
Abstract
The cellular thermal shift assay (CETSA) was introduced in 2013 as a means to assess drug binding in complex environments such as cell lysates, live cells, and even tissues. The assay principle relies on the well-proven biophysical concept of ligand-induced thermal stabilization of proteins, which in CETSA applications is measured as a persistent presence of soluble protein at elevated temperatures. Given its recent development, we have just started to learn about the benefits and pitfalls of the method as it is applied to a growing number of protein target classes, the majority of which are intracellular soluble proteins. One of the early technology developments concerned the transfer of the original assay procedure from PCR tubes and Western blot detection of soluble protein to a homogeneous assay in high-density microplates. A move to high-throughput formats is essential for a more systematic application in drug discovery settings, as well as in academic efforts for validating chemical probes through studies of structure–activity relationships. This perspective aims at providing an overview of knowledge gained in microplate formatting of CETSA and makes an attempt at forecasting future applications.
Keywords
Introduction
Pharmacological validation of a target protein requires highly selective chemical probes for which the affinities to the intended target, as well as relevant off-targets, are known.1-4 If these prerequisites are not sufficiently well established, there is a significant risk that functional and phenotypic cell assay results are driven by off-target pharmacology. It is well known that polypharmacology can be useful from a clinical standpoint,5-7 but when validating the pharmacological importance of an individual target protein, an understanding of the chemical probe selectivity and its impact on the results is essential.
The potential influence of off-target pharmacology is commonly addressed using a battery of in vitro assays for target-related proteins, as well as broader unbiased selectivity panels. Although an understanding of in vitro selectivity is useful, such panels are not sufficient for exclusion of all possible off-target contributions and neglect influences of metabolic processing of the small molecule in vivo. Evidence that a desired phenotypic effect results from pharmacological modulation of a given target protein is instead inferred from the concurrent appearance of functional readouts more closely linked to the target protein function, such as measuring an intracellular product of an enzyme of interest. As it may be challenging to find unique readouts for a given target protein, it is essential to also link the degree of target occupancy with the extent of phenotypic response to further strengthen the validation. 2 The concept of quantitatively comparing structure–activity relationships (SARs) for intracellular target occupancy with those of well-designed, “close-to-target” functional, as well as phenotypic, readouts is appealing and would be particularly useful as an additional inclusion criterion for well-validated chemical probes. 3
Quantitative coupling of target-mediated response to target occupancy levels is also critical for target-based drug discovery efforts. Recent metadata from clinical trials indicate that proof of target engagement, alongside the concurrent presence of drug candidate in the desired tissue and validated functional and phenotypic outcomes, is essential for successful clinical trial outcomes.8,9 Although it is highly relevant to include routine measurements of target engagement during preclinical and clinical development, this parameter has proven difficult to establish, especially in living cells and organisms.8,9
The significant need for measurements of target engagement in live cells and organisms has naturally motivated the development of new technology, and this topic is well covered in two recent reviews.10,11 Several of the described methods rely on proximal biomarkers that can be measured using proteomic or metabolomic approaches. Other efforts are based on modified small molecules (tracers) and engineered proteins with signaling tags that can be applied for measuring target occupancy in model systems. Some of these are well suited for application in high-density microtiter plates and will thus be useful to support optimization and characterization of chemical probes and drug candidates. Examples include the use of enzyme fragment complementation (EFC)12,13 or time-resolved fluorescence resonance energy transfer (TR-FRET) 12 to measure ligand-induced changes in intracellular protein stability and, as a consequence, altered levels of target protein. An alternative approach is based on bioluminescence resonance energy transfer (BRET), in which the simultaneous application of a luminescent protein and a cell-permeable fluorescent tracer allows for direct studies of the kinetics of target engagement in live cells. 14
This early perspective is focused on the recent observation that measurements of target engagement can be achieved in live cells and tissues with the cellular thermal shift assay (CETSA). 15 As this methodology rests on ligand-induced changes of an inherent biophysical property of the investigated target protein, there is no requirement for either ligand or protein modification, meaning it has the potential to translate to clinical samples from treated patients. Furthermore, since the ligand is applied “as is,” it enables studies of SARs in academic chemical probe discovery programs, as well as drug discovery programs. This prospect has motivated us to pursue CETSA in microplate format, an area that is just emerging and is likely to develop significantly in the coming years. Herein, we will share knowledge gained from these efforts ranging from technical troubleshooting to potential impact of the technology.
Basic Principles of Microplate-Based CETSA
Assay Principle
Thermal shift assays16-18 and isothermal denaturation assays19-21 are routinely applied in microplate format for studies of ligand interactions with isolated proteins. The underlying thermodynamic basis of these assays is identical in that selective ligand binding to the native protein results in its thermal stabilization. This is manifested as either a shift of the apparent melting temperature (Tm) to higher temperatures or a reduced rate of denaturation when the protein is challenged at temperatures approaching Tm.
CETSA is based on the discovery that ligand-induced stabilization of proteins can also be investigated in more complex environments, including cell lysates, live cells, and tissues following ex vivo and in vivo treatment. 15 Practically, CETSA measurements are possible when heat-induced protein unfolding is tightly coupled with irreversible aggregation, such that the remaining amount of soluble protein reports on the proportion of folded protein. The assay principle is schematically illustrated in Figure 1A and involves sample treatment with ligand, a transient heat challenge to induce unfolding and subsequent irreversible aggregation of unliganded protein, and finally detection of remaining levels of soluble protein following cell lysis. Most CETSA experiments are based on either the establishment of temperature of aggregation (Tagg) curves by heating identical samples to increasing temperatures ( Fig. 1B ) or concentration–response experiments in which the ligand is titrated to examine its impact on soluble protein levels following an isothermal temperature challenge ( Fig. 1C ). The latter experiments are referred to as isothermal dose–response fingerprints (ITDRFCETSA) to clearly signal the apparent nature of these responses. Generally, triplicate measurements are sufficient to accurately establish the Tagg or EC50, although this can vary depending on the variation in the cell material and assay setup.
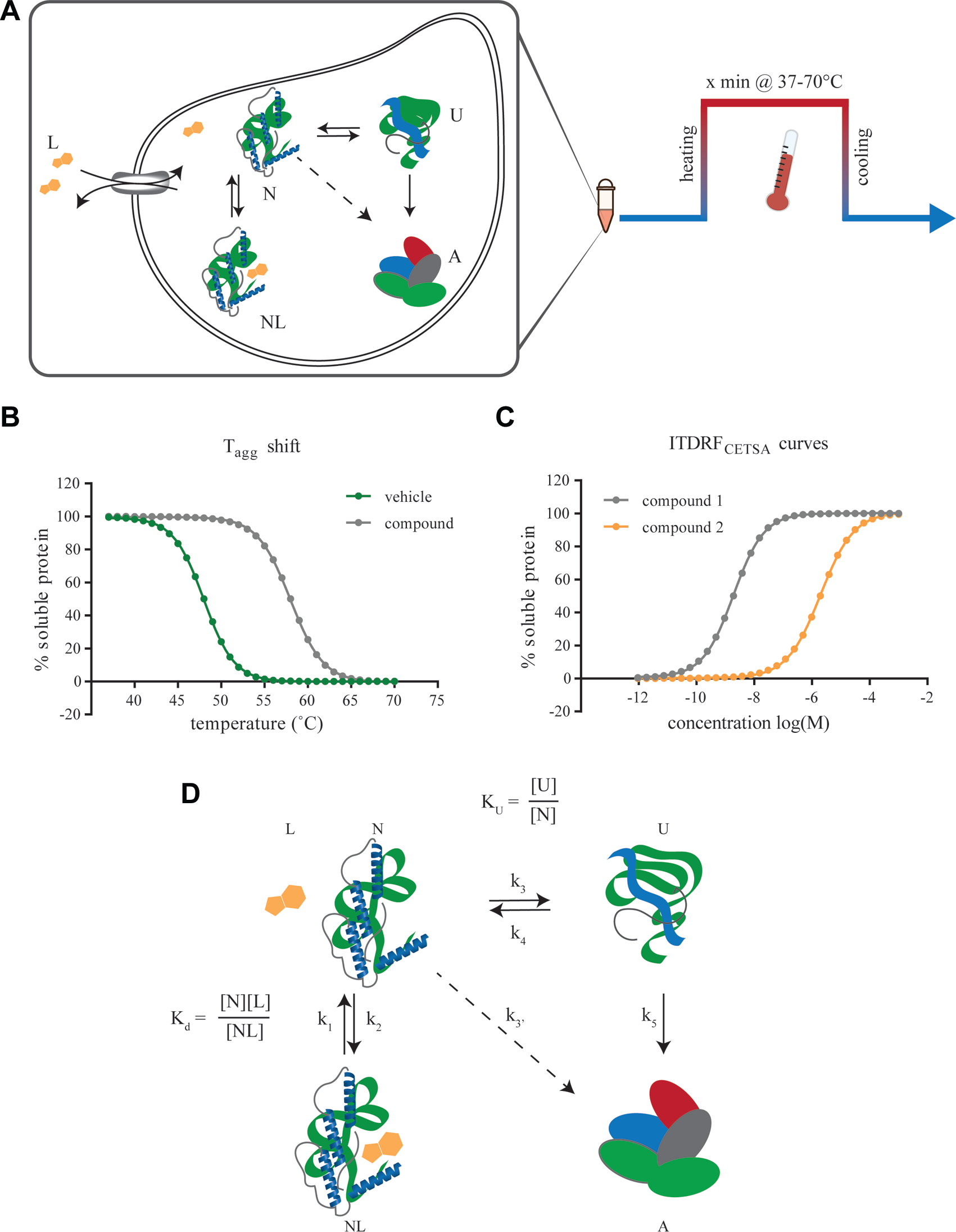
(
It is important to emphasize that CETSA experiments are not performed at equilibrium, and hence the measured apparent parameters are dependent on the experimental setup. Of practical importance for the readout is the rapid aggregation of unfolded protein that occurs in cell lysates and intact cells, likely driven by the appearance of multiple hydrophobic surfaces when several proteins unfold simultaneously. The extent to which the investigated ligand binding and protein folding/unfolding process have time to first reequilibrate at the elevated temperature, and then deviate from equilibrium over time as a result of protein aggregation, will naturally depend on the length of the transient heating period and its relation to the rate constants for the interaction, folding, and aggregation ( Fig. 1D ). This is discussed in more detail in a following section on data quantification. Importantly, the common use of a 3 min heat challenge has been shown to not acutely affect cell membrane integrity up to temperatures of approximately 60–65 °C.15,22 Furthermore, depending on protein of interest, the heating period can be lowered to below 30 s and still yield a sufficient difference in signal to allow experiments. 23
Detection Modalities
There are numerous ways to measure remaining levels of soluble protein, including but not limited to quantitative Western blot (WB), 15 dot blot, 15 and a broad range of different enzyme-linked immunosorbent assay (ELISA) and other immunoassay-based formats.22,24 The use of mass spectrometry (MS) as a detection modality has been adopted in a technique called thermal proteome profiling (TPP) or thermal stability profiling, which allows for the concurrent measurement of the entire melting proteome.25-28 As recently reviewed, 23 the majority of early adopters have applied WB detection for interrogation of individual target proteins in low-throughput format, as described in the original protocol.
High-throughput measurements require adaptation to a high-density, microplate format, and so far we have implemented and published on the use of AlphaScreen immunoassays for unmodified proteins.22,24 CETSA applications based on tagged protein to support, for example, EFC or luminescence-based readouts, remain to be published in peer-reviewed journals, but are emerging at conferences (e.g., the Society for Laboratory Automation and Screening [SLAS] Fifth Annual International Conference and Exhibition). All of these approaches are compatible with measurements of target protein levels in the presence of debris from cell lysates, such that the extensive sample workup required for WB- and MS-based approaches can be avoided. This is important to ensure sufficient throughput, but a reduction of the number of assay steps also serves to minimize variability between wells in the microplate.
It is outside the scope of this perspective to cover an extensive list of additional tentative readouts for quantification of soluble protein levels, whether these are based on affinity reagents or tagged proteins. There are pros and cons with any of these detection modalities, and the choice in a given laboratory will likely be pragmatically based on the experiences of the user and equipment available in the laboratory. Such decisions will also be governed by assay purpose, throughput needs, and choice of input material in terms of primary cells versus engineered cell lines, and we will come back to these issues in the following sections.
Challenges in Microplate Adaptation
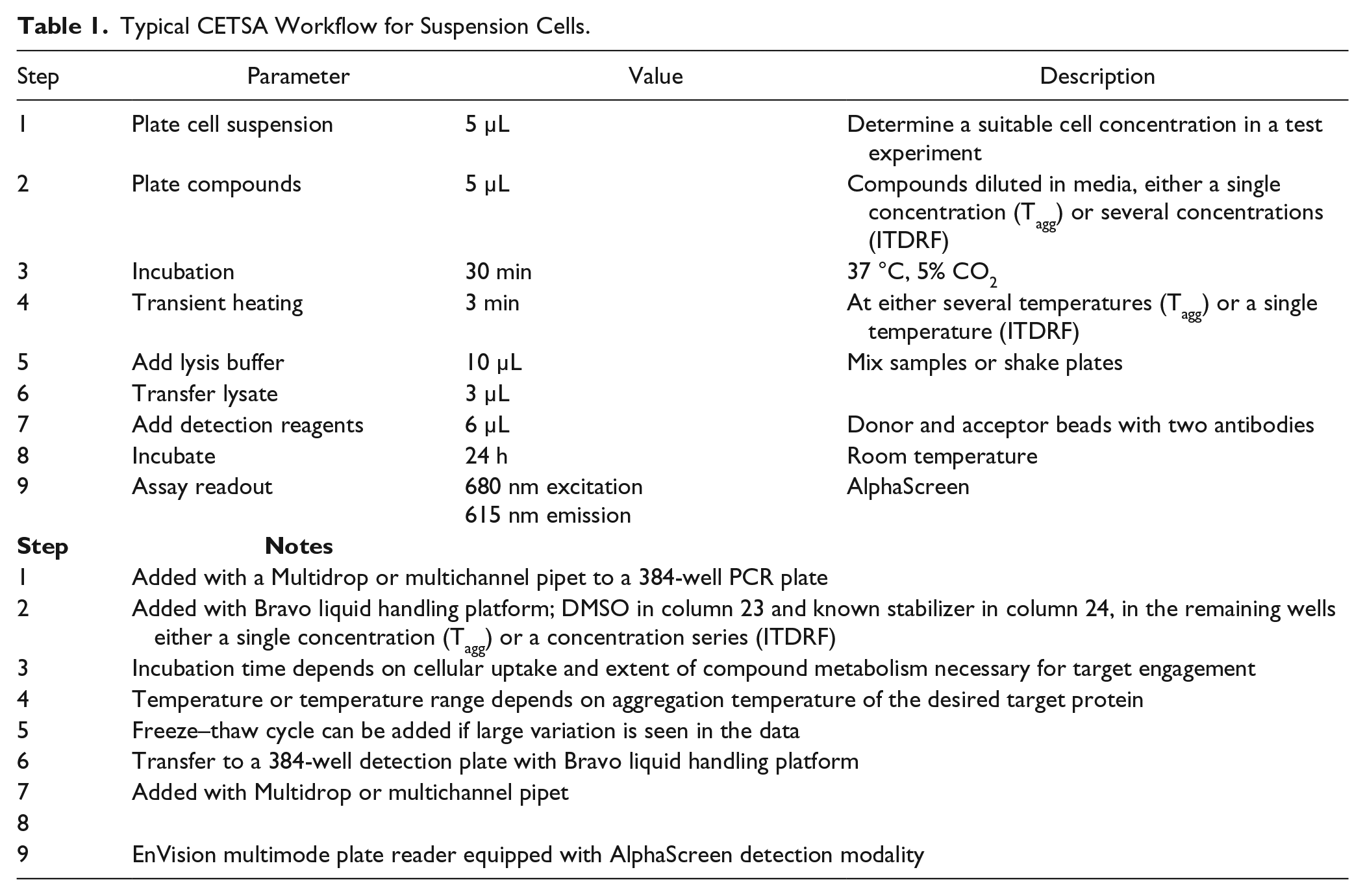
The original CETSA workflow 15 for studies of target engagement in whole cells involved sample treatment in cell culture flasks and included both wash steps and sample workup prior to WB detection. Hence, to enable high-throughput assays, there was a need to change to a microplate-compatible readout for protein quantification and to reduce the number of assay steps to improve sample throughput as described in detail previously. 22 A schematic illustration of the original assay workflow is provided alongside a fully screen-compatible workflow in Figure 2 . An example of an optimized workflow for an example microplate-based assay is also provided in Table 1 . No major concerns were expected in moving the assay from PCR tubes and manual pipetting to 384-well PCR plates and automated liquid handling, although a few details had to be addressed. We will go through them in order of appearance in the assay workflow, starting with the compound treatment step. The focus is on application of the assay to live cells, as this is associated with more significant obstacles and potential variability than the use of cell lysates.
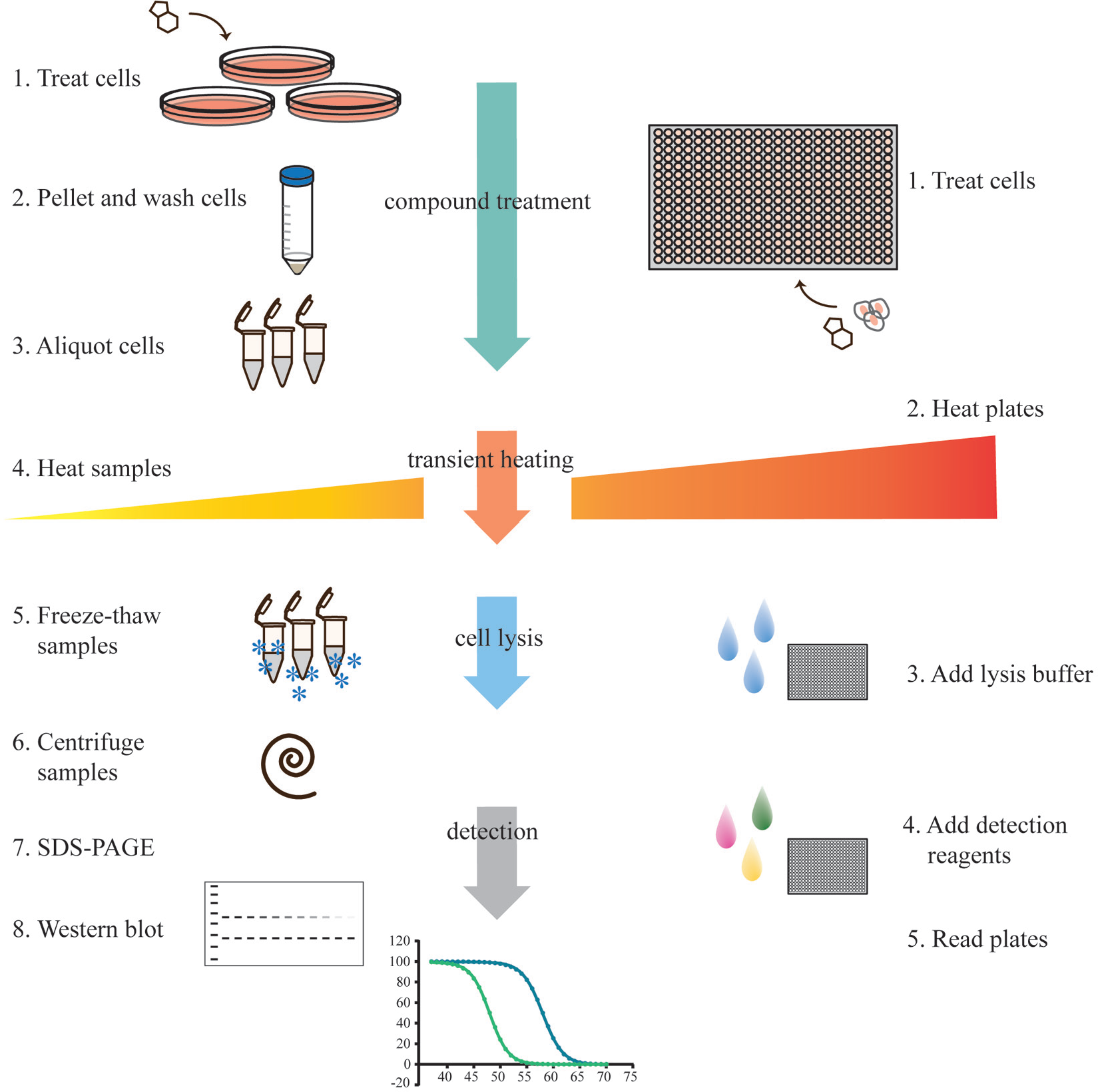
Cartoon illustration of the experimental workflow for CETSA. The left-hand panel depicts the steps for the original WB-based protocol, and the right-hand side shows the corresponding workflow for a fully screen-compatible assay. In the WB-based protocol, when adherent cells are investigated, it is also necessary to trypsinize cells at step 2. The detection using WB (steps 7 and 8) also comprises several steps that are not outlined in the figure. For screen-compatible experiments, additional steps for transferring samples from different plate types are sometimes necessary (i.e., lysate from PCR plate to a detection-compatible plate), lengthening the procedure. In an ideal setup, such transfers would be avoided. For some detection methods, cell lysis is not necessary, thereby shortening the overall assay procedure.
Typical CETSA Workflow for Suspension Cells.
Sample treatment
In the original protocol, cells were first treated in tissue culture flasks and washed prior to the transient heat challenge, probably driven by a concern that the heating would alter cell permeability for the compounds and thus potentially perturb the results. Although this is likely to occur, the extent depends on the kinetics involved and the length of the heat challenge. This concern should be weighed against the added steps required to wash the cells in microplate format. In addition, washing of cells will also alter the linked equilibria and comes with the risk of favoring ligands with a longer off-rate that stay bound to the target protein even after the excess ligand is removed (i.e., smaller k1, Fig. 1D ). As with other thermal shift assays, it may also be necessary to saturate the protein target to achieve a detectable Tagg shift,29,30 and this should be taken into account when selecting a relevant compound concentration.
Published microplate-based applications of CETSA are hitherto restricted to model systems that allow the use of cells grown in suspension during the treatment step. We have deliberately avoided the use of adherent cells, as their use in suspension may alter the biology of interest, even if incubation times following detachment are kept to a minimum. Consequently, their ideal application in CETSA requires the adherent cells to remain attached to a suitable matrix during ligand treatment and the subsequent transient heating. Although there are several ways to achieve this, a straightforward solution would be to use suitable tissue culture–treated 384-well PCR plates. Such plates are needed to allow an even seeding of adherent cells in all wells and, at the same time, ensure excellent thermal contact with the heating block in the PCR machine. A less than perfect contact with the samples could otherwise lead to plate-edge effects or result in the need for increased heating times. On this note, one challenge in the screen adoption of CETSA has been to identify PCR plates that are compatible with the transient heating with minimal skewing of the plate after return to room temperature. This is essential for allowing either an even transfer of the heated and lysed samples to a suitable detection plate or reliable plate stacking and detection in a microplate reader.
Another challenge associated with the treatment step in 384-well PCR plates was the random appearance of a lack of contact between compound solutions dispensed to the bottom of the wells by means of acoustic dispensing and the cell suspension added with a Multidrop. This was traced to the presence of an air gap between the compound solution and the cell suspension, which sticks to the surface of the V-shaped well because of surface tension. To solve this issue, the dispensing could be performed either in the reverse order, which in our lab becomes more challenging for reasons of timing, or by centrifugation of the plate without pelleting of the cells. We have also solved this by more traditional dispensing of diluted compounds prior to the addition of the cell suspension.
Cell lysis
A further screen adaptation concerned the use of lysis buffer instead of repeated freeze thawing in the original protocol to achieve cell lysis. Our initial concern was that the use of detergents in the lysis buffer would resolubilize irreversible aggregates of denatured proteins, thus negatively affecting our ability to accurately measure the proportion of soluble protein. This must be carefully examined during assay development for each individual case, for example, by comparing Tagg curves and concentration–response curves with those observed using the original WB protocol.22,24 A recent study carefully investigated the impact of a variety of different detergents and showed that the irreversible aggregates were not readily solubilized for a broad range of different proteins, 26 with the exception of high concentrations of sodium dodecyl sulfate (SDS), which should be avoided. While the resolubilization has not been an issue in the case studies investigated so far, a more significant problem has been the lack of reliable and consistent cell lysis, which is achieved using lysis buffers. As this has a very significant impact on the assay variability, it is highly recommended that this is kept under careful control. For example, to ensure successful cell lysis, it is important that there is sufficient mixing of the sample with the lysis buffer. If the resulting data still have high variation, a higher concentration of lysis buffer can be applied, or a freeze–thaw cycle can be added after the addition of lysis buffer. We have also found that the effectiveness of the lysis buffer should be monitored over time.
Protein detection
Additional simplification of microplate-based protocols concerns the option to leave out the centrifugation step that removes irreversible aggregates of denatured proteins (see the comparison of workflows in Fig. 2 ). This is possible when using detection modalities that allow for selective quantification of stabilized protein against a background of denatured and aggregated proteins. Such measurements require the signaling entities, whether they are epitopes for antibody recognition, as in our Alpha-Screen setups, or introduced tags allowing for luciferase or EFC-mediated signals, for example, to remain buried and inaccessible to detection reagents following cell lysis.
One of the biggest and somewhat unexpected challenges when moving to a fully homogeneous assay with immunoassay detection in microplate format was the observation of significant ligand-induced signal suppression.22,24 This phenomenon was also observed in the absence of heating, and hence was not related to the CETSA approach, but resulted from the loss of antibody recognition in the presence of the ligand. We have observed this for all proteins tested so far, such that the identification of a suitable antibody pair requires careful testing of ligand-induced signal perturbation or conditions that suppress this effect.22,24,31
This issue is less likely to be encountered in assays that are not dependent on affinity recognition of epitopes on native proteins, provided the alternative signaling tags are positioned in such a way that their availability is not affected by ligand binding. As a consequence, the use of generic tags avoids the requirement to test multiple antibodies to find a suitable pair, thus facilitating and shortening assay development. As already mentioned, microplate-based applications of CETSA based on tagged proteins are just emerging, and it is likely that other approaches will follow. The decision to opt for tagged or unmodified proteins will naturally depend on the assay purpose and the cells under investigation. Our focus has been on the identification of workflows and detection modalities that allow for quantitative, high-throughput studies in primary cells, with the intent to enable such studies directly in patient cells.
Example Applications
The outline of the original CETSA publication was largely focused on demonstrating potential utility for the drug discovery process and included concentration–response experiments of multiple drugs, selectivity studies, examples of intracellular drug activation, blocking of transporters to slow drug uptake and action, and in vivo studies of target engagement. 15 For more efficient application in drug discovery programs, however, the assay logistics must be changed from a WB format to a higher-throughput format.
Library Screening
We recently outlined the prerequisites for transferring CETSA to a microtiter-plate format, including a small feasibility study based on target engagement of the human kinase p38α (MAPK14) in live HL60 cells in microtiter plates. 22 As this was a pilot study, the goal was to compare the results from a homogeneous immunoassay readout (AlphaScreen) with those observed using the original WB protocol. Agreement was demonstrated with regards to both Tagg shifts and ITDRFCETSA data for two well-characterized and cell-permeable p38α inhibitors in a 96-well-based assay. This study also included an experiment where these two inhibitors were seeded into a randomly chosen screening plate within our primary screening set to ensure that they were selectively identified as actives in the context of a library screen. Encouraged by these results, we next moved to a more realistic setting for a primary screening campaign, targeting human thymidylate synthase (TS) in intact K562 cells. 24
There are several reasons why TS serves as an excellent model system for the investigation of screen feasibility of CETSA in live cells. First, it is a well-validated drug target with several different classes of drugs in clinical use,
32
many of which were present in our primary screening set. The majority of these drugs are dependent on intracellular enzymatic activation to enable potent binding to TS, such that the screen had to be conducted in live cells to mediate a positive signal of target engagement.
15
Besides serving as quality controls for the screen outcome, the activating mechanism of these drugs also allowed for a comparison of the time dependence of the appearance of target engagement, as illustrated in
Figure 3
. While 5-fluorouracil (5-FU) remains a mainstream treatment, its intracellular activation in vitro in K562 cells is considerably slower than what we observe for structurally related floxuridine, bearing a 2-deoxy-
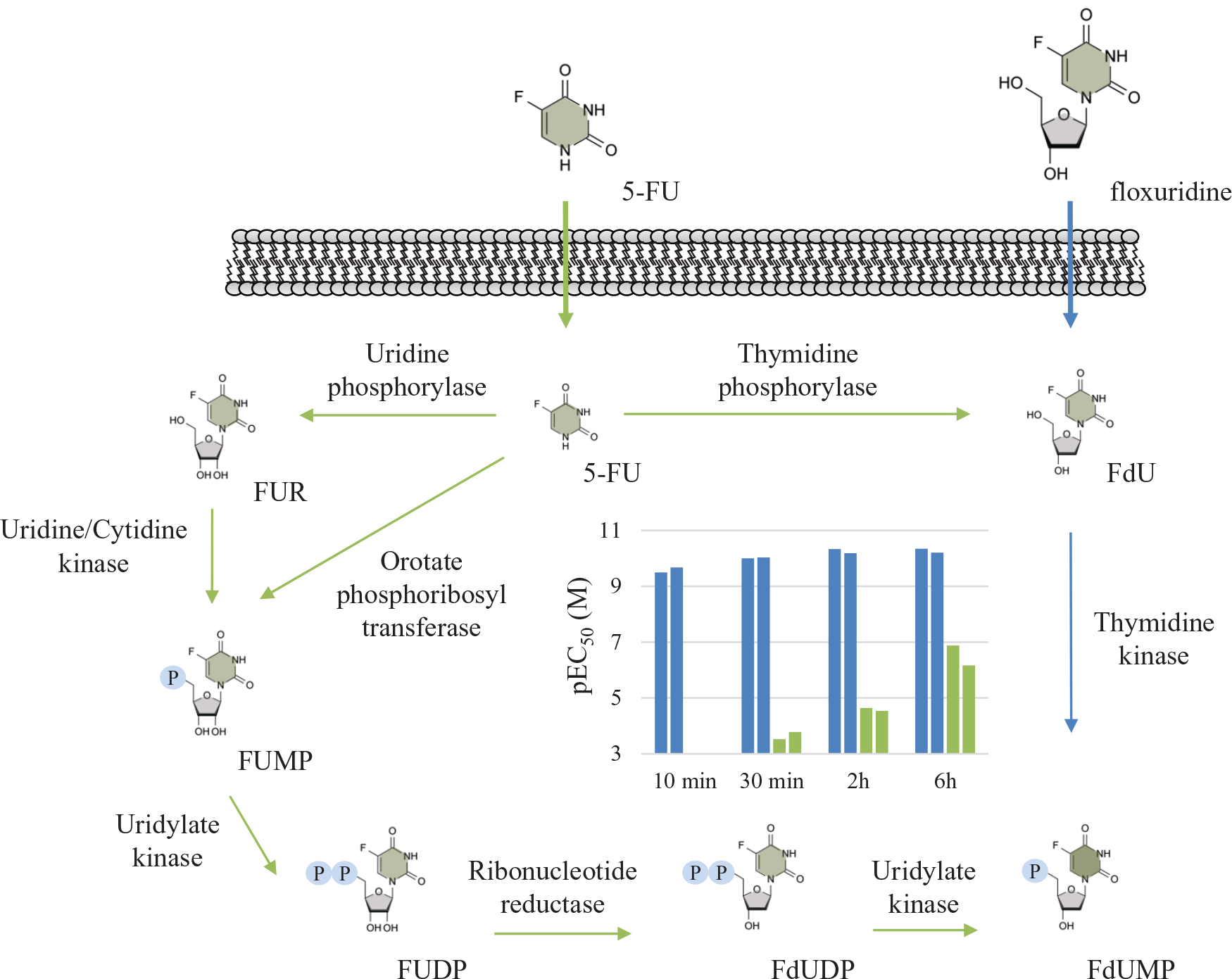
Schematic outline of the cellular uptake of the two drugs floxuridine and 5-FU and their intracellular enzymatic conversion to the active species FdUMP, which is a prerequisite for potent binding to TS. At the center of the scheme is a graph showing the apparent EC50 values from ITDRFCETSA experiments in live K562 cells, with data provided from two independent experiments in which the drugs were incubated for different times with the cells prior to the transient heating. 24 As can be seen from the data, there is a significant difference in the rate of uptake and metabolic conversion, with a near immediate target engagement observed for floxuridine (left bars), while 5-FU (right bars) takes 6 h to reach apparent submicromolar potency.
The pilot screen identified not only all known TS inhibitors within the screening set, serving to demonstrate a low rate of false negatives, but also previously unknown inhibitors, including an intracellular metabolite of the drug decitabine. A more systematic investigation as to the potential appearance of false negatives was done by means of a search for compounds with structural resemblance to known inhibitors, and our preliminary conclusion is that the false-negative, as well as false-positive, rate is low. Clearly, more data from additional screens are required to further qualify this statement. Using a combination of recombinant enzymes and cotreatment experiments with inhibitors thereof, we were able to identify both deoxycytidine kinase (DCK) and deoxycytidylate deaminase (DCTD) as crucial enzymes in the intracellular conversion of decitabine to the active species inhibiting TS. Interestingly, binding of decitabine to TS was not observed when tested on the recombinant protein itself, and hence this finding would have been missed in a traditional in vitro screen based on the isolated enzyme. Although metabolic processing of hit compounds would not be considered as the norm, the results make a case for the complementary use of target engagement studies in live cells for primary screening, especially for projects in search of prodrugs.
SAR Studies and Chemical Probe Validation
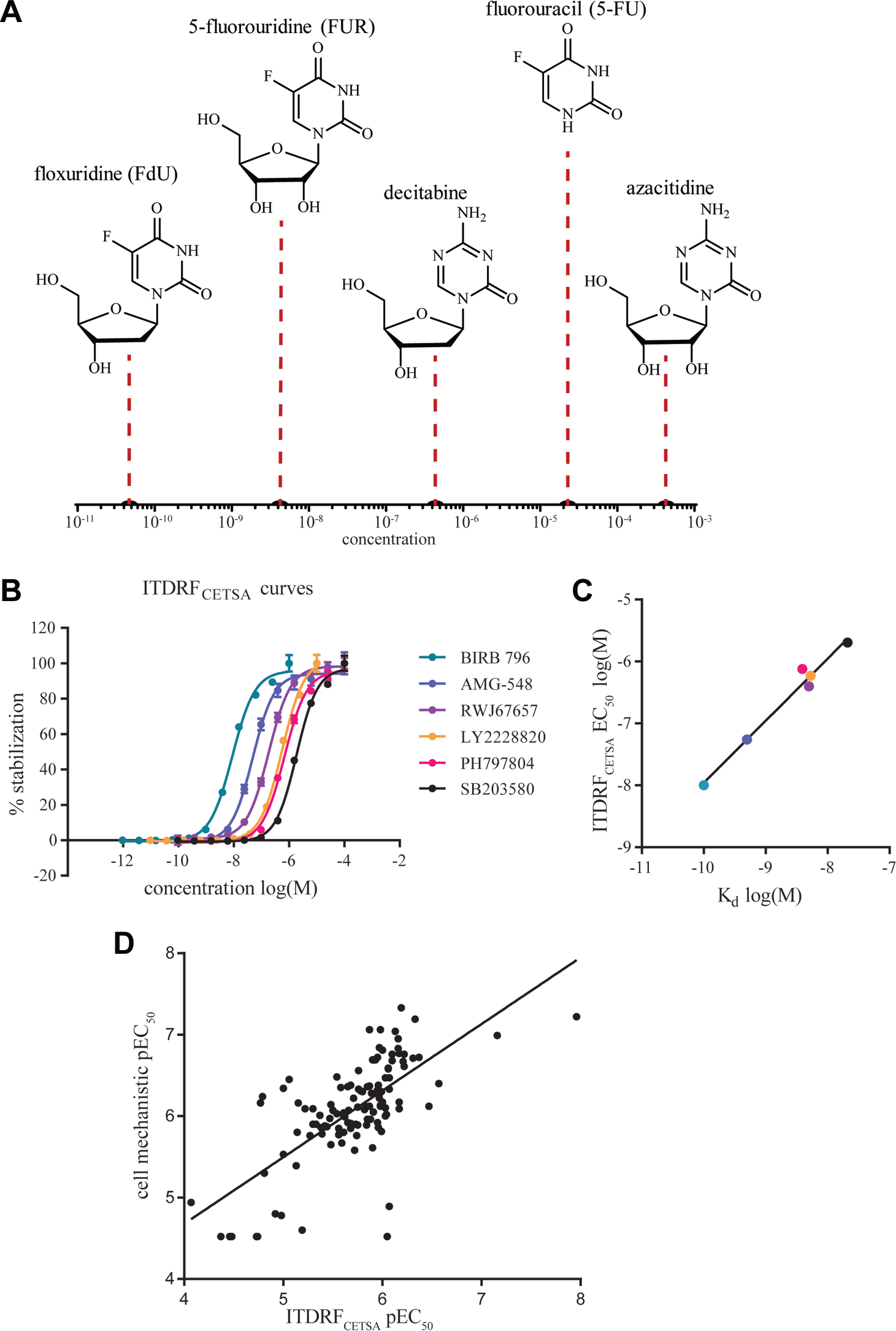
The follow-up work downstream of the TS screening campaign involved confirmatory studies of all hits in concentration–response experiments with several replicates at multiple assay occasions. Given the composition of the screen library, it also became an investigation of the SARs for a substantial number of active pyrimidine-based nucleosides. In this particular case, the SAR is unusually complex, as it includes different intracellular metabolic activation mechanisms (some of which are highlighted in Fig. 3 ), besides the normal aspects of a cellular assay with serum binding, cell permeability, and intracellular protein binding, and the differential binding to the target itself. Despite the complexity, the results largely reflected our expectations, with the potent deoxyuridine-like drugs ending up at the top of the list and with a clear preference for the deoxy forms. A crude illustration of the composite SAR is provided in Figure 4A .
(
To further investigate whether the CETSA response was concurrent with the appearance of the known active species of these drugs, we employed MS/MS methodology to measure their intracellular concentrations33,34 as a function of time. The CETSA responses were largely concurrent with the appearance of active metabolites, thus illustrating a feasible approach for coupling a given target engagement with the intracellular presence of the active compounds. Although this example came from a hit confirmation activity, the same approach could be taken as a secondary assay during hit selection and expansion. As alluded to in the introduction, SAR studies based on target engagement in live cells are valuable, as they allow for comparisons with the SAR observed for proximal functional and phenotypic responses in the same cells and under the same conditions. An excellent example of such a comparison was recently published for histone deacetylase (HDAC) inhibitors using BRET technology instead of CETSA to measure target engagement in live cells. 14 Observation of a correlation between target engagement of specific HDAC isoforms and the antiproliferative action of the compounds builds confidence that the observed pharmacology is primarily target mediated.
Additional examples of such comparative efforts are emerging, and recently, two different approaches based on EFC and TR-FRET were compared for measurement of intracellular ligand-induced stabilization of the bromodomain protein 4 (BRD4). 12 The comparison was done for a broad set of inhibitors covering more than two orders of magnitude in potency, with excellent agreement between the two readouts. These responses were also compared with data from a biochemical assay, resulting in a more moderate correlation, presumably due to aspects related to compound availability and permeability in the cellular assays. Using CETSA, we have similarly examined the SAR for a small subset of p38α inhibitors in cell lysates ( Fig. 4B ). 31 A comparison with reported biochemical data shows a strong correlation ( Fig. 4C ; r2 = 0.98), likely reflecting the maturity of these optimized inhibitors. Of note, however, is the apparent loss of potency in the CETSA EC50 values compared with the measured Kd values. This shift can be attributed to the fact that the CETSA experiments were conducted at a temperature (52 °C) well above the apparent Tagg of the protein, thus resulting in a need to saturate the interaction (see below for further details). Furthermore, in a collaboration with scientists at Uppsala University and GlaxoSmithKline (GSK), we have continued to investigate these correlations for a larger subset of inhibitors, demonstrating excellent agreement between live-cell CETSA responses and the observed impact on the tumor necrosis factor (TNF)-α production. 35 These results are in line with the well-validated involvement of p38α signaling in proinflammatory responses.36,37
Unfortunately, the initial use of CETSA for measuring target engagement has often been restricted to a few selected compounds, likely because of the limited throughput of the WB format. Although demonstration of target engagement in relevant cells is highly encouraging, it does not rule out additional contributions from off-target pharmacology. If it can instead be applied to larger series of compounds, similar to what was presented for the BRET approach, 14 and shown to correlate with phenotypic responses, it will increase the pharmacological validation of a given target protein considerably. We consider the risk that off-target effects would also match the SAR for a given target engagement as small, especially if the compound series span several orders of magnitude in potency. Hence, these types of correlations would undoubtedly bring significant value to a well-validated chemical probe.2,3
The lack of peer-reviewed publications on the use of CETSA for systematic investigations and comparisons of SAR likely reflects the short time period for which microplate-based assays have been available. Data are, however, emerging, and one example was presented by Marc Holbert of GSK at the SLAS Fifth Annual International Conference and Exhibition in 2016. 38 Using a CETSA approach based on EFC to report on levels of soluble protein, McNolty et al. systematically examined the concentration–response of >100 compounds for an undisclosed target protein and compared this with the SAR from a cellular mechanistic assay closely related to the target function. The compound potencies spanned over nearly three orders of magnitude, and the correlation between the two assays was good for the majority of compounds ( Fig. 4D ; r2 = 0.48). As the availability of generic readouts for CETSA in microplates increases, we expect to see a significant rise of these types of comparative studies, both for the purpose of compound prioritization and for pharmacological validation of chemical probes. Ideally, these comparisons are made with the full range of functional and phenotypic readouts available to the chemical probe or drug discovery project, further improving the understanding of target protein involvement in the measured pharmacologies.
Data Interpretation and Quantification
In order to use CETSA data successfully in hit selection and optimization, it is important to be able to rank candidate compounds by their affinities to the target protein. However, there are still very limited data publicly available where CETSA data are compared with other measures of potency across large series of compounds. Such systematic studies are thus urgently needed. It is also of interest to investigate selectivity by comparing the affinities of a single compound across a range of protein targets. These activities also require a quantitative framework that allows for translation of CETSA-derived concentration–response data to affinities.
Moving toward a more quantitative interpretation of CETSA results, it is of interest to first compare these data with data from thermal shift assays on isolated proteins, as they rely on the same underlying principle, and hence are likely to be governed by the same thermodynamic principles. A comparison with data from traditional thermal shift assays using differential static light scattering (DSLS) was done for three inhibitors of PARP-1, demonstrating agreement between the measured Tagg, as well as the magnitude and ranking of the shifts between the compounds. 15 A reasonable correlation has also been demonstrated between measured Tagg values for eight human and herpes proteins overexpressed in Escherichia coli using CETSA in live bacteria and Tm values established from thermal shift assays on the purified proteins. 39 These studies indicate that there could be portions of the proteome for which we can apply the same models that are used for analysis of traditional thermal shift assays.
However, when comparisons were instead made across the melting proteome using TPP, there was a relatively poor correlation between observed Tagg shifts for binding of staurosporine to known kinase targets with measured potencies using an affinity pulldown assay. 25 The pulldown was done using the kinobead approach, which is based on immobilized broad-spectrum kinase inhibitors, followed by MS/MS detection of captured proteins. From the standpoint of fully understanding compound selectivity or identifying potential new target proteins with TPP, it is troublesome that 17 kinases, which demonstrate significant binding in the kinobead assay, were nonresponsive in the CETSA approach. Although there could be several confounding factors when comparing data from two different technologies, the authors accurately point out that the size of the Tagg shift is likely to differ between proteins and ligand binding modes.
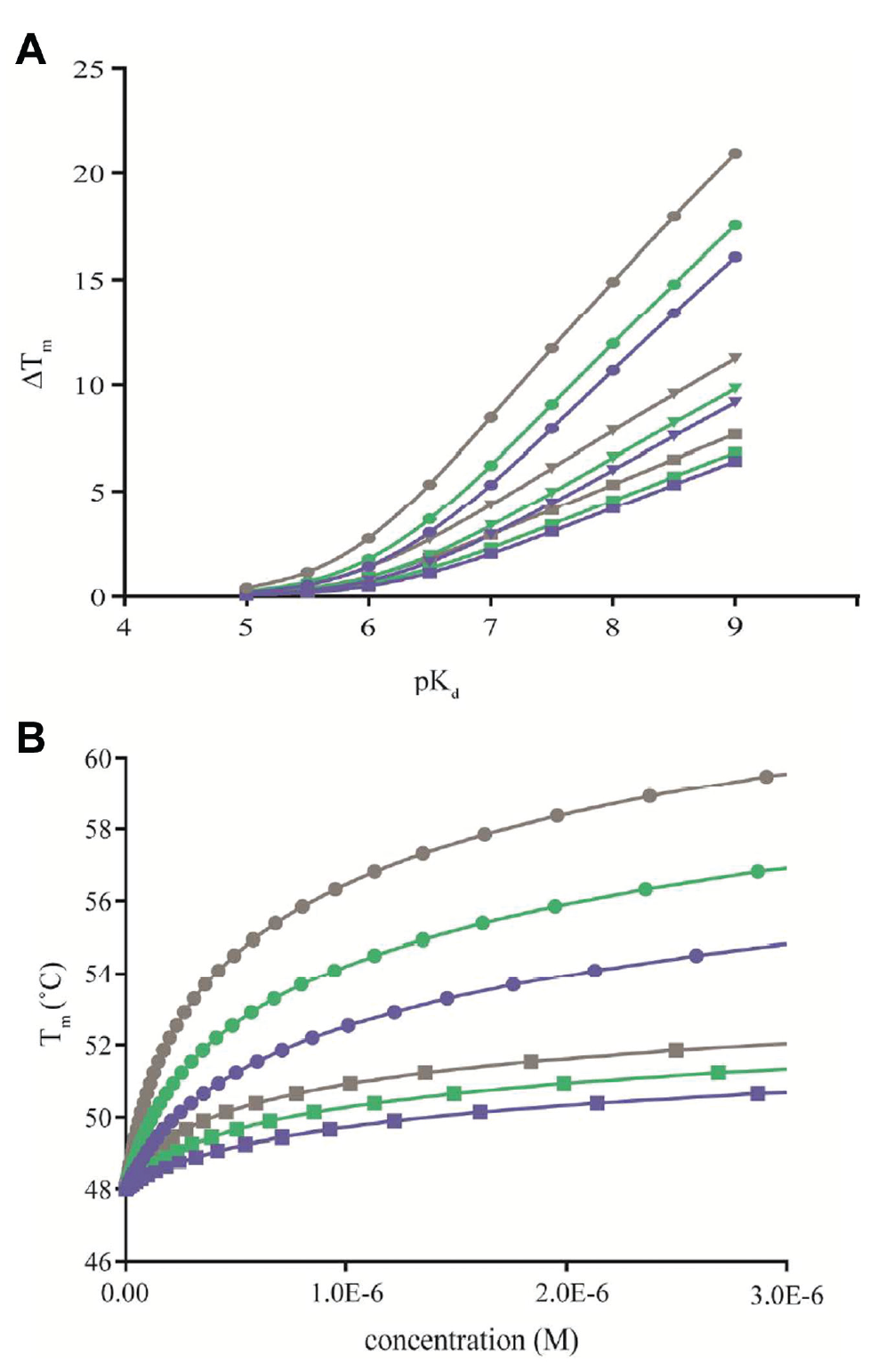
The key difference between Tm shift assays and most other binding assays is that the Tm shift, and thereby the interaction affinity, is measured at the shifted Tm and extrapolations are needed to compare affinities at a common reference temperature (e.g., 37 °C). It is thus well established in the field that the magnitude of Tm shifts is not only dependent on the ligand affinity at the reference temperature, but also influenced by ligand binding and protein unfolding thermodynamics.30,40,41 This dependency is illustrated in Figure 5A for a scenario that assumes equilibrium conditions, a single site interaction exclusively with the native protein and a two-state protein unfolding transition. Although not specifically addressed here, the reader should also be aware of the possibility that the ligand can also bind the unfolded state, which requires the application or models that also take account of the additional equilibria. 42
(
Using such simulations, it can clearly be seen that a given Tm shift is not exclusive for a certain binding affinity (at the reference temperature), but instead represents a broad range of different affinities when extrapolated to, for example, 37 °C to enable comparisons with other binding experiments. In fact, one weakness of Tm shift assays is that there is a bias toward entropically driven interactions,40,41 as these become more potent with increasing temperature, while enthalpically driven interactions move in the opposite direction. This is unfortunate considering that there are rational arguments for why we should strive toward compounds with improved enthalpic efficiency.43,44 Regardless of these considerations, the main point to consider is that any comparison of binding to different proteins, or classes of ligands, using thermal shift assays must take account of differences in thermodynamics to avoid a skewed view of the relative potencies. On a more positive note, it is clear from literature that significant correlations are observed when studying a single protein and ligand binding enthalpies remain similar, 17 whereas weaker correlations are observed when comparisons are done across similar target proteins.45,46 Although it remains to be seen to what extent CETSA data comply with these expectations, we anticipate them to broadly follow the same principles as traditional thermal shift assays. Careful studies addressing these matters in detail are needed.
A significantly better correlation was observed when the TPP-derived Tagg data were replaced by CETSA-derived isothermal concentration–response data for a comparison with affinities for the kinase inhibitor GSK3182571. 25 Similarly, in a follow-up study ITDRFCETSA for the HDAC inhibitor panobinostat ranked the binding to different HDAC isoforms in agreement with kinobead-derived data. 28 These data are more in line with the correlations observed for TS, p38α, and an undisclosed target protein, as described in the previous section. The improved correlation for concentration–response data is expected because contrary to the magnitude of the Tagg shift, the inflection point of these curves is less dependent on ligand binding and protein unfolding thermodynamics ( Fig. 5B ). This gives the impression that the alternative use of ITDRFCETSA instead of Tagg shifts may improve selectivity measures from TPP experiments, but as described below, care must be taken to instead account for changes in potency with experimental temperature as Tagg differs between proteins.
Although the correlations with kinobead data improved when comparisons were based on ITDRFCETSA data, there are still discrepancies in the absolute magnitudes, with the curves shifted to higher ligand concentrations in the CETSA experiment. This observation is also in agreement with expectations from studies on isolated proteins, where the application of isothermal denaturation techniques at temperatures below the Tm is known to be more sensitive and respond to lower concentrations of ligand than thermal shift assays. 21 As CETSA is commonly applied at or above Tagg to ensure detectable levels of protein denaturation and aggregation, we expect a similar loss of sensitivity. From the equilibrium two-state model, it can be shown that increasing concentrations of ligand are required to stabilize the protein as the temperature is progressively increased above Tm ( Fig. 5B ), and our unpublished data on p38α are largely in accordance with these expectations. 31 A similar trend with decreasing apparent potencies with increasing temperature, although with the relative ranking between the compounds remaining intact, was also reported for the undisclosed target studied by GSK, as described above. 38 Contrary to these observations, ITDRFCETSA data for dasatinib obtained at three different temperatures remained largely invariant with experimental temperature, 25 thus leaving us with the need for additional data before we can better understand how to build models and quantitatively interpret CETSA data.
It is important to emphasize that the theoretical arguments and illustrated scenarios assume conditions where the ligand–protein interaction and protein unfolding remain at equilibrium throughout the melting experiment. In CETSA experiments, the thermal unfolding is not induced using a slow temperature scan rate to ensure equilibrium conditions, but instead, these experiments are more similar to isothermal denaturation experiments where the temperature is raised instantly and the thermal denaturation is followed over time.19-21 Presently, CETSA is employed as an endpoint assay with the key experimental parameters being the temperature and length of the transient heating step. These can be optimized for each system to ensure sufficient irreversible aggregation to allow detection of reduced levels of soluble protein. Furthermore, the heat challenge in CETSA experiments is commonly set to 3 min, which is a much shorter time frame than the length of most temperature scans employed in thermal shift assays. The transient nature of this step thus comes with an increased risk of encountering “frozen” equilibria. 29 Put in other words, the transient heating may be too short to allow for the system to reestablish equilibrium at the higher temperature, for example, if the rate constant for the ligand dissociation is very low in relation to the heating time. In these instances, CETSA may yield exaggerated Tagg shifts and concentration–response curves shifted toward higher apparent potencies.
As a final note, it is important to point out a few other known confounding factors that could influence the interpretation of CETSA data. In live-cell experiments, the protein under investigation may already be stabilized by endogenous ligands, and hence these may prevent a further stabilization of the target protein or even result in an apparent destabilizing effect when the natural ligands are displaced. 23 In such cases, stabilization may instead be achieved in a lysate, where cell lysis leads to an effective dilution of the endogenous ligands. 23 In line with this, ATP binding proteins were generally shown to be stabilized in intact cells compared with cell lysates, 25 likely as a result of intracellular ATP binding. Importantly, if experiments are done in cell lysates, instead of live cells, conditions must be established such that the actively binding form of the target protein is retained following cell lysis, for example, by including protease or phosphatase inhibitors to prevent unwanted changes in posttranslational modifications.
Perceived Applications in Drug Discovery Programs
Recent meta-analysis on the requirements for successful progress through clinical trials has led to the establishment of the “four pillars” or, alternatively, the “five R’s.”2,8,9 These guidelines outline the following criteria: the choice of an appropriate target protein (1) that translates to useful clinical effects when modulated by drugs; demonstration of drug candidate presence (2) with concurrent target engagement (3) in the right tissue; and finally, the translation of drug-induced target modulation into the desired functional effects (4) and phenotypic responses (5). The need for target engagement measurements in patient cells has fueled recent technology developments,10,11 and as these become more readily available, it appears relevant to ask the question as to where they fit in the drug and chemical probe discovery processes. 47 Below, we will speculate on this matter, but will restrict the discussion to foreseen applications in microplate format. For a discussion on later stage applications in in vivo models and the tentative application in patient cells, the reader is referred to recent reviews on the subject.10,23
As already described, there are several technologies emerging that could support the use of live-cell target engagement measurements already at the stage of primary screening, whether these are based on, for example, EFC, TR-FRET, BRET, or CETSA.12-14,24 The necessity to apply such technologies at this early stage depends on the nature of the target protein and to what extent it can be expressed and purified in an active and relevant form to allow a traditional screen on the isolated target. It will also depend on the availability of suitable in vitro assays for the chosen targets, which in turn relies on whether it belongs to an established target class or is an orphan. Besides technical and financial considerations when comparing these approaches, the decision will likely also reflect the different cultures between labs, such as whether a complex SAR that takes account of, for example, serum binding, cell permeability, and intracellular activation mechanisms is considered beneficial for bias toward discovery of hits with suitable properties. At the very least, it offers a generic possibility for target proteins that are not amenable to other screening approaches.
Given the expertise and work procedures built around expression and purification of target proteins and established routines for screening of many target classes, it is perhaps more likely that these assays will be applied at the stage of hit profiling, when compounds in the 102 to 104 range are to be triaged. The prevalence of hit compounds with undesirable characteristics, including pan-assay interfering substances (PAINS) 48 and promiscuous aggregators, 49 creates a great need for selection procedures that easily identify such false positives. Thermal shift assays are generally considered effective in promoting hits with sound mechanisms, as they select for protein stabilizers, although it should be noted that thermal shift assays can also report on destabilizing compounds. 42 These can result from a simultaneous (or exclusive) interaction of the ligand with the unfolded protein. In live cells or in concentrated cell lysates, the observation of an apparent destabilizing effect can also result from the displacement of an endogenous ligand with different ligand binding thermodynamics.
The role of target engagement measurements during lead generation was recently revisited, pointing out the need to complement traditional in vitro measures of target potency; selectivity; and administration, distribution, metabolism, excretion, and toxicity (ADMET) properties, ideally in cells reflective of the target tissue. 47 As the target engagement assays can be performed in live cells, they naturally enable alternative examinations of these properties by varying, for example, the serum concentration or blocking of transporters, while monitoring the impact on intracellular target protein binding. In addition, the kinetics of uptake and interaction, 14 as well as potential intracellular compound metabolism, 24 could already be followed at this stage. In such studies, it is important to preincubate the ligand and the cell material to ensure sufficient time for the compound to penetrate the cell membrane or undergo metabolic processing. Inadequate preincubation length can lead to the appearance of false negatives.
The natural progress through the drug discovery process normally involves the gradual application of more primary cells, and thus more predictive material over time. Although there is currently a strong drive toward the application of more advanced cell models and even organoids directly in primary screening,50,51 in an effort to early on select for the most suitable starting points, this has been hampered by both the lack of sufficiently specific and screen-amenable technologies for handling these cells and the costs and feasibility involved in scaling up these systems to support screening of sufficiently sized libraries. 52 We foresee that CETSA will be applied in a more traditional manner, that is, starting with engineered cell lines or even in cell lysates, and then progress toward more primary material and even patient cells, but also that efforts will be attempted at which primary cells are moved all the way up to the initial stages of a drug or chemical probe discovery effort. On this note, we have preliminary unpublished data demonstrating excellent performance of the p38α CETSA assay in human primary neutrophils (Hanna Axelsson, personal communication).
We anticipate that as the field matures, more studies will investigate selectivity for a small set of compounds with TPP.25-28 The use of MS-based experiments for broad measures of selectivity on native proteins in live cells represents a great complement to in vitro panels of selectivity assays. Given the costs involved, this technology will, in its current setting, likely be applicable only to selected compounds, and thus not support SAR investigations on selectivity. However, one can also envision smaller selectivity panels centered on related target proteins and known important off-targets for a given project. Following our successful application of immunoassays for a few selected target proteins, a natural next step would be to attempt simultaneous measurements for multiple proteins in the same well of a microplate, using detection methodologies that allow for multiplexing.
A tentative approach inspired by the AlphaScreen setup would be to move to a multiplexed particle-based flow cytometric assay, 53 where one of the target-directed antibodies is immobilized to the beads and the second antibody is labeled with a suitable fluorophore. The use of two selective antibodies toward each target protein helps to improve specificity, but a simpler variant would be a competition format of the same assay based on a highly selective antibody with a matched fluorescently labeled peptidic antigen. In addition, the application of generic detection formats based on tagged proteins allows for the construction of, at least in principle, near proteome-wide selectivity panels in microplate formats, with each protein construct in a single well, as already outlined for an approach that investigates drug-induced changes in protein levels. 54 These types of target engagement formats, when applied to live cells, will allow for selectivity studies that take into account the interplay between different target proteins and the intracellular environment.
Discussion
The appearance of several new technologies for measurements of intracellular target engagement significantly improves our ability to link desirable phenotypic effects to a given target occupancy. While these studies used to be cumbersome, often requiring radiolabeling or other chemical modification of each ligand, they seldom allowed for studies of more than a few selected compounds. With the arrival of microplate-based methodologies, these studies can now be applied to full screening libraries or alternatively support SAR examinations to aid in the selection and prioritization of compounds. Such studies over complete compound series, instead of a few representative compounds, are likely to have a significant impact on the validation of chemical probes.
One of these technologies is based on the novel finding that thermal shift assays can be used to study drug binding in living systems. While systematic studies are still needed to fully develop a quantitative framework that allows our accurate interpretation of affinities and selectivities, it is already clear that the CETSA will be useful for ranking of compounds. So far, the methodology has been applied either to a select number of well-defined target proteins or to the full protein meltome, while it is likely that future developments will also include intermediately sized panels of tagged proteins to address selectivity within target classes or otherwise selected proteins. Alternatively, such selectivities could also be assessed by the application of detection modalities that allow for significant multiplexing in the same well.
The further translation of the methodology into patient cells, and potentially even into the clinic as a biomarker for drug responses, requires appropriate choices of robust and reliable detection methodologies that allow for detection of native unmodified proteins. Given our challenges with drug-induced loss of antigen recognition, this may require the development of alternatives to standard immunoassays or assay conditions that eliminate these issues. The prospect of potential clinical application may also serve as a significant driver for the development of novel protein detection methodologies that are compatible with drug presence.
Footnotes
Acknowledgements
Karolinska Institutet, SciLifeLab, and the Swedish Research Council (Vetenskapsrådet) are acknowledged for funding of Chemical Biology Consortium Sweden. B.S.L. acknowledges the Royal Institute of Technology. Hanna Axelsson and Helena Almqvist are gratefully acknowledged for their generous sharing of experimental details and know-how on CETSA in microplate format and for careful reviewing of the manuscript. The majority of our CETSA work as described herein was conducted in close collaboration with the research groups of Professor Pär Nordlund at the Karolinska Institutet and Nanyang Technological University in Singapore. Marc Holbert of GSK is acknowledged for sharing nonconfidential CETSA data from his poster at the SLAS Fifth Annual International Conference and Exhibition in San Diego in 2016.
Declaration of Conflicting Interests
The authors declared no potential conflicts of interest with respect to the research, authorship, and/or publication of this article.
Funding
The authors received no financial support for the research, authorship, and/or publication of this article.