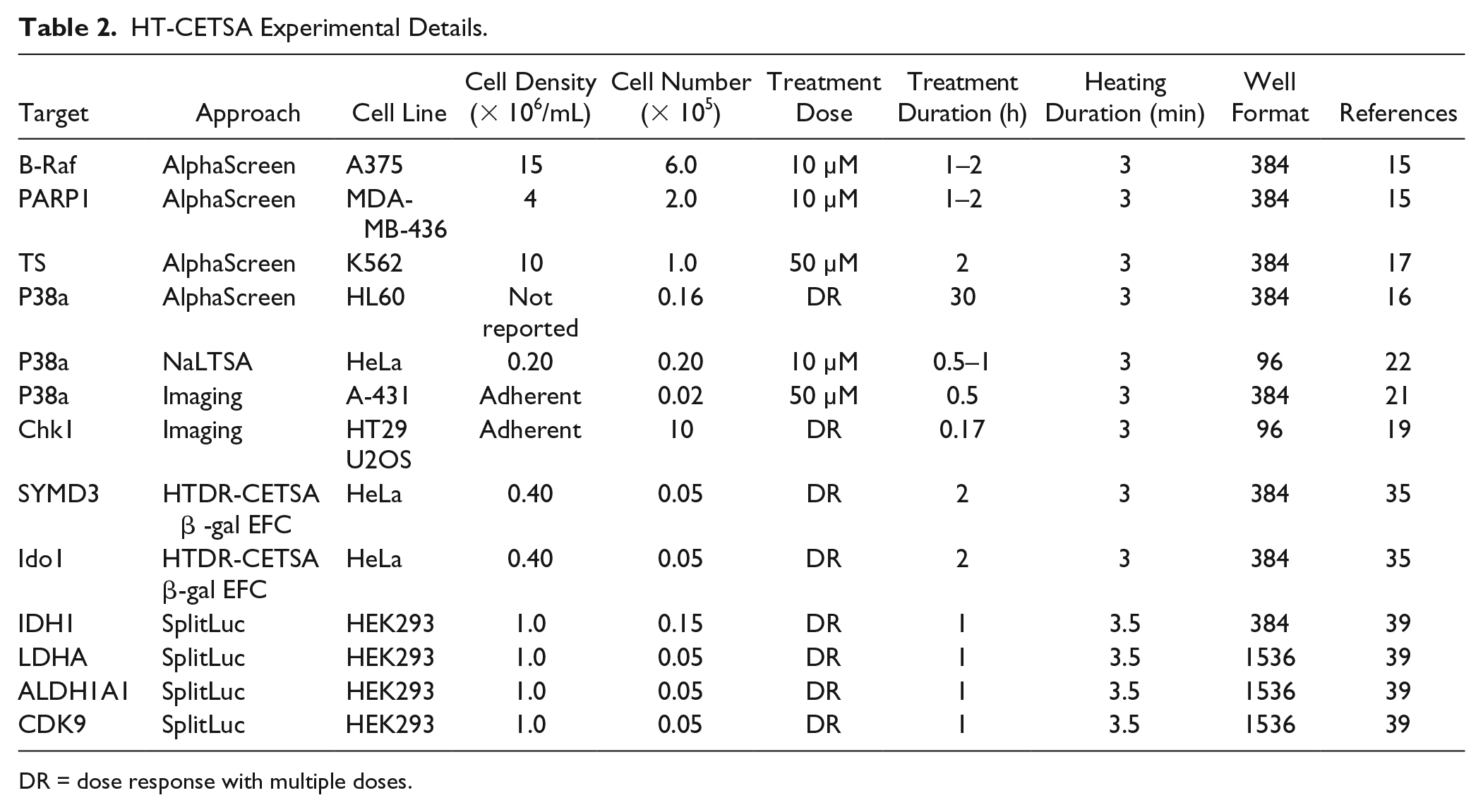
DR = dose response with multiple doses.
HT-CETSA Approaches
HT-CETSA can be separated into two general categories, where either the endogenous protein or a reporter-tagged protein is measured. Assays that measure endogenous proteins maximize physiologic relevance as (1) expression is regulated from the natural locus, (2) the protein sequence is unmodified, and (3) variants resulting from alternative splicing can be captured. Endogenous protein assays require independent validation of reagents for each target being studied, and in some cases satisfactory reagents are not commercially available. Reporter-based technologies provide an advantage in the flexibility of the system through the use of protein tags that are appended to the targets, allowing detection reagents and procedures to remain relatively fixed. In some cases, reporters can afford an increase in sensitivity by combining bright reporters with overexpression of targets, particularly when the endogenous protein cannot be detected in an HTS format. Notably, some of these advantages can also be considered caveats. For instance, overexpression of the protein can alter function, localization, or protein–protein interactions. Additionally, the reporter tag can have undesirable effects by impacting function or aggregation temperature (Tagg). Therefore, a comprehensive assessment of the tagged protein’s behavior relative to its endogenous counterpart is prudent to gain confidence in the system, but this can be a time-consuming task. Between endogenous and reporter approaches are hybrid approaches, where a reporter tag can be engineered into the endogenous locus using genome editing tools such as CRISPR/Cas9. Several groups have reported HT-CETSA strategies to study target engagement, using both endogenous and reporter-based approaches, as discussed below. The detection system approaches are depicted in Figure 2 .
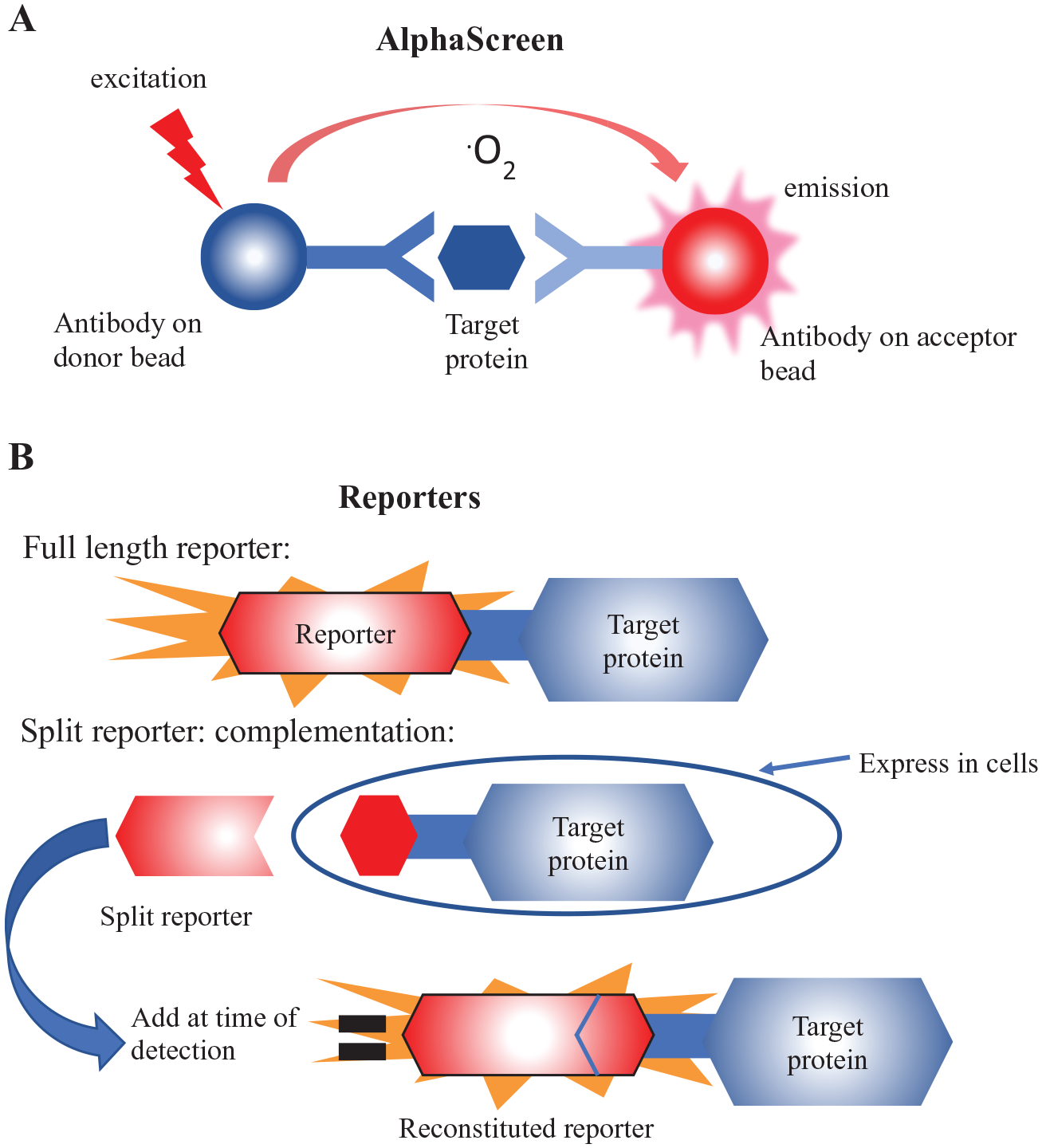
The high-throughput target protein detection methods that have been reported for CETSA. (
HT-CETSA: Endogenous Protein
Antibodies are commonly used to quantitate individual proteins from complex cellular lysates. High-throughput versions of immunoassays have been developed, making them adaptable to small-molecule screening. Homogeneous assays are often favored, where reagents are added directly to cells or lysates and read using a plate reader without any aspiration or wash steps. One common homogeneous assay format for HTS is the AlphaLISA/AlphaScreen technology (
CETSA using AlphaScreen was also reported for p38α and thymidylate synthetase. In the p38α study, 16 35 compounds were tested in dose–response and a gap between biochemical activity and cellular inhibition of TNFα expression was observed, with an average 10-fold difference in potency between the assays. To understand this discrepancy, the authors considered the intracellular bioavailability of the compounds (Fic). By taking a more holistic view and incorporating measurements of biochemical potency (IC50) and intracellular bioavailability (Fic), it was possible to predict target engagement, which was confirmed by HT-CETSA measurement. For thymidylate synthetase, 17 a diverse collection of 10,928 compounds was screened, yielding 65 hits that passed the stabilization cutoff of 11.7%, the majority of which were analogs of pyrimidine nucleosides. Of the compounds with the greatest magnitude of stabilization (>30%), 90% were confirmed upon retesting.
To date, all reports of HT-CETSA that have measured endogenous proteins in cellular lysates have used Alpha-bead technologies. Other assay formats, particularly homogenous assays such as time-resolved (TR) FRET, are expected to be compatible with HT-CETSA. Depending on throughput requirements, other antibody detection methods could also be adaptable, for example, dot blots performed with acoustic transfer of lysate 18 or semiautomated capillary-based Western blots. For all immunoassays, antibody specificity is of utmost importance. Antibody specificity can be ascertained using knockout cell lines or lysates, expression knockdown studies, overexpression experiments, or a combination of these approaches.
More recently, HT-CETSA approaches using immunofluorescence coupled with high-content imaging have been described. The method quantifies “soluble” protein in adherent cells, as the antigenic epitope becomes inaccessible upon heating and aggregation. Massey reported such an assay, named HCIF-CETSA, 19 and demonstrated heat-dependent aggregation and loss of immunofluorescence for the serine/threonine protein kinase Chk1. Interestingly, treatment with the Chk1 inhibitor V158411 resulted in a more rapid loss of immunofluorescence upon heating, in contrast to thermal stabilization observed with this compound by traditional CETSA; 20 this discrepancy is not fully understood but highlights the potential for CETSA detection modalities to show discordant results. This result could indicate, for example, that the compound induced a conformational change that increased thermal stability, while also masking the epitope binding site used for immunofluorescence detection. A second imaging-based CETSA was published concurrently with HCIF-CETSA: 21 Axelsson et al. examined p38α aggregation in adherent cells and screened a kinase-focused collection of 1120 compounds, yielding 14 hits that stabilized the protein >30%. The high-content imaging approaches provide several advantages over traditional CETSA. First, in some cases the imaging format requires fewer cells, as highlighted by the p38α imaging assay that used eight times fewer cells than the AlphaScreen-CETSA for the same target. Second, imaging provides an opportunity to capture heterogenous effects, such as changes in thermal stability that occur in a subset of cells or within specific subcellular compartments.
Each of the aforementioned HT-CETSA strategies for endogenous proteins requires specific antibodies. Other tools that provide specific binding to proteins, for example, nucleic acid aptamers, could be considered as alternate detection modalities. Label-free detection using mass spectrometry has also been demonstrated to be a powerful technique for examining proteome-wide thermal stability of cellular proteins, in an approach known as thermal proteome profiling (TPP). 13 Mass spectrometry-based CETSA for individual targets has not been reported in a high-throughput fashion but might be feasible using high-throughput sample analysis (e.g., RapidFire or acoustic droplet ejection/injection), if the protein of interest can be effectively detected and quantified.
HT-CETSA: Reporters
A variety of protein-based reporters have been used in conjunction with CETSA. To date, HT-CETSA strategies have primarily used luminescent readouts, with full length or reconstituted enzyme reporters ( Fig. 2 ). Dart et al. 22 demonstrated HT-CETSA using NanoLuciferase, a 19 kDa protein with bright and stable luminescent output, to track thermal aggregation profiles for kinases, bromodomains, and histone deacetylases. In this approach, also known as NaLTSA, NanoLuciferase was fused to the N- or C-terminus of targets and expressed in HeLa cells by transiently transfecting the cytomegalovirus (CMV)-driven reporter. NaLTSA was used to characterize target engagement for p38α, examining a set of 80 kinase inhibitors. The assay was expanded to a set of 38 additional NanoLuciferase-tagged kinases (20 tyrosine kinases, 18 serine/threonine kinases) to examine the binding profile of ponatinib, a multikinase inhibitor approved for chronic myeloid leukemia (CML) and Philadelphia chromosome-positive acute lymphoblastic leukemia (ALL). The study identified 19/20 known targets of ponatinib, with JAK2 as the lone false negative. Additional kinases that had not previously been identified as ponatinib targets were stabilized in the NaLTSA assay; whether these represent bona fide target engagement or false positives is unclear at this time.
Other full-length reporters could potentially be used for HT-CETSA; however, the melting temperature of the reporter is required to be greater than that of the protein of interest. For this reason, some common reporters such as firefly luciferase (Tm ~37 °C), 23 Renilla luciferase (Tm ~42 °C), 24 and β-galactosidase (Tm ~59 °C) 25 could be problematic for CETSA. Reporter variants with increased thermal stability, such as firefly luciferase YY5, mCherry (Tm ~90 °C), 26 enhanced green fluorescent protein (eGFP) (Tm ~78 °C), 27 or ultrastable GFP 28 might be more suitable.
To circumvent the limitations of using full-length reporters, several groups have pursued protein complementation approaches, where reporters are split into two fragments and activity is reconstituted upon interaction of the fragments when recombined. In the context of CETSA, this allows a small tag to be appended to the protein of interest and the larger fragment can be delivered at the detection stage, after heating. Complementation systems have been developed for luciferase enzymes including Renilla luciferase, 29 firefly luciferase, 30 Gaussia luciferase, 31 and NanoLuciferase. 32 Fluorescent complementation systems GFP (splitGFP) 33 and β-galactosidase (ProLink, ProLabel) 34 are also available.
McNulty et al. 35 described high-throughput dose–response (HTDR)-CETSA, where a 42-amino acid peptide derived from β-galactosidase (enhanced ProLink [ePL]) was appended to the histone methyltransferase SYMD3 or dioxygenase IDO1. After heating and lysing cells, the large fragment of β-galactosidase and a luminescent substrate were added, allowing quantitation of the protein by measuring light output. Baculovirus (BacMam) was used to deliver the ePL-tagged vectors and transduction was titrated to match expression with endogenous protein levels. The SYMD3-ePL assay was used to examine 123 compounds, which were previously identified as potent inhibitors in a biochemical assay. The compounds were tested in duplicate, revealing favorable assay reproducibility as 109 of 123 compounds had a percent coefficient of variation of <15% and 12 of the 14 remaining compounds were weak or inactive (pEC50 < 4). HTDR-CETSA was compared with MEKK2 methylation, as an orthogonal readout of cellular SYMD3 activity for 97 compounds. Activities in the two assays showed high correlation, supporting the use of HTDR-CETSA to assess cellular inhibition of SYMD3.
The β-galactosidase complementation technology has also been used to examine target engagement at physiologic temperatures. In this approach, also known as InCellHunter, small-molecule binding is detected by a stabilization of the protein under standard growth conditions (without additional heat). This is not CETSA per se, but exhibits significant conceptual similarities as small-molecule engagement drives protein stabilization. High-throughput InCellHunter assays have been described for BRD, 36 PRMT3, 37 and MEK1. 38 Notably, other detection modalities, such as TR-FRET, can be used in place of enzyme fragment complementation, as was demonstrated for BRD4. 36 For some targets, CETSA and physiologic stabilization could be used as complementary approaches to examine target engagement.
SplitNanoLuciferase has also been implemented for HT-CETSA. In this approach, a 15-amino acid fragment of NanoLuciferase (HiBiT variant; aka 86b) was fused to proteins of interest and the large fragment of NanoLuciferase (11S) and the substrate furimazine were added after heating, to quantify the amount of nonaggregated protein. 39 Martinez et al. performed a multidose screen for small-molecule binders of lactate dehydrogenase A (LDHA) utilizing an oncology-focused library of 1850 compounds. A single LDHA-stabilizing hit was identified, GSK2837808A, an advanced LDHA inhibitor. In contrast, screening the library using a cell-based readout of LDHA activity, in which lactate output was measured, resulted in 376 active compounds, many of which were expected to be off-target as they did not alter LDHA activity in a biochemical assay. The SplitLuc CETSA was also used to examine a set of 15 LDHA inhibitors identified through a MedChem campaign, with comparisons to either a cell-based lactate readout or LDHA biochemical assay; the two cell-based assays showed better agreement in activity measures. Similar observations were made for aldehyde dehydrogenase A1, although potencies in the SplitLuc-CETSA were right-shifted 1 log unit compared with an alternate cell-based readout. Furthermore, the SplitLuc CETSA assay was used to examine cyclin-dependent kinase 9 (CDK9), for which a kinase-focused set of 977 compounds was screened for binders. The CDK9 HT-CETSA screen identified three active compounds (0.3%), all with previously reported CDK9 activity. A corresponding binding assay using recombinant CDK9 protein yielded 89 hits (~9%), highlighting the differences in activity that can be observed for purified protein versus cell-based systems.
Additional reporter tags that facilitate protein quantitation are amenable to CETSA. For example, NUDT7 target 40 engagement was demonstrated using a FLAG-tagged protein. Likewise, Cdk2 and cyclin E1 CETSAs were reported for hemagglutinin (HA) and V5-tagged proteins, respectively. Other small epitope tags, such as Myc or StrepII, should also be suitable for CETSA. To date, epitope tags have not been used in HT-CETSA, but coupling with Alpha or TR-FRET technologies may be feasible since antibody-based reagents are available for these and other common epitope tags.
The diversity of HT-CETSA approaches published to date indicates that there is some flexibility when approaching assay design for new targets. Notably, a systematic comparison of endogenous versus reporter approaches for a single target has not been reported. In our view, it is unlikely that general rules and principles will apply when selecting an appropriate HT-CETSA strategy for novel targets, because each target and assay methodology will require unique conditions and considerations.
Observations and Considerations
The transition from traditional CETSA to HT-CETSA methods comes with several technical considerations. First, physical separation of insoluble aggregates by centrifugation is challenging for high-density plates, as centrifugal forces used in standard CETSA cannot be achieved. Second, heating with consistency is critical. Hardware designed for precision heating, such as PCR blocks, is well suited for the job because individual wells are physically mated to the heat source. Thermal cyclers are available in 384- and 1536-well format; however, these are not available in many laboratories. Flat heating blocks (1536- and 384-well imaging) and water baths (384-well imaging) have also been used successfully, albeit with an expected loss in temperature precision and reduced rate of heat transfer. Neither PCR thermal cyclers nor flat heating blocks offer ultrafast solutions for heating large numbers of plates at once unless the equipment is scaled. Third, HT-CETSA lysis buffers need to be compatible with a homogenous no-spin assay, and therefore ionic detergents such as SDS should be avoided to prevent resolubilization of aggregated proteins.
There are also practical considerations when using reporter fusions for HT-CETSA. The risk of deleterious effects being imparted by the reporter can be minimized by (1) assessing protein behavior using a functional assay, (2) confirming similar aggregation profiles of the reporter fusion with endogenous protein by Western blot, and (3) assessing subcellular localization of the reporter versus endogenous protein by immunofluorescence. Small-molecule or peptide controls, when available, can provide confidence in the HT-CETSA approach and demonstrate the capacity for a protein to be stabilized. However, for many proteins, compounds with previously characterized binding in the cellular environment are not available, requiring experiments with such targets to proceed at risk.
CETSA can be performed using intact cells or lysates, with live cell assays enriching for compounds that have sufficient membrane permeability. In some instances, a control molecule that engages the target of interest is not cell permeable and must be tested in a lysate setting. For example, stabilization of spindlin1 41 with a control peptide required CETSA to be performed in lysates. For lysate-based CETSA, subcellular compartmentalization and molecular concentrations (cofactors, proteins, nucleic acids) will be altered, which may impact CETSA results. The effect of cofactor concentration on target engagement was exemplified for ALDH1A1, where small-molecule binding only occurred in lysates with addition of the cofactor NAD+. Similar observations have been made for kinases, where ATP concentration impacts target engagement profiles. 42
HT-CETSA assays are typically run in isothermal mode after determining the Tagg in preliminary experiments (e.g., choosing a temperature at which 50%–90% of the protein is aggregated). For some proteins with Tagg above 60 °C, cell membrane integrity may be disrupted during the heating phase. Several studies have characterized membrane permeability during a 3–3.5 min heating step used in CETSA and observed loss of membrane integrity between 63 and 70 °C for a variety of cell lines. Therefore, HT-CETSA at temperatures near or exceeding 65 °C may reflect an environment more representative of lysates, rather than intact cells.
Other aspects of the HT-CETSA protocol can be optimized during assay development. While a heat duration of 3 min has been used for most CETSA experiments presented in the literature, optimizing the heating duration for an HT-CETSA screen might improve assay performance. Similarly, the compound treatment phase of CETSA is typically 1–4 h, to allow sufficient time for compounds to traverse the membrane and engage the target, while limiting indirect effects (e.g., transcriptional responses) that could confound interpretation. The treatment duration selected for HT-CETSA could impact the hit rate and number of false positives or negatives. Lastly, compounds are usually added to cells in suspension, even for cell lines that prefer to grow in an adherent state, as V-shaped PCR tubes are not designed for cell attachment. Transitioning cells from adherent to suspension culture has the potential to alter cell physiology and protein behavior, which could impact target engagement. Methods to heat flat multiwell plates are available, but they lack the temperature precision afforded by conventional PCR plates.
Interpretating Experimental Outcomes
Target engagement often results in protein stabilization, but ΔTagg can occur in either positive or negative directions. The magnitude of stabilization (Emax) is protein dependent and is not an indicator of the utility of the approach for assessing compound binding. For example, NaLTSA accurately detected 19 of 20 known kinase targets for ponatinib, with ΔTagg values ranging from 2.05 to 9.06 °C; however, the larger shifts in Tagg did not necessarily indicate better target engagement. 22 When comparing across protein targets, potency (EC50) provides a more dependable metric of cellular engagement. Notably, the apparent Tagg in a given system represents an average of all protein states within the cell. This becomes particularly relevant when a small molecule binds to a subpopulation of the target (e.g., within a protein complex or a subpopulation containing posttranslational modification) as the Tagg shift will become difficult to detect if only a small portion of protein is engaged. Furthermore, not all ligands produce a thermal shift when they bind to protein targets. For instance, a thermal shift in BCR-ABL was not detected for the extensively characterized inhibitor dasatinib. Therefore, a lack of activity in HT-CETSA should not be interpreted as lack of engagement. On the other hand, few examples of false positives have been reported for HT-CETSA.
When comparing biochemical assays with CETSA, various factors including membrane permeability, serum binding, off-target binding, intracellular bioavailability, metabolism, and compound efflux will contribute to activity. Unsurprisingly, discrepancies in cellular versus biochemical potency have been reported for many targets. Another factor that can affect CETSA potency is target concentration within cells, which in many cases is vastly different than concentrations used in biochemical assays. Oftentimes, the absolute concentration of a target protein is not empirically determined for a cellular model, so expectations about potency based on biochemical readouts or other cell-based assays may be misleading. Target concentration can also vary between different cell-based assays. These discrepancies in protein expression are particularly relevant in the context of overexpression systems, such as CMV-driven reporters. When using reporters, titrating expression to match endogenous levels is desirable, as was demonstrated for SYMD3 using BacMam. 35 Alternatively, this can be accomplished by engineering reporter tags into the endogenous locus.
In addition to the aforementioned biological factors, experimental factors may also affect apparent potencies in CETSA. For example, cell density can impact measurements of EC50. This was demonstrated for Chk1, where a cell-based phosphorylation assay showed 43-fold greater potency than CETSA. Notably, these assays were run at different cell densities, with CETSA using a 67-fold greater concentration of cells. When the phosphorylation assay was tested at the higher cell density, the EC50 became comparable to that of CETSA. Other assay conditions will alter CETSA potency, for instance, the heating temperature. This was demonstrated for SYMD3 inhibitors, 35 where the EC50 for a set of inhibitors was right-shifted ~1 log unit as the temperature increased from 46 to 52 °C; however, rank order was maintained. Decreased potency with increasing temperatures has also been demonstrated for p38α, where temperatures ranging from 49.5 to 61.5 °C increased the pEC50 of SB203580 nearly 2 logs. 43 Heating duration also affects apparent potency. For example, an LDHA inhibitor was examined with heat applied for 5–50 min, with a corresponding 35-fold right-shift in EC50, but compound rank order was maintained. 39
In this context, it is important to note that the rate of heating may also influence the measured effect of different compounds. Drawing from the wealth of knowledge accumulated during studies of targets and compounds using biochemical thermal shift methods, the behavior of p38a inhibitor classes with differing enthalpic and entropic contributions to p38α binding was examined. 43 The authors noted that the relative shifts in potencies and ranking could be rationalized based on the distinct thermodynamic parameters associated with each specific compound scaffold.
For isothermal mode screens, an increase in soluble protein could indicate (1) thermal stabilization, (2) an increase in protein expression, or (3) artifact related to reporter or assay reagents. Hits that do not reflect engagement can be removed by comparison to an unheated control sample for each compound (i.e., each compound plate is replicated for both heated and unheated assays), which would flag artifacts, but at the cost of twice the number of test samples. HT-CETSA is typically a gain-of-signal assay, and therefore artifacts from the assay components will be expected to be less common than for a similar loss-of-signal assay. False positives can occur, though, as highlighted by the study of NLuc-MAPK14, where Lavendustin, an epidermal growth factor receptor (EGFR)-specific inhibitor, showed an apparent stabilization of both the fused and unfused NanoLuc control, indicating a stabilization of the reporter itself. 22 For split NanoLuciferase or other enzyme fragment complementation systems, compound effects on protein complementation or enzyme activity can be counterscreened using recombinant protein fragments spiked into cellular lysates (untransfected, to match screening conditions). For AlphaScreen CETSA, TrueHits is marketed by the vendor specifically to flag false positives associated with singlet oxygen. For most detection formats that involve luminescent or fluorescent readouts, compound addition after the heating step can identify artifacts unrelated to protein engagement and stabilization.
Confirmation experiments and counterscreens provide confidence that a small-molecule induced ΔTagg is real, which, in many cases, indicates direct target engagement. Binding, however, is not equivalent to functional inhibition and CETSA results will not necessarily align with other biochemical or cellular assays. This can be exemplified by a study of PARP1, where a set of 19 compounds showed binding in both FP and CETSA assays but were not active in a cellular PARylation assay. In addition to compound-mediated effects, thermal stability can also be altered by posttranslational modifications, glycosylation, 44 redox status, 45 or protein–protein interactions, 11 and therefore a Tagg shift does not ensure engagement. Similar to other assay formats, activity in HT-CETSA needs to be confirmed using independent assays.
Applications in Research and Drug Discovery
HT-CETSA has several strengths as a screening platform. First, it provides an opportunity to move quickly from primary screening to a set of compounds enriched for membrane permeability and the capacity to engage the target within a complex cellular environment. Second, targets that are challenging from an HTS assay development perspective may be amenable to HT-CETSA. For instance, this includes targets that require multicomponent complexes to reconstitute physiologically relevant activity, which can be challenging to re-create in a biochemical setting. Furthermore, for many targets, cell-based phenotypic readouts are not available or rely on nonspecific endpoints. Third, HT-CETSA is well positioned for the study of rare monogenic diseases, where it provides an assay platform that can be rapidly developed to screen for small-molecule binders (e.g., chemical chaperones) of a known protein target. Fourth, HT-CETSA provides a platform to identify molecules that bind, but not necessarily inhibit, a target. This could be advantageous for targets that share high homology within the catalytic site to antitargets, where binding to a less conserved part of the protein is favorable. Recent advances in proteolysis-targeting chimeras (PROTACs) may pave the way toward developing binders identified in HT-CETSA into functional inhibitors. While HT-CETSA is still in its early stages, there are numerous opportunities for the development and implementation of this approach, in both research and drug discovery settings.
Discussion
Significant progress has been made in the industrialization of CETSA as a platform to support early drug discovery, as well as in understanding how CETSA potency (EC50 value) quantitively translates to target occupancy and intracellular binding. The development of multiple HT-CETSA formats has resulted in the generation of CETSA EC50 values for hundreds of compounds spanning a number of target classes. These data sets provide evidence for a general correlation between CETSA potencies for both biochemical activity and on-target cellular mechanistic readouts for a target of interest. The examples reviewed in this perspective begin to build confidence in the use of CETSA for hit qualification, for confirmation of target engagement, and as a useful assay to contribute to SAR studies, resulting in the prioritization of compounds for further lead optimization. However, it remains to be seen if CETSA potency can be used by medicinal chemists to directly influence SAR-driven ligand design and provide more than a semiquantitative measure of target engagement to help rank-order compounds of interest. There are no reports where CETSA has been used to disqualify compounds from either hit-to-lead or lead optimization small-molecule drug discovery campaigns. Extending the experimental path outlined for quantitative interpretation of p38α CETSA data 43 to other target classes will be an important step in realizing CETSA-driven lead optimization for small-molecule discovery programs.
As noted in this review, there are examples of compounds where shifts in potencies between biochemical activity measurements, CETSA-generated cellular target engagement values, and cellular mechanistic potencies are not easily explained by either compound physiochemical properties or poor pharmacokinetic parameters. In these cases, the ability to run HT-CETSA in both whole-cell and lysate formats can be followed by a classical Western-based CETSA in a disease-relevant cell line or tissue sample. One could then readily probe if cellular target engagement (as measured by a CETSA EC50) can be used to build a translational pipeline showing that a compound with biochemical potency engages the desired target in cells and results in modulation of a disease-linked phenotype. In some instances, CETSA can actually (in)validate a given target hypothesis resulting in early attrition, significant cost savings in time and resources expended, and greater understanding of disease biology. Two examples where CETSA played a significant role in (in)validating targets in oncology for GlaxoSmithKline and Bayer are SMYD3 35 and MTH1. 46 In both examples, structurally distinct, potent, cell-permeable, soluble, and selective inhibitors for SMYD3 and MTH1 were developed. CETSA EC50 values showed correlation with both biochemical potency and target-dependent functional cellular readouts, yet none of these properties translated into antiproliferative efficacy. This highlights the potential (at least in an oncology indication) for CETSA to serve as a bridge between biochemical potency and modulation of a desired phenotype. If CETSA could be applied and help remove the need for extensive cellular mechanistic assay development and transcriptional profiling, it has the potential to decrease the cycle times in a typical lead optimization small-molecule drug discovery program.
As we look to the future, primary diversity screening by HT-CETSA to identify small-molecule leads in early drug discovery is still in its infancy. To date, the largest set of compounds screened to identify novel chemical starting points was for thymidylate synthase, where 10,928 compounds were screened by HT-CETSA (AlphaScreen) in K562 cells. 17 HT-CETSA has been adopted to a 1536-well microplate using split NanoLuciferase demonstrating the feasibility for millions of compounds to be screened by CETSA. We readily admit that not all target classes are amenable to CETSA screening, but there is experimental evidence that more than 5000 proteins have quantifiable melting profiles in K562 cells, which provides validation that HT-CETSA screening is feasible for a subset of the human proteome. 11 For proteins that are not expected to demonstrate a thermal shift with compound binding, one could design reporter–domain fusion constructs, which would be of a smaller size, and therefore more amenable to a measurable melt profile and compound-induced thermal stabilization. The reported ease of use of CRISPR/Cas9 for the insertion of tags into more disease-relevant cell lines and even primary cells for HT-CETSA applications offers another avenue for the deployment of HT-CETSA that has not yet been realized. Finally, the identification of compounds that destabilize a given protein target in an HT-CETSA assay could provide starting points for the optimization and development of PROTAC molecules, an alternative modality to modulate disease phenotypes. 47
Footnotes
Declaration of Conflicting Interests
The authors disclosed the following potential conflicts of interest with respect to the research, authorship, and/or publication of this article: Marc A. Holbert and Lorena A. Kallal were employed by GlaxoSmithKline at the time of the work on the article and their research and authorship of this article was completed within the scope of this employment.
Funding
The authors received no financial support for the research, authorship, and/or publication of this article.