
Abstract
Focal adhesion kinase (FAK) is a promising cancer drug target due to its massive overexpression in multiple solid tumors and its critical role in the integration of signals that control proliferation, invasion, apoptosis, and metastasis. Previous FAK drug discovery and high-throughput screening have exclusively focused on the identification of inhibitors that target the kinase domain of FAK. Because FAK is both a kinase and scaffolding protein, the development of novel screening assays that detect inhibitors of FAK protein–protein interactions remains a critical need. In this report, we describe the development of a high-throughput fluorescence polarization (FP) screening assay that measures the interactions between FAK and paxillin, a focal adhesion–associated protein. We designed a tetramethylrhodamine (TAMRA)-tagged paxillin peptide based on the paxillin LD2 motif that binds to the focal adhesion targeting (FAT) domain with significant dynamic range, specificity, variability, stability, and a Z’-factor suitable for high-throughput screening. In addition, we performed a pilot screen of 1593 compounds using this FP assay, showing its feasibility for high-throughput drug screening. Finally, we identified three compounds that show dose-dependent competition of FAT–paxillin binding. This assay represents the first described high-throughput screening assay for FAK scaffold inhibitors and can accelerate drug discovery efforts for this promising drug target.
Introduction
Focal adhesion kinase (FAK) is a 125 kDa nonreceptor tyrosine kinase that is overexpressed in a variety of human malignancies, including melanoma, glioblastoma, and breast, colon, ovarian, and pancreatic cancers. 1 We were the first to show overexpression of FAK in human tumor samples, 2 and years of experimental validation have led to the conclusion that FAK is a critical component for human cancer progression. From a biological standpoint, FAK is involved in cell motility, invasion, angiocrine signaling, lymphangiogenesis, metastasis, and the epithelial-mesenchymal transition (EMT).3,4 FAK also sequesters and inactivates proapoptotic proteins, such as p53 and receptor-interacting protein (RIP), to enhance the survival signals necessary for a cancer to invade and metastasize.5,6 FAK’s role in cancer has been confirmed by multiple methodologies, including clinical prognostic studies, genetically engineered mouse models, gene knockout studies, and site-directed mutagenesis.7–9
FAK functions as both a kinase and scaffolding protein, and it has three major domains: the N-terminal 4.1, ezrin, radixin, moesin (FERM) domain; the central kinase domain; and the C-terminal focal adhesion targeting (FAT) domain. FAK knockdown results in robust activation of apoptosis and growth arrest in cancer cells, with minimal effects in normal cells.10,11 Conversely, FAK-kinase inhibitors have a partial effect on apoptosis and tumor growth,12,13 and it is hypothesized that the FAK scaffold is the major modulator of FAK-dependent antiapoptosis. 14 FAK directly binds p53 to suppress p53-mediated apoptosis, 5 and disruption of the FAT domain by adenoviral FAK–carboxyl (C)-terminal domain of FAK (FAK-CD) was shown to induce apoptosis in cancer cells. 15 We showed that FAK-kinase inhibitors do not drastically inhibit FAK phosphorylation at autophosphorylation residue Y397, and receptor tyrosine kinases (RTKs) transphosphorylate FAK as a drug resistance mechanism. 16 Furthermore, FAK-kinase inhibitors have shown limited efficacy in Phase I and II clinical trials.17,18
Recent attention has focused on the FAT domain of FAK as an alternative approach to target FAK in cancer.
19
The FAT domain is a four-helical bundle at the C-terminus of FAK that contains key residue Y925 and is involved in multiple protein–protein interactions at the focal adhesion site. Numerous data have emerged showing the importance of the FAT domain as the initiator of FAK activation through its multiple interactions with paxillin, leupaxin, CD4, and DCC (Deleted in Colorectal Cancer).20–22 The integrity of the FAT domain is essential for localization of FAK to focal adhesions, association with integrins and RTKs, and downstream FAK signaling.
23
More specifically, FAK localization to the focal adhesion is mediated by the FAT–paxillin interaction, and mutation of the binding site was shown to have drastic effects on FAK phosphorylation, paxillin phosphorylation, focal adhesion turnover, cell adhesion, migration, and invasion.24,25 Paxillin contains alpha helical binding motifs termed LD motifs, named after the first two amino acids of the consensus sequence (LDXLLXXL). Two such LD motifs (LD2 and LD4) are required for binding to the FAT domain of FAK.
26
The FAT–paxillin LD2/LD4 interaction has been well characterized (
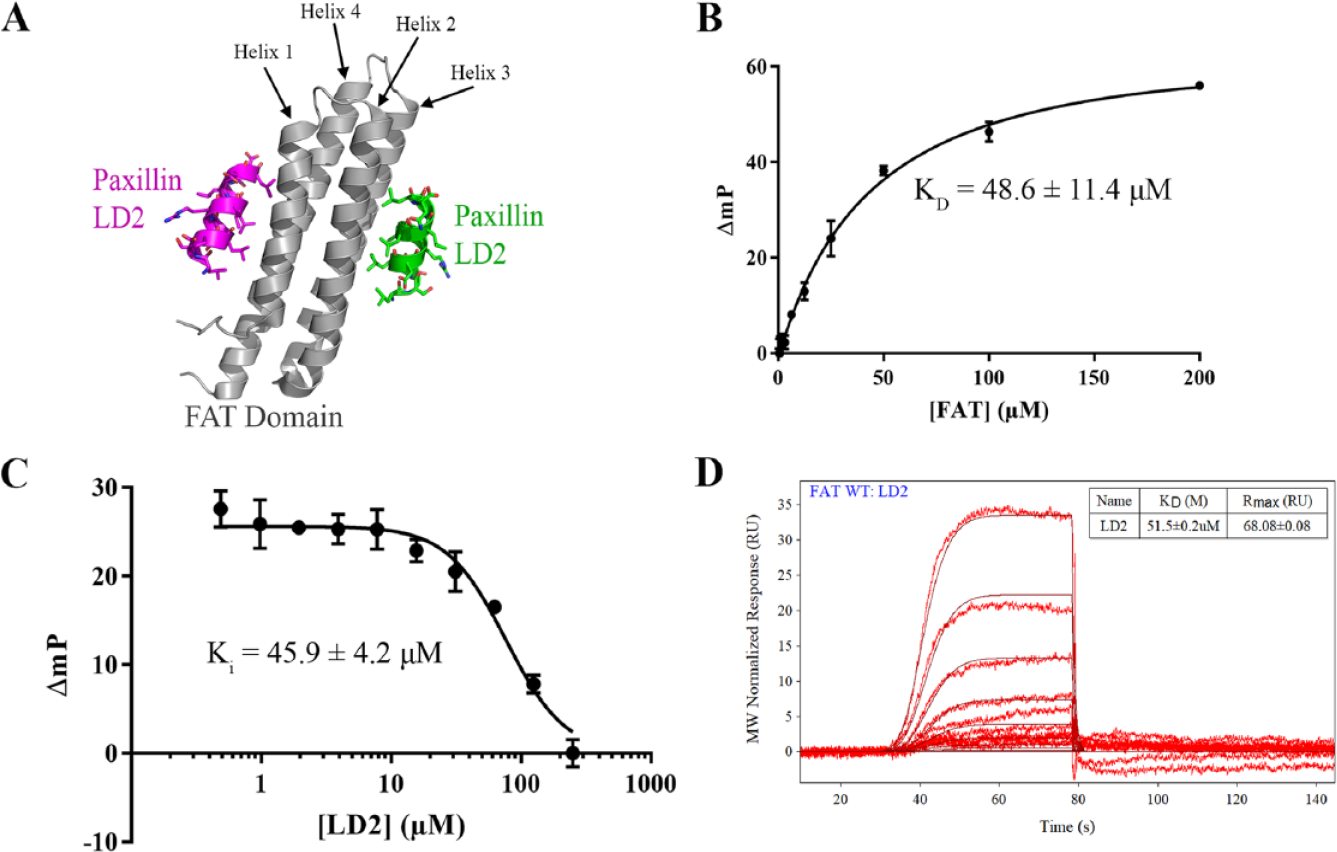
Characterization of the focal adhesion targeting (FAT)–paxillin interaction through modeling, surface plasmon resonance (SPR), and fluorescence polarization (FP).
Here, we report the development of a high-throughput FP assay to measure FAK–paxillin interactions and use in small-molecule drug-screening efforts. We show the structure-based optimization of a tetramethylrhodamine (TAMRA)–paxillin LD2 probe that binds to both the Helix 1-4 and Helix 2-3 binding sites on the FAK FAT domain. We use SPR as well as site-directed mutagenesis to validate the specificity of the signal. We evaluate the high-throughput characteristics of the assay and perform a pilot screen using the National Cancer Institute (NCI) Diversity Set V (Div5) 1593-compound library and automated liquid handling systems. Finally, we identify three compounds that display dose-dependent inhibition of FAT–paxillin binding. These results demonstrate a novel drug-screening approach for the identification of potential FAK scaffold inhibitors.
Methods
Reagents, Chemicals, and Peptides
TAMRA-tagged and unlabeled peptides were acquired as lyophilized powder through Biomatik (Cambridge, ON, Canada). High-performance liquid chromatography (HPLC) and mass spectrometry data confirmed proper identity and purity >95% for each peptide. Peptides were dissolved at 1–10 mM in 40 mM Tris (pH 8.1). For assay optimization studies, 384-well plates were acquired from Thermo Fisher (Nunc cat. nos. 264576, 242764, 262260, and 267461; Waltham, MA), Greiner (cat. nos. 781209 and 784900; Monroe, NC), and Corning (cat. nos. 3575 and 4514; Corning, NY). The NCI Diversity Set V library of 1593 compounds was graciously provided by Dr. David Azorsa (University of Arizona).
Expression and Purification of His-Tagged FAT and Avitag-FAT
The FAT domain of FAK (residues 892–1052) was cloned into the pET15b vector to produce an N-terminally His(6)-tagged FAT domain. The peT15b–FAT vector was transformed into Escherichia coli BL21(DE3) chemically competent cells (Thermo Fisher). Bacteria were grown in LB broth at 37 °C to an absorbance of 0.8 at wavelength 600 nm. To induce protein expression, 0.2 mM of IPTG (isopropyl β-D-1-thiogalactopyranoside) was added. Bacteria were further incubated for 4 h at 37 °C, after which cells were collected through centrifugation at 5000 ×g for 10 min. Cell pellets were resuspended using lysis buffer [20 mM Tris (pH 8.0), 200 mM NaCl, 5 mM β-mercaptoethanol (β-me), 10 mM imidazole, and 1× Halt protease inhibitor cocktail; Thermo Fisher] and frozen overnight at −80 °C.
On thawing, cells were further lysed through sonication (mild sonication for 2 min, 50% cycle, 5 s per cycle), and insoluble components were then removed through centrifugation at 12,000 rpm for 15 min. The cell lysate was incubated with HisPur Ni-NTA resin (Thermo Fisher) overnight while gently shaking at 4 °C. The resin was previously equilibrated with lysis buffer. For purification, the nickel resin with bound His-tagged FAT was transferred to a spin column and washed with 20 mM Tris (pH 8.0), 200 mM NaCl, 5 mM β-me, and 25 mM imidazole. The protein was eluted using 20 mM Tris (pH 8.0), 200 mM NaCl, 5 mM β-me, and 250 mM imidazole. The eluted protein was buffer exchanged using Zeba spin desalting columns (Thermo Fisher) into 20 mM Tris pH 8.0, 200 mM NaCl, and 5 mM β-me. Coomassie-stained SDS-PAGE (sodium dodecyl sulfate–polyacrylamide gel electrophoresis) analysis of the purified protein demonstrated around 90% purity.
Fluorescence Polarization Assay
The FP assay buffer used was 20 mM Tris, 200 mM NaCl, 0.05% β-me, 0.1% Triton X-100, 5% glycerol, and 1× Halt protease inhibitor cocktail. All final FP reactions were placed into a 384-well plate (Nunc cat no. 267461; Thermo Fisher) at 30 µL and shaken for 3 h at room temperature to reach equilibrium. The plates were read on a PerkinElmer EnVision plate reader with software EnVision Manager version 1.13 (PerkinElmer, Waltham, MA). A boron-dipyrromethene (BODIPY) tetramethylrhodamine (TMR) FP optical module (2100-4100) was used as the mirror. The excitation filter (2100-5830) used a wavelength of 531 nm, and both emission filters (2100-5800 and 2100-5810) were at wavelengths of 579 nm. The baseline mP of TAMRA–LD2–L10D (leucine 10 to aspartic acid conversion) only was set to 15 mP through the assay optimization wizard on the EnVision Manager software. The assay optimization set the measurement height to 6.5 mm, excitation light to 100%, G-factor to 1.01, detector gain to 300, and number of flashes per well at 25.
Saturation Binding Assays
For KD determination for TAMRA–LD2 and TAMRA–LD2–L10D, FAT was titrated from 0.2 µM to 200 µM into FP buffer containing 0.1 µM TAMRA–LD2 or TAMRA–LD2–L10D for a final volume of 30 µL. Wells with no FAT were used as a baseline value that was subtracted from the raw values to produce ΔmP values. The saturation binding data were processed through GraphPad Prism (GraphPad, San Diego, CA) using the “One site − Total” model to produce a saturation curve and a calculated KD with standard error (SE).
Competition Assay
For IC50 determination of unlabeled LD2 as an inhibitor, LD2 was titrated from 1 µM to 250 µM into FP buffer containing 20 µM FAT and 0.1 µM TAMRA–LD2 or TAMRA–LD2–L10D. Wells with no FAT were used as a baseline value that was subtracted from the raw values to produce ΔmP values. The plate was read at every hour for 4 h to test for time differences in IC50. The titration data were processed through GraphPad Prism to produce a dose–response curve and a calculated IC50 with SE. A four-parameter dose–response inhibition model was used, and the bottom fit was constrained to the lower plateau of the curve. Ki was determined from the following calculation: 30
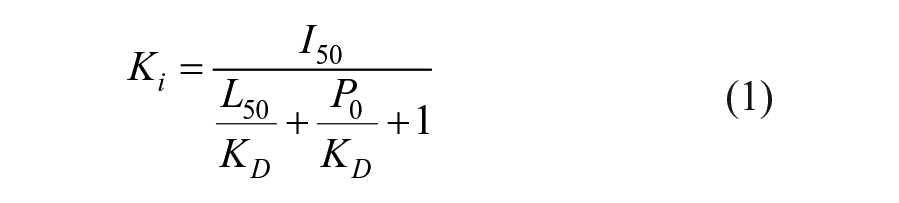
where I50 is the concentration of free inhibitor at 50% inhibition, L50 is the concentration of free ligand at 50% inhibition, P0 is the concentration of free protein, and KD is calculated from the saturation curve in
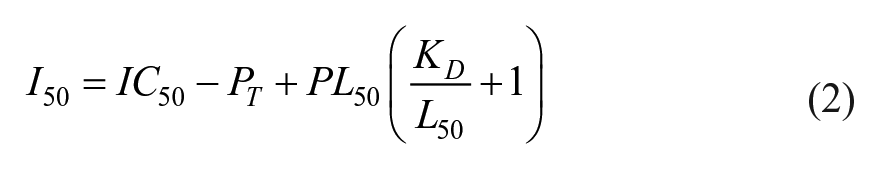
where PT is the total protein concentration, LT is the total labeled-ligand concentration, P0 is the positive root of P02 + (KD + LT) × P0 − PT, PL50 = PL0 / 2 (where PL0 = P − P0), and L50 = LT − PL50 (where L0 = LT − PL0). The compounds for the dose–response curves were serially diluted one to one from a concentration of 5 mM to 5 µM in DMSO. Then, 2 µL of compound at each respective concentration was added to 30 µL of a master mix containing 20 µM FAT and 0.1 µM TAMRA–LD2–L10D for a final DMSO concentration of 6.25% in each well, and for a final compound concentration range of 312.5 µM to 0.31 µM. The dose–response curves were fitted on GraphPad Prism, and the Ki was calculated using Equations (1) and (2). The SE of the Ki was calculated similarly by inputting IC50 SE into Equations (1) and (2). Commercially available compounds were purchased for follow-up studies at higher concentrations.
Surface Plasmon Resonance
SPR binding studies were performed on a ForteBio Pioneer FE SPR system (ForteBio, Fremont, CA). In brief, a SADH (streptavidin in dextran hydrogel) biosensor (ForteBio) was docked onto the flow cell and preconditioned with two injections of 10 mM NaOH, 1 M NaCl for 1 min at 50 µl/min. Subsequently, biotinylated Avitag-FAT protein was diluted in running buffer (20 mM Tris-HCl, 200 mM NaCl, and 0.05% Tween-20) and injected at 10 µl/min to achieve approximately 1000 RU of immobilized protein on channel 1. Empty channel 2 served as the reference control. After achieving a stable baseline, a concentration series (200 µM–0.01 µM) of paxillin LD2 wild-type (WT) and L10D peptide was prepared in final running buffer (20 mM Tris-HCl, 200 mM NaCl, 0.05% Tween-20, and 5% DMSO) and injected using the OneStep gradient injection method at a flow rate of 75 µl/min. 3% sucrose was used as a bulk standard control for OneStep injection, and a DMSO calibration curve was performed using a concentration range of 3.5% to 6.5% DMSO. Raw SPR data were appropriately processed in Qdat software (ForteBio) by normalizing the baseline prior to injection, aligning the channels, subtracting the reference channel, and blank subtraction. Kinetic data were fitted to a pseudo-first-order 1:1 interaction binding model used to calculate KD. In addition, a steady-state model and Req data points were used to validate the binding affinity. Visual inspection of the SPR sensograms was performed to verify appropriate model fitting, lack of mass transport effects, return to baseline, and lack of irregular kinetics.
High-Throughput Screening
A small-molecule diversity set library, Div5, was obtained through the NCI. Two microliters of each small molecule in DMSO were aliquoted into 384-well Nunc plates using the Acoustic Transfer System Gen 4 from Biosero (San Diego, CA). A master mix of 20 µM His-tagged FAT and 0.1 µM TAMRA–LD2–L10D in FP buffer was added into each well by a Tecan Liquid Handling machine using the MCA96 pipetting head (Tecan, Zurich, Switzerland), for a final drug concentration of 312.5 µM. A negative control of 20 µM FAT, 0.1 µM TAMRA–LD2–L10D, and 6.25% DMSO; a positive control of 0.1 µM TAMRA–LD2–L10D and 6.25% DMSO; and a positive control of 20 µM FAT, 0.1 µM TAMRA–LD2–L10D, 250 µM unlabeled LD2, and 6.25% DMSO were added to each plate for baseline referencing and Z’-factor calculation. Data were processed through GraphPad Prism to produce a hit plot. Z’-factor calculations for high-throughput screening (HTS) efficiency were calculated by Equation (3): 31
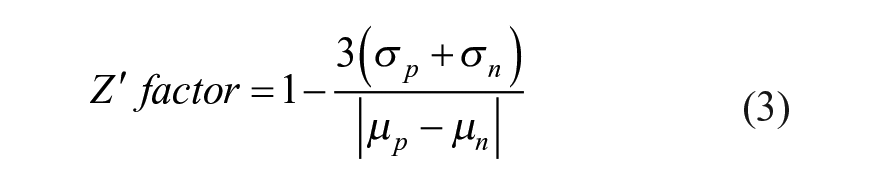
where
Molecular Modeling
The PyMOL molecular graphics program (Schrödinger, New York, NY) was used for general structural representation and examination of the FAK FAT–paxillin LD2 complex [Protein Data Bank (PDB) 1OW8]. Residues that were proximal to the interaction interface (<5Å) and showed key intermolecular contacts were selected for mutagenesis. The PyMOL mutagenesis wizard was used to predict the effects of mutations on FAT–LD2 interaction, protein folding, and steric clashes. Publication-quality images were generated using the ray-trace command.
Site-Directed Mutagenesis of the FAT Domain
Mutation primers were designed through the Agilent QuickChange Primer Design website (Agilent, Santa Clara, CA) and ordered through Invitrogen (Waltham, MA). Primer sequences are listed in
Results
Development of the Initial TAMRA–LD2 Probe and Establishment of the Fluorescence Polarization Assay
To develop a high-throughput FP assay capable of detecting inhibitors of the FAK–paxillin interaction, we first started with the design of a fluorescently labeled paxillin probe to be used in the assay. We used the X-ray crystal structure of the FAK FAT–paxillin LD2 complex (PDB 1OW8) to guide design efforts. As shown in
Optimization of the TAMRA–LD2 Probe and FP Assay
On further inspection of FP saturation and competition binding curves, we noticed that the maximum binding (Bmax) level was relatively lower compared to other published similar FP assays.32,33 Given the high hydrophobicity of our TAMRA–LD2 probe, we hypothesized that the peptide could be self-associating or nonspecifically binding to the plate surface, therefore preventing maximum binding to FAK FAT protein. To test this, we examined the X-ray crystal structure of the FAT–paxillin LD2 complex (PDB 1OW8) and identified leucine 10 as a potential residue for hydrophobic-to-hydrophilic substitution (
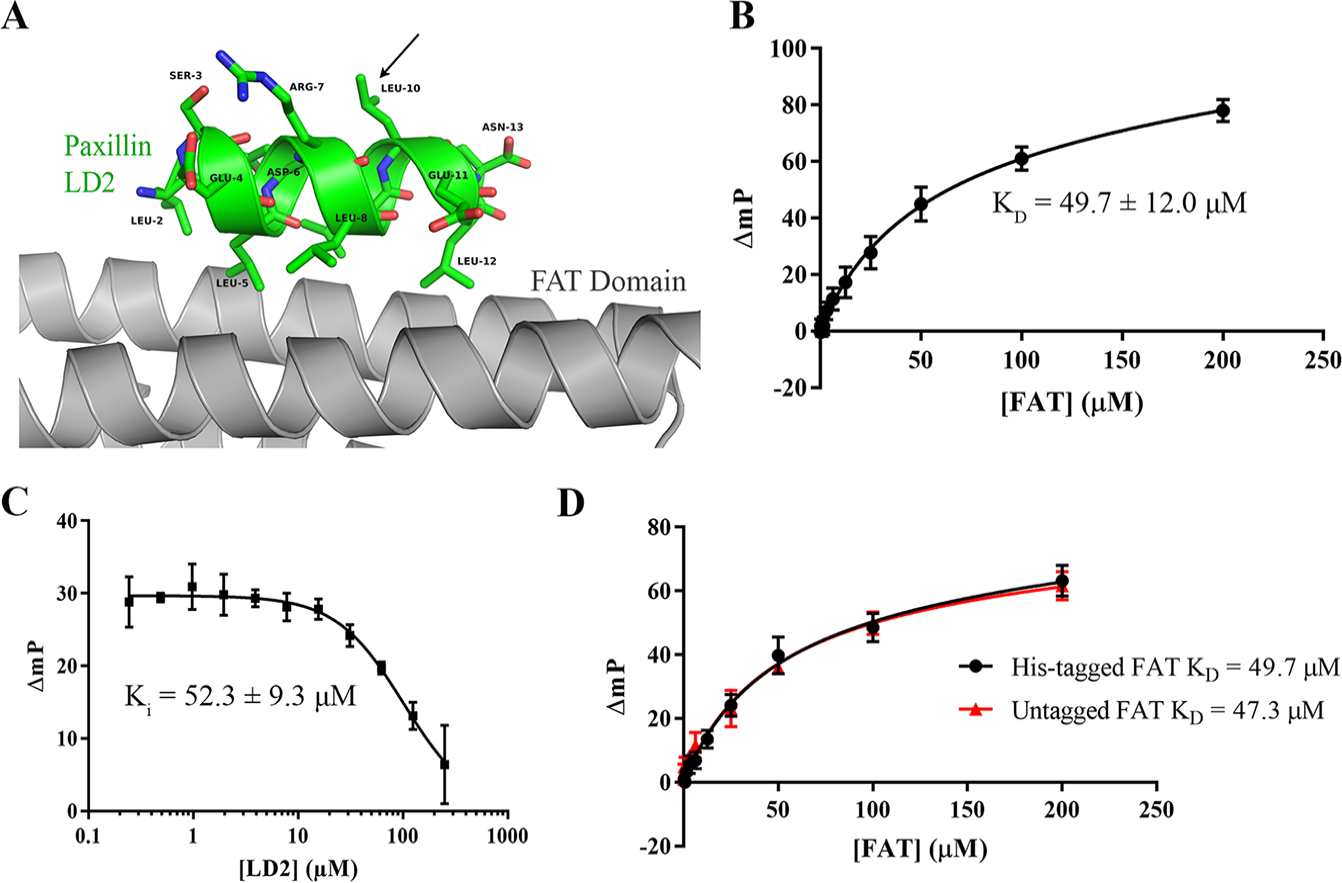
Peptide and protein optimization for the fluorescence polarization (FP) assay. (
Validation of Binding Specificity by Site-Directed Mutagenesis
To validate that the FP binding signal was in fact due to specific binding at the FAT–paxillin interface, we designed a series of mutant FAT proteins at both the Helix 1-4 and Helix 2-3 binding sites. In addition, mutant proteins could be used to characterize Helix 1-4 or Helix 2-3 selective binding of future small molecules. We used the X-ray crystal structure of the FAT–paxillin LD2 complex (PDB 1OW8) as well as a previously described report
27
to select a series of surface residues at each binding site suitable for mutation (
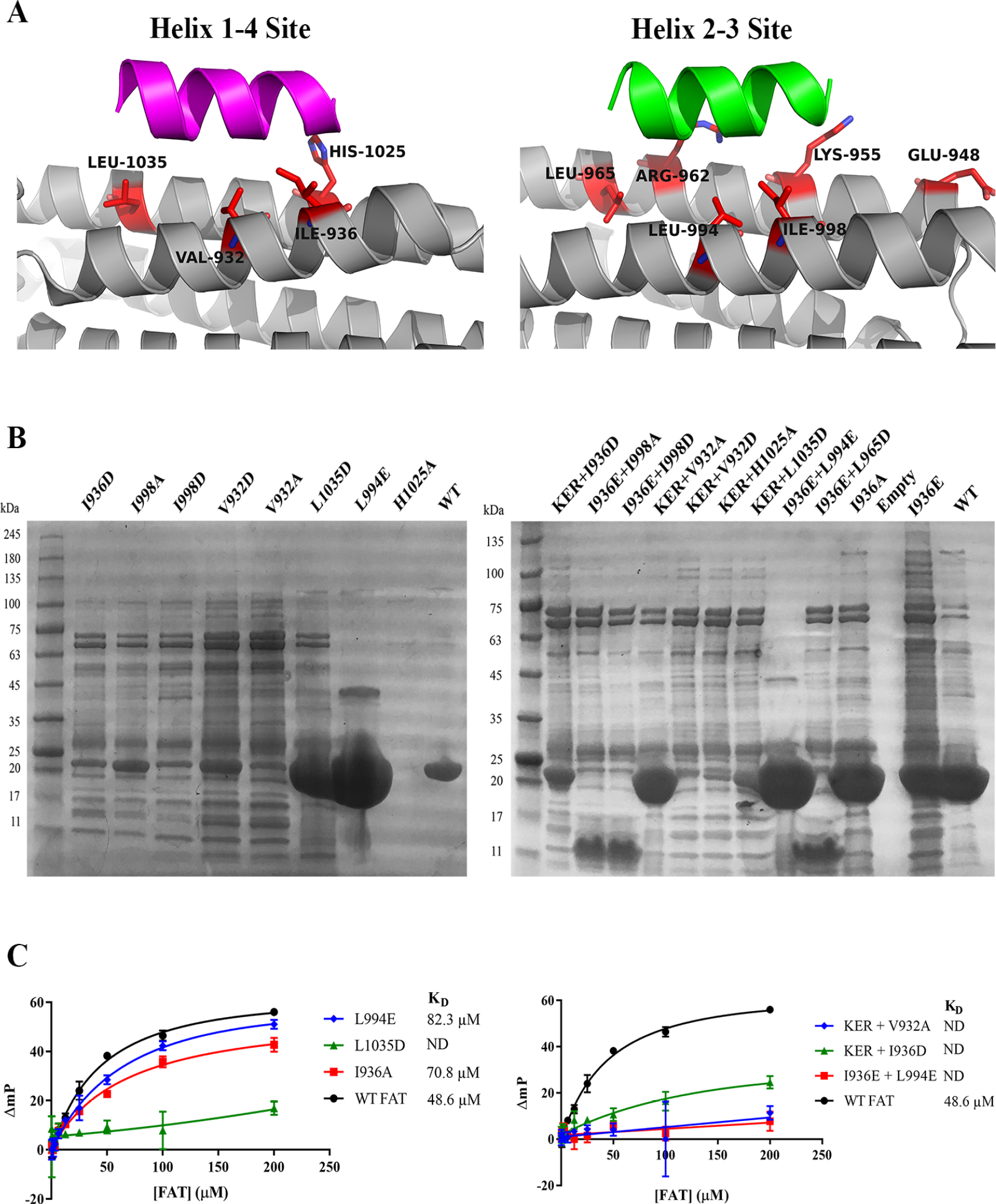
Assay validation through mutagenesis of focal adhesion targeting (FAT). (
Optimization and Validation of High-Throughput Characteristics
Next, we sought to optimize the 384-well plate material and vendor to achieve the best possible assay window and signal variability. We tested eight different 384-well plates that varied in plate material (polypropylene, polystyrene, or nonbinding surface), well volume, well shape (round, square, flat bottom, or round bottom), and vendor (
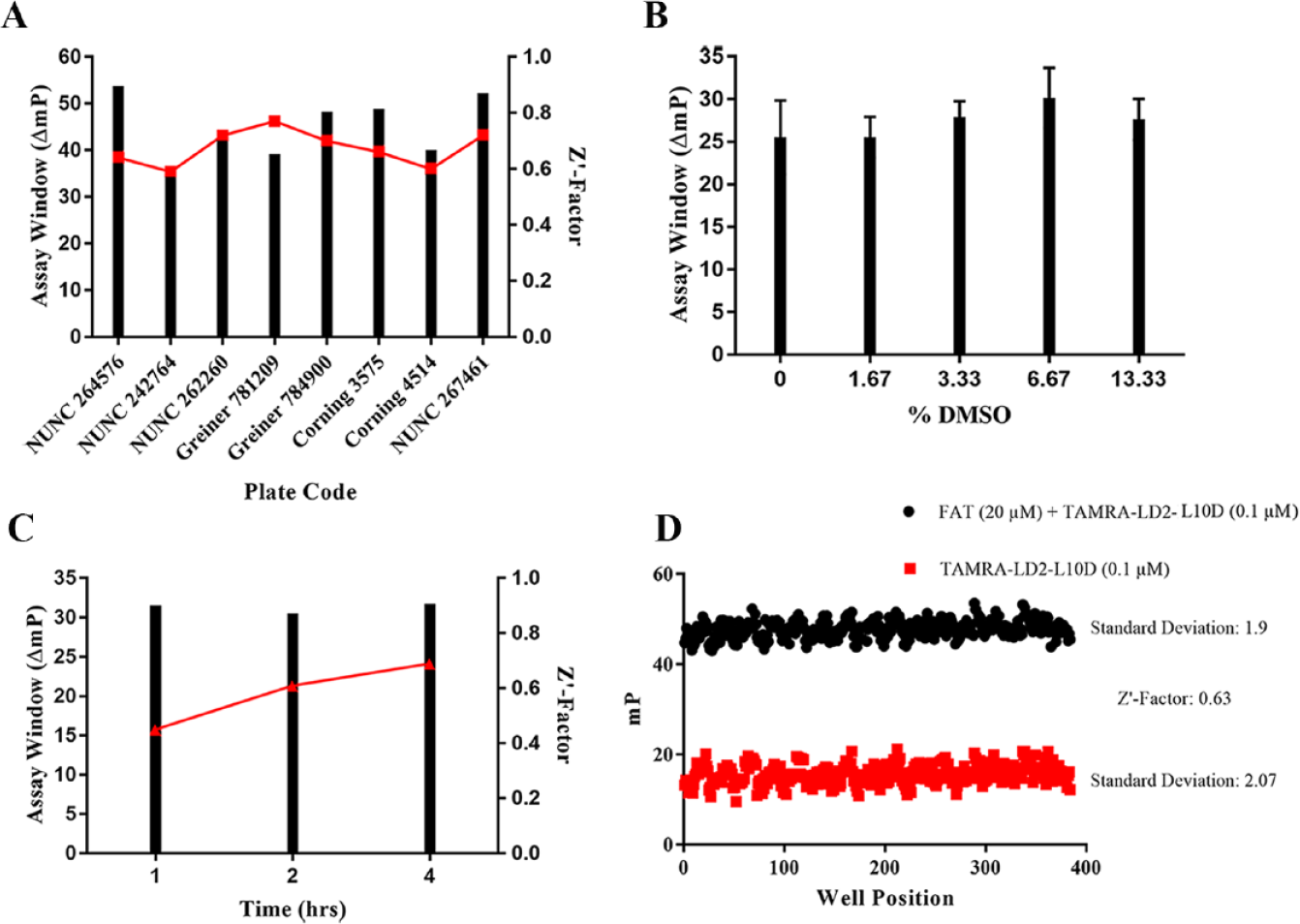
High-throughput screening metric optimization. (
Pilot Screen
To further validate our FP assay for high-throughput drug screening, we proceeded with a pilot drug screen using our Tecan Freedom Evo 100 liquid handling robot. Tecan scripts were programmed using aqueous buffer and DMSO as test solutions. We selected Div5, the Diversity Set V library of 1593 compounds available from the NCI, to use for our pilot screen. Compounds were screened at a final concentration of 312.5 µM (6.25% DMSO) in 384-well format (
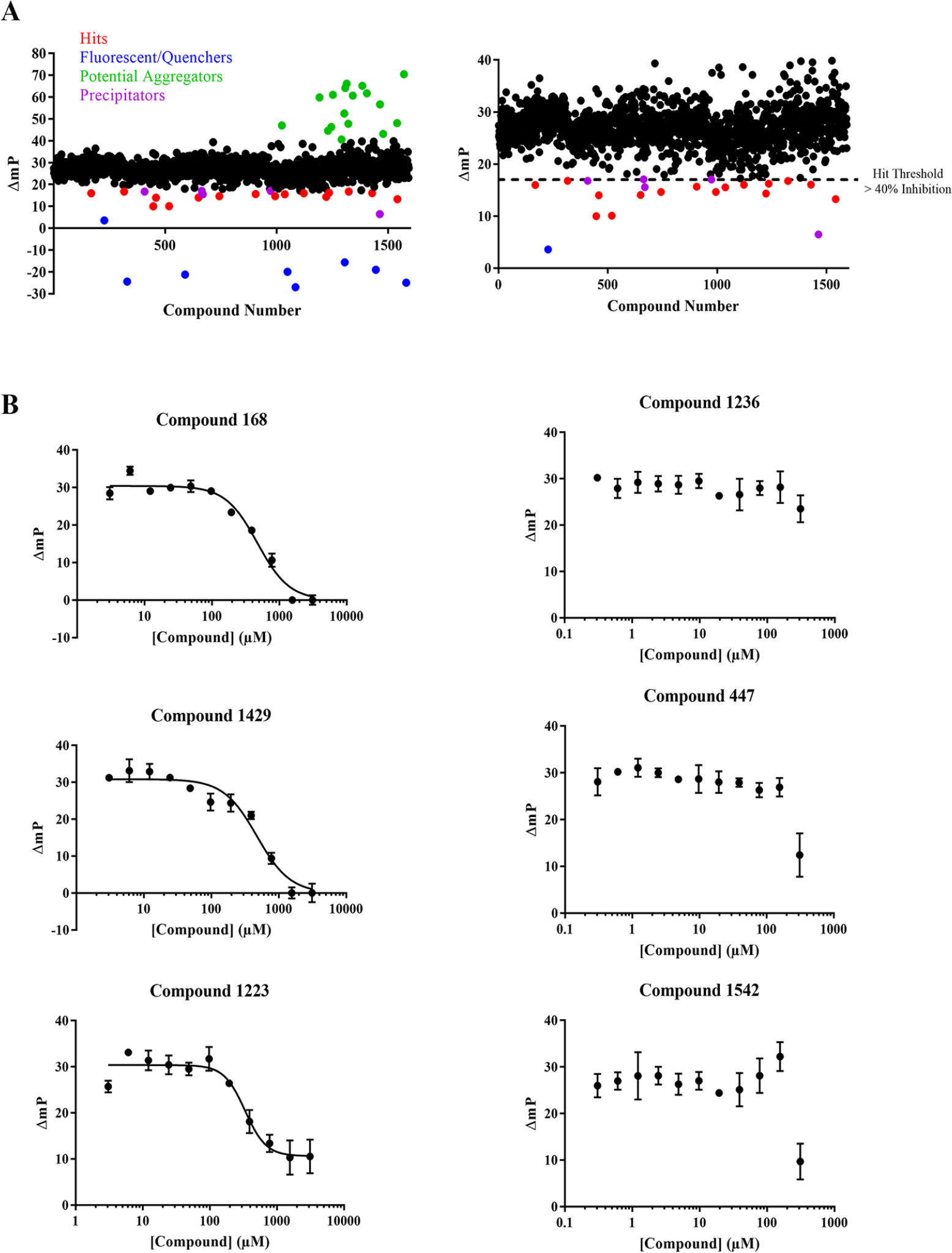
Pilot screen using National Cancer Institute (NCI) Diversity Set V. (
Dose–Response Inhibition Data for Validated Hits.

Imax indicates the maximum inhibitory effect relative to the tetramethylrhodamine (TAMRA)–LD2–L10D and TAMRA–LD2–L10D + focal adhesion targeting (FAT) controls. Calculated IC50 and Ki ± standard error (SE) values are shown. Results show only 3 out of 16 compounds are true validated hits; the rest had ND (nondeterminable) Ki. Values represent data from three independent experiments, each performed in technical triplicates. FP, Fluorescence polarization; SE, standard error.
Discussion
FAK is widely expressed in multiple different tumor types and is a central node to multiple oncogenic signaling pathways.1,36 Despite FAK’s biological importance in cancer, drug discovery efforts have solely focused on targeting the kinase domain and have not addressed the critical scaffolding functions of the protein.37,38 This report represents a significant advancement to the FAK drug discovery field because we disclose the first described HTS assay for noncatalytic FAK inhibitor discovery. The FAK FAT–paxillin interaction is gaining traction as an alternative site for FAK inhibitor discovery due to its importance in regulating invasion, metastasis, and apoptosis, as well as the breadth of X-ray crystallographic information available.19,20 Furthermore, traditional adenosine triphosphate (ATP)-competitive FAK–kinase inhibitors have had limited success in clinical trials, and resistance mechanisms have emerged.17,18 These studies highlight the need for FAK–paxillin inhibitors to alternatively target FAK in cancer.
To develop a FAK–paxillin screening assay, we performed significant assay validation and optimization, including validation by SPR, site-directed mutagenesis, competition studies, plate optimization, and high-throughput metrics. In addition, we performed structure-guided optimization of the TAMRA–LD2 probe to promote lower background mP signal and a greater assay window. Intriguingly, when we tested small-scale protein purification of 18 FAT mutants, only a small amount had sufficient yield and purity for FP studies. We speculate that specific FAT mutations may confer low bacterial expression, affect protein folding, decrease protein stability, or increase proteolytic cleavage. Our results further emphasize the need to empirically test multiple mutant constructs when attempting a site-directed mutagenesis approach. Another observation was that our FP assay was very stable at high concentrations of DMSO (>5%). This DMSO tolerability may be due to the high thermostability of four-helical bundle proteins like the FAK FAT domain that have a high degree of hydrophobic packing and electrostatic interactions to stabilize 3D structure. 39 Note that in plate optimization studies, polypropylene plates seemed to correlate best with assay performance, perhaps due to the amphipathic nature of the LD2 peptide. Future studies may include further optimization of the TAMRA–LD2 probe to reduce potential aggregation or modification of the FAK protein construct (e.g., length) to allow greater mP change on binding.
On processing the data from our FP pilot screen, we applied a computational PAINS (pan-assay interfering substances) filter on the entire NCI Div5 library to identify compounds that had potentially reactive or promiscuous moieties. 40 This process is standard in our laboratory as well as in many HTS labs. Unexpectedly, we found that 163 of 1593 total compounds (10.2%) were positive for PAINS moieties after substructure filtering. These included known reactive or promiscuous structures such as Michael acceptors, azo compounds, quinones, and others. These findings caution the use of publicly available compound libraries such as the NCI Div5 library for drug discovery applications and highlight the importance of flagging compounds that are positive for PAINS moieties. Furthermore, we emphasize the careful consideration of rigorous follow-up assays to rule out nonspecific inhibition, compound aggregation, and pan-assay interference.
The major purpose of the pilot screen with the NCI Div5 library was to validate signal variability from plate to plate, establish a method for counterscreening and hit triage, and establish a reasonable hit rate. The use of our FAT–paxillin FP assay in a pilot drug screen has established the feasibility of this assay for a potential larger drug screen using a larger library such as a 3D diversity set or a protein–protein interaction-focused library. We obtained three hits from the pilot screen that showed valid dose-dependent inhibition and intend to follow up on these compounds using secondary assays such as SPR, saturation transfer difference (STD) NMR, heteronuclear single quantum coherence (HSQC) NMR, X-ray crystallography, aggregation studies, and various cell-based assays. Future screening studies may include development of a SPR- or NMR-based FAT domain assay for fragment-based drug discovery.
In conclusion, we have presented a high-throughput fluorescence polarization assay that measures FAK–paxillin interactions in 384-well format for drug discovery applications. To the best of our knowledge, this represents the first described high-throughput assay focused on the detection of FAK scaffold inhibitors as opposed to FAK kinase inhibitors. Through structure-based design of the TAMRA–paxillin LD2 probe, we were able to design an assay that had a sufficient assay window, low variability, a Z’-factor of 0.63, a stable 384-well signal, and straightforward robotic automation. Future goals include the screening of additional small-molecule libraries to identify potent FAK–paxillin inhibitors that will be tested as an alternative approach to target FAK in cancer.
Supplemental Material
DS_DISC874313 – Supplemental material for Development of a High-Throughput Fluorescence Polarization Assay to Detect Inhibitors of the FAK–Paxillin Interaction
Supplemental material, DS_DISC874313 for Development of a High-Throughput Fluorescence Polarization Assay to Detect Inhibitors of the FAK–Paxillin Interaction by Timothy Marlowe, Carlos Alvarado, Andrew Rivera, Felicia Lenzo, Rohini Nott, Dena Bondugji, Justin Montoya, Alana Hurley, Matt Kaplan, Andrew Capaldi and William Cance in SLAS Discovery
Footnotes
Acknowledgements
We would like to thank Dr. David Azorsa for use of the NCI Diversity Set V for pilot screening studies.
Supplemental material is available online with this article.
Abbreviations
focal adhesion kinase (FAK)
focal adhesion targeting (FAT)
protein data bank (PDB)
Declaration of Conflicting Interests
The authors declared no potential conflicts of interest with respect to the research, authorship, and/or publication of this article.
Funding
The authors disclosed receipt of the following financial support for the research, authorship, and/or publication of this article: National Cancer Institute (Grant No. R01 CA065910 to William Cance and Timothy Marlowe).
References
Supplementary Material
Please find the following supplemental material available below.
For Open Access articles published under a Creative Commons License, all supplemental material carries the same license as the article it is associated with.
For non-Open Access articles published, all supplemental material carries a non-exclusive license, and permission requests for re-use of supplemental material or any part of supplemental material shall be sent directly to the copyright owner as specified in the copyright notice associated with the article.