
Abstract
DNA mismatch repair (MMR) deficiency has been associated with a higher risk of developing colorectal, endometrial, and ovarian cancer, and confers resistance in conventional chemotherapy. In addition to the lack of treatment options that work efficaciously on these MMR-deficient cancer patients, there is a great need to discover new drug leads for this purpose. In this study, we screened through a library of commercial and semisynthetic natural compounds to identify potential synthetic lethal drugs that may selectively target MLH1 mutants using MLH1 isogenic colorectal cancer cell lines and various cancer cell lines with known MLH1 status. We identified a novel diarylpentanoid analogue, 2-benzoyl-6-(2,3-dimethoxybenzylidene)-cyclohexenol, coded as AS13, that demonstrated selective toxicity toward MLH1-deficient cancer cells. Subsequent analysis suggested AS13 induced elevated levels of oxidative stress, resulting in DNA damage where only the proficient MLH1 cells were able to be repaired and hence escaping cellular death. While AS13 is modest in potency and selectivity, this discovery has the potential to lead to further drug development that may offer better treatment options for cancer patients with MLH1 deficiency.
Introduction
Germline mutations in DNA mismatch repair (MMR) genes give rise to an autosomal disorder manifesting as hereditary nonpolyposis colorectal cancer (HNPCC). HNPCC, also known as Lynch syndrome, accounts for approximately 3% of all colorectal cancers (CRCs), which is the third most common cancer in men and women worldwide.1,2 Lynch syndrome also predisposes patients to >80% increased risk of developing CRC and other tumors due to inheritance of the defective MMR allele. 3 Many studies have reported that DNA sequencing of affected patients was indeed associated with MLH1 (42%) and MSH2 (33%), with a minor proportion attributed to MSH6 (18%) and PMS2 (7.5%) deficiency. 4 Interestingly, approximately 15% of all CRCs contain somatic defects in several of these MMR genes, where 60% were due to epigenetic silencing of MLH1 rather than acquired mutations,5,6 hence implying a divergent molecular origin of hereditary versus sporadic CRCs. 7
The standard of care for CRC patients includes colectomy, radiotherapy, chemotherapy, or a combination of these. More commonly, chemotherapeutic drugs are administered after surgery to eliminate remaining cancer cells in which the initial backbone treatment is known as FOLFOX—a combination of 5-fluorouracil (5-FU), leucovorin, and oxaliplatin. 8 However, the therapeutic activities of these drugs are often nonselective, where they also target normal dividing cells and hence lead to many unwanted side effects. 7 Furthermore, there is a growing body of evidence suggesting strong correlation between MMR-deficient CRC patients and drug resistance of several chemotherapeutic agents, such as platinum-containing drugs, methylating and alkylating agents, topoisomerase inhibitors, and antimetabolites such as 5-FU.9,10
In recent years, there has been a fundamental shift toward novel therapeutic approaches that minimize side effects and at the same time offer a more selective targeting of genetic defects underlying tumor initiation and progression. The concept of synthetic lethality takes advantage of an aberrant gene by targeting a second gene partner causing cell lethality, whereas inhibition of only one of these genes is compatible with viability. 11 This approach has been reported by seminal studies of cancer cells harboring BRCA1/2 dysfunction, which were found to be more sensitive to poly ADP-ribose polymerase (PARP) inhibitors compared with cells with normal BRCA function.12,13 Noteworthy, the FDA has approved olaparib, a PARP inhibitor for the treatment of advanced ovarian cancer with deleterious germline BRCA mutations. 14
Similar to BRCA and PARP, the first synthetic lethal relationship between MMR components and DNA polymerases was identified in a Saccharomyces cerevisiae yeast model, 15 suggesting possible treatment of MMR-deficient cancer patients with DNA polymerase inhibitors. Recent studies have further extended these observations to demonstrate MLH1 synthetic lethal relations with DNA polymerase gamma (POLG) 16 and pan-MMR deficiency with PTEN-induced putative kinase 1 (PINK1)17,18 in human cancer lines utilizing RNA interference. Subsequent screening effort identified several drugs, such as menadione,16,19 cytarabine, 18 ethinyl estradiol, 20 and desferrioxamine, 20 to be MLH1-deficient selective. However, none of the identified drugs have entered clinical trials. With the recent development of immunotherapy, pembrolizumab was approved for the treatment of solid tumor with MMR deficiency 21 in 2017. Nonetheless, immunotherapy drugs work by sensitizing the immune system to recognize the tumor, thus still offering an opportunity of combination with drug leads targeting mutant MLH1 tumor suppressors.
In this study, we report the discovery of a novel diarylpentanoid analogue, 2-benzoyl-6-(2,3-dimethoxybenzylidene)-cyclohexenol, coded as AS13, which was observed to selectively target MLH1-deficient CRC cells but not its proficient counterpart. Subsequent evaluation using a panel of MLH1-deficient cancer cells further confirmed the observed selectivity. The possibility of AS13 also targeting other MMR genes including MSH2 warrants further validation. The following mechanistic studies suggested that AS13 confers selectivity through oxidative stress that only MLH1-proficient cells are able to repair, hence escaping cellular death. Findings from this study suggested possible treatment options that could be further developed for cancer patients with MLH1 deficiency.
Materials and Methods
Synthesis of Diarylpentanoid Analogues
The synthesis of diarylpentanoid analogues (AS1–32) has been described previously by Leong et al. 22 In general, analogues were prepared by a series of chemical reactions involving benzoylation of cyclohexanone and aldol condensation of aromatic aldehydes. Briefly, cyclohexanone (20 mmol; 1.86 mL) was first reacted with pyrrolidine (20 mmol; 1.23 mL) in toluene (30 mL) to afford N-(1-cyclohexenyl)pyrrolidine, the essential enamine intermediate for benzoylation. The intermediate was then reacted with benzoic anhydride (20 mmol) in toluene (20 mL) to afford 2-benzoylcyclohexanone crude product, which was further purified using column chromatography prior to further reaction with appropriate benzaldehydes in a 1:1 ratio (5 mmol each) under acidic aldol condensation conditions to prepare the desired analogues AS1–32.
Cell Lines and Culture Conditions
HT29 (colon adenocarcinoma), DLD-1 (colon adenocarcinoma), LoVo (colon adenocarcinoma), PC3 (prostate adenocarcinoma), and MCF7 (breast) human cancer cell lines and HEK 293T (embryonic kidney) were obtained from the American Type Culture Collection (ATCC, Manassas, VA). KM12 (colon adenocarcinoma) and DU145 (prostate) human cancer cell lines were kindly provided by Professor Chee-Onn Leong (International Medical University [IMU], Bukit Jalil, Malaysia). HCT116 (colon) MLH1-defective (–/–) and MLH1-proficient (+/–) lines were purchased from Horizon Discovery (UK). The HCT116 MLH1 (+/–) was validated as a variant of HCT116 that has lower authenticity (less than 80% similarity from its source) using short tandem repeat (STR) genotyping. All lines were maintained in RPMI (Invitrogen, Grand Island, NY) supplemented with 10% fetal bovine serum (Invitrogen) and 100 U/mL penicillin and streptomycin solution (Invitrogen). Primary human fibroblast cells (NPC252) established from human tissue biopsies with ethic approval and informed consent were maintained in Dulbecco’s modified Eagle’s medium (DMEM), high glucose (Invitrogen) supplemented with 10% FBS, and 100 U/mL of penicillin and streptomycin solution. Immortalized human primary nonmalignant nasopharyngeal epithelial cell line (NP460) was kindly provided by Professor Tsao (University of Hong Kong) and cultured in keratinocyte serum-free medium (Invitrogen), supplemented with bovine pituitary extract, 5 ng/mL EGF, and 100 U/mL penicillin and streptomycin solution. All cell lines were maintained at 37 °C with 5% carbon dioxide. Cell lines were authenticated (>80% similarity to their source) and were profiled by STR genotyping at the Centre for Translational Research and Diagnostics (CTRAD), National University of Singapore (NUS).
Natural Product and Small-Molecule Screening
HCT116 MLH1 isogenic pairs were seeded in 384-well format at ~1000 cells per well with 1× concentration of nonlytic bioluminescent substrate and enzyme mix (RealTime-Glo; Promega, Madison, WI). After ~24 h incubation, cells were treated with compounds (2.5 µL) semirobotically (epMotion 96; Eppendorf, Hamburg, Germany) to give a final concentration of 10 µM. The chemical library for primary screening consists of ~1400 structurally diverse and cell-permeable compounds comprising (1) new chemical entities, (2) compounds in an advanced phase of clinical development, and (3) FDA-approved compounds for the treatment of cancer and other diseases. All commercially available compounds were obtained from Selleckchem with nuclear magnetic resonance (NMR) and high-pressure liquid chromatography (HPLC) validation to ensure high purity. Another ~1000 compounds were from natural and synthetic compound libraries (IMU and its collaborating associates), and synthetic natural product analogue collections were from Universiti Putra Malaysia (UPM, Selangor, Malaysia). Cell viability was determined 48 h after treatment (Molecular Devices SpectraMax M3), and percentage cell viability was calculated for each compound by normalizing to vehicle controls (1% DMSO). Positive hits were defined as compounds that reduced the relative viability of mutant MLH1 versus wild-type MLH1. Positives hits and their structural derivatives were selected for secondary screening to determine dose responses and structure–activity relationships (SARs). Stock solutions of compounds (10 mM) were made in analytical-grade DMSO (Sigma Aldrich, St. Louis, MO).
Dose–Response Cellular Viability Assay
Secondary screening of AS13 and its derivatives was assessed by MTT-based colorimetric assay to determine the half maximal inhibitory concentrations (IC50). All cancer cell lines were seeded into 96-well format at 3000–4000 cells per well (80 µL) while primary human fibroblast and NP460 noncancer cell lines were seeded at 6000 cells per well (80 µL), due to their slow growth rate. After ~24 h incubation, cells were treated with compounds (20 µL) in a concentration range of 0.04–100 µM by serial dilution and cultured for an additional 72 h. Next, MTT solution (20 µL, 6 µg/mL; Amresco, OH) was added and further incubated at 37 °C for 2 h. Formazan produced was dissolved using 100 μL of analytical-grade DMSO (Fisher Scientific, Loughborough, UK) and the absorbance was measured at 570 nm (Synergy H1M Multi-Mode Microplate Reader; BioTek, Winooski, VT). Dose–response curves were normalized to vehicle controls (DMSO 1%, v/v) and generated to calculate the IC50 values using four-parameter variable-slope dose–response inhibition in GraphPad Prism 6 (GraphPad Software, Inc.). All stock solutions were prepared at 10 mM in analytical-grade DMSO.
Generation of HT29 MLH1 Knockout Isogenic Lines
Envelope glycoprotein (pVSVg) was a gift from Bob Weinberg (Addgene plasmid no. 8454). Packaging vector (psPAX2) was a gift from Didier Trono (Addgene plasmid no. 12260). LentiCRISPR plasmid was a gift from Feng Zhang (Addgene plasmid no. 52961).
Cloning of MLH1 Target Sequence into LentiCRISPR-Cas9 Vector
Three single-guide (sg)RNA-targeting MLH1 genes were designed, each 5′ to 3′:
sg1: CACCGGCAGCCACTTCAGCAGGGGC and AAACGCCCCTGCTGAAGTGGCTGCC
sg2: CACCGTGAAGTTCACTTCCTGCACG and AAACCGTGCAGGAAGTGAACTTCAC
sg3: CACCGTCACACAGCCCACGAAGGAG and AAACCTCCTTCGTGGGCTGTGTGAC
LentiCRISPR plasmid (3.4 µg, 1.5 µL) was digested with restriction enzyme BsmBI (NEB, Ipswich, MA; 5.0 µL) in 10× NEBuffer 3.1 (NEB; 2.0 µL) and milliQ water (11.5 µL) and incubated at 55 °C (15 min) and then equilibrated to 37 °C. Shrimp alkaline phosphatase (rSAP; NEB, 5 µL) was added to the mixture to prevent religation and incubated further (30 min). The rSAP enzyme was then heat inactivated at 80 °C (20 min). Next, lentiviral vector (~11 kb) was purified using a QIAquick Gel Extraction Kit following the manufacturer’s instructions. Concurrently, the sgRNA oligo pairs were phosphorylated and annealed. Briefly, each oligo (1 µL ea, 100 µM) was added to 2× Quick ligase buffer (NEB; 5 µL), T4 PNK (NEB; 0.5 µL), and milliQ water (2.5 µL) and placed in a thermocycler at 37 °C (30 min) and then 95 °C (5 min). After, the sgRNA annealed pairs were cooled at a rate of 5 °C/min to 25 °C. The oligo duplex was then diluted by a factor of 200. Next, the annealed sgRNA oligo duplexes are ligated to the BsmBI-digested lentriviral vector. Briefly, the vector (50 ng, 0.5 µL) and diluted oligo duplex (1 µL) were added to 2× Quick ligase buffer (5 µL), milliQ water (3.5 µL), and Quick ligase (NEB; 1 µL) and incubated at room temperature (rt) for 10 min.
Amplification of Plasmid
Luria broth (LB) agar with ampicillin (100 µg/mL) was prepared beforehand and stored at 4 °C. Competent DH5-alpha (in glycerol) stored at −80 °C was thawed on ice. Plasmid (50 ng) was added to DH5-alpha (200 µL) and incubated for 30 min. The cells were then heat shocked at 42 °C for 45 s and further incubated on ice for 3 min. Sterile LB media (500 µL) was added and incubated with shaking (300 rpm) for 1 h. Cells were spun down and resuspended in LB media (100 µL) followed by plating onto the LB agar plates. Plates were incubated at 37 °C overnight. Single colonies for each sgRNA plasmid were selected and incubated in LB media (5 mL) with ampicillin (100 µg/mL) at 37 °C (6 h; 300 rpm). After, the culture (1 mL) was added into fresh LB media (200 mL) and left to grow overnight (37 °C; 300 rpm).
Purification of Plasmid
Plasmids in DH5-alpha cells were purified using the Plasmid Midi Kit (Qiagen, Germany, Hilden) following the manufacturer’s instructions.
Production of Lentiviral Particles
HEK 293T cells were seeded in a six-well platform to 60%−70% confluency. Each well was treated dropwise with serum and antibiotic-free DMEM (500 µL) containing the purified lentivector plasmid (3 µg), packaging vector (2 µg; psPAX2), envelope glycoprotein (1 µg; pVSVg), and Turbofect (6 µg; Fermentas, IL) that had been incubated for 20 min. After 24 h, the transfection media was replaced with fresh media without antibiotic (2 mL). After an additional 24 h, the supernatant was filtered (0.45 µm) to give the lentiviral particle solution.
Editing of MLH1 Gene in HT29 Cells
Concurrently, HT29 cells were seeded in a six-well platform to 60%–70% confluency. The lentiviral particle solution (1 mL) was then infected to HT29 cells for 24 h. After, fresh media was added and further incubated until confluency. Cells were then cultured under antibiotic selection containing 1 µg/mL puromycin (Gibco, Carlsbad CA).
Selection of MLH1 Knockout Single Colonies
After a week of continued selection in puromycin, the HT29 cells were then seeded at 15 cells/mL to produce single colonies. When colonies become observable under the microscope, they were picked and trypsinized with sterile filter paper. These single colonies were then grown separately until confluency under continued puromycin selection. Protein lysates of these single colonies were extracted to determine the status of MLH1. Confirmed MLH1 knockouts were DNA amplified at their respective sgRNA region to determine their MLH1 mutational profile by Sanger sequencing (1st Base, Apical Scientific, Malaysia). DNA extracts of two single colonies with MLH1 knockouts HT29(MLH1-)2a and HT29(MLH1-)2b were collected and authenticated to match HT29 with more than 80% similarity.
Immunoblotting
Protein extraction and immunoblotting analysis were performed as previously described by Velaithan et al. 23 Primary antibodies used were anti-MSH2, anti-MLH1, and anti-human PARP (BD Pharmingen, Franklin Lakes, NJ); anti-γ-H2A.X (S139), anti-cdc2, anti-p-cdc2 (Y15), anti-p-cdc2 (T14), anti-p-cdc2 (T161), anti-cleaved caspase 3, and anti-p21Waf1/Cip1 (Cell Signaling, MA); anti-β-actin (Merck Millipore, Palo Alto, CA); and anti-p53 (Novocastra, NE, UK). Anti-β-actin monoclonal antibody was used to normalize loading.
Click-iT Cell Proliferation Assay
The effect of compound treatment on DNA synthesis were assessed using the Click-iT EdU Alexa Fluro 647 Imaging Kit (Thermo Fisher, Eugene, OR) as previously described by Velaithan et al. 23
Quantification of 8-Oxoguanine
Both floating and attached cells were collected and genomic DNA was extracted using the QIAamp DNA Blood Mini Kit (Qiagen) following the manufacturer’s instructions. DNA samples were standardized, converted to single stranded (95 °C, 5 min), rapidly cooled on ice, and digested with nuclease S1 (Thermo Fisher; 37 °C, 2 h) according to the manufacturer’s instructions. The pH was then adjusted to pH 7–8 with Tris-HCl (1.0 M, pH 8.8) and was converted to nucleosides with rSAP (5 units; 37 °C, 1.5 h) in CutSmart buffer (NEB) and then denatured. 8-Oxoguanosine (8-oxodG) levels were quantified using an 8-oxodG-specific enzyme-linked immunosorbent assay (ELISA) in 96-well plate format (Cell Biolabs, Inc., San Diego, CA) according to the manufacturer’s instructions. Absorbance was quantified at 450 nm (Synergy H1M Multi-Mode Microplate Reader) and standard curves were generated from serial dilutions of 8-oxodG standards using quadratic regression as the line of best fit.
Cell Cycle Analysis
Both floating and attached cells were collected, washed with phosphate-buffered saline (PBS), and fixed with 70% ethanol on ice and stored at −20 °C until analysis. The fixed cells were washed with PBS, stained with propidium iodide (10 µg/mL; Sigma-Aldrich) containing RNase (20 µg/mL; Sigma-Aldrich), and incubated for 30 min in the dark. The effect of test compounds on cellular cycle distribution was analyzed by measuring the DNA content, performed using BD FACSCanto II with BD FACSDiva software (BD Biosciences). Cell cycle distribution profiles were generated using ModFit version 4 (Verity Software House).
Detection and Quantification of DNA Damage γ-H2A.X (S139)
Immunofluorescence
Cells were grown onto coverslips to 60%–70% confluency followed by compound treatment for 24, 48, and 72 h. Cells were then fixed (4% methanol-free formaldehyde) and permeabilized (0.5% Triton X-100 in PBS) before overnight incubation (4 °C) with anti-γ-H2A.X (S139; 1:200 dilution in PBS with 1% bovine serum albumin (BSA) and 0.3% Triton X-100). Cells were washed with 3% BSA in PBS (×3) and incubated (2 h, rt) with the corresponding secondary antibody tagged with Alexa Fluor 568 (1:200 dilution in PBS with 1% BSA and 0.3% Triton X-100). Actin filaments were stained with Oregon green 488 Phalloidin (Thermo Fisher) in PBS with 3% BSA. The stained cells were washed before mounting on microscope slides and viewed using a fluorescence microscope (IX81 Olympus). Double-stranded DNA breaks (γ-H2.AX) were visualized under fluorescence with an in-built green excitation filter, and actin stain was visualized with a blue excitation filter.
Flow Cytometry
Quantitative analysis of DNA double-stranded breaks (DSBs) was assessed using flow cytometry. Both floating and attached cells were collected, fixed in 4% formaldehyde, permeabilized with ice-cold methanol (30 min), and stored at −20 °C until analysis. The cells were washed with PBS and incubated (1 h, rt) with anti-γ-H2A.X (S139; 1:800 dilution in PBS with 1% BSA). Next, cells were washed once with 3% BSA in PBS and incubated (1 h, rt) with the corresponding secondary antibody tagged with Alexa Fluor 568 (1:800 dilution in PBS with 1% BSA). Finally, cells were again washed with 3% BSA in PBS before resuspending in PBS. The effect of test compounds on fluorescence detection of γ-H2A.X (S139), indicating the formation of DNA DSBs, was measured using BD FACSCanto II. The geometric mean of fluorescence intensity distribution was determined using ModFit LT 5.0.
Measurement of Intracellular ROS Levels
Cells were grown onto coverslips to 60%–70% confluency and were treated with 10 µM 2′,7′-dichlorofluorescin diacetate (DCFDA) for 1 h. Cells were washed with PBS (twice) and were treated with selected compounds for 48 and 72 h. Cells were harvested, washed, and resuspended in PBS. The intensity of the formed DCF as a result of carboxy-DCFDA hydrolysis was analyzed with a flow cytometer with excitation and emission wavelengths of 488 and 525 nm, respectively.
Statistical Analysis
All experiments were conducted in triplicates. Results were recorded as mean ± SEM. Differences between treatment groups with vehicle controls were analyzed using an unpaired Student t test with GraphPad Prism software, whereby * (p < 0.05) and ** (p < 0.01) indicated significant differences.
Results
Identification of a Diarylpentanoid Analogue, 2-Benzoyl-6-(2,3-Benzylidene)-Cyclohexenol, by Synthetic Lethality Screening
We performed a high-throughput synthetic lethal screen (48 h) to identify compounds that could selectively kill HCT116 MLH1 (–/–)-deficient cells but not its proficient counterpart, HCT116 MLH1 (+/–). The parental HCT116 MLH1 (–) cells carry a hemizygous point mutation (C755A) in exon 9 of the MLH1 gene, 24 resulting in a premature stop codon and loss of protein function, whereas the isogenic counterpart has a heterozygous knock-in of wild-type MLH1 allele (+/–) 25 ( Fig. 1a ). Using an ATP-based cell viability assay (CellTiter-Glo), we screened more than 2500 compounds (10 µM) and identified AS13 that exhibited selective activity toward HCT116 MLH1 (–/–) cells compared with HCT116 MLH1 (+/–) by ~5-fold difference in cell viability (HCT116 MLH1 –/–: 78.4%; HCT116 MLH1 +/–: 15.4%) ( Fig. 1b,c ). Dalcetrapib, A-769662, and GSK3787 were also noted to have ≥2.5-fold difference. The novel diarylpentanoid analogue, 2-benzoyl-6-(2,3-benzylidene)-cyclohexenol, AS13 ( Fig. 1d ), was synthetically designed with an asymmetric diarylpentanoid backbone and a cyclohexyl ring inserted in the active methylene site.
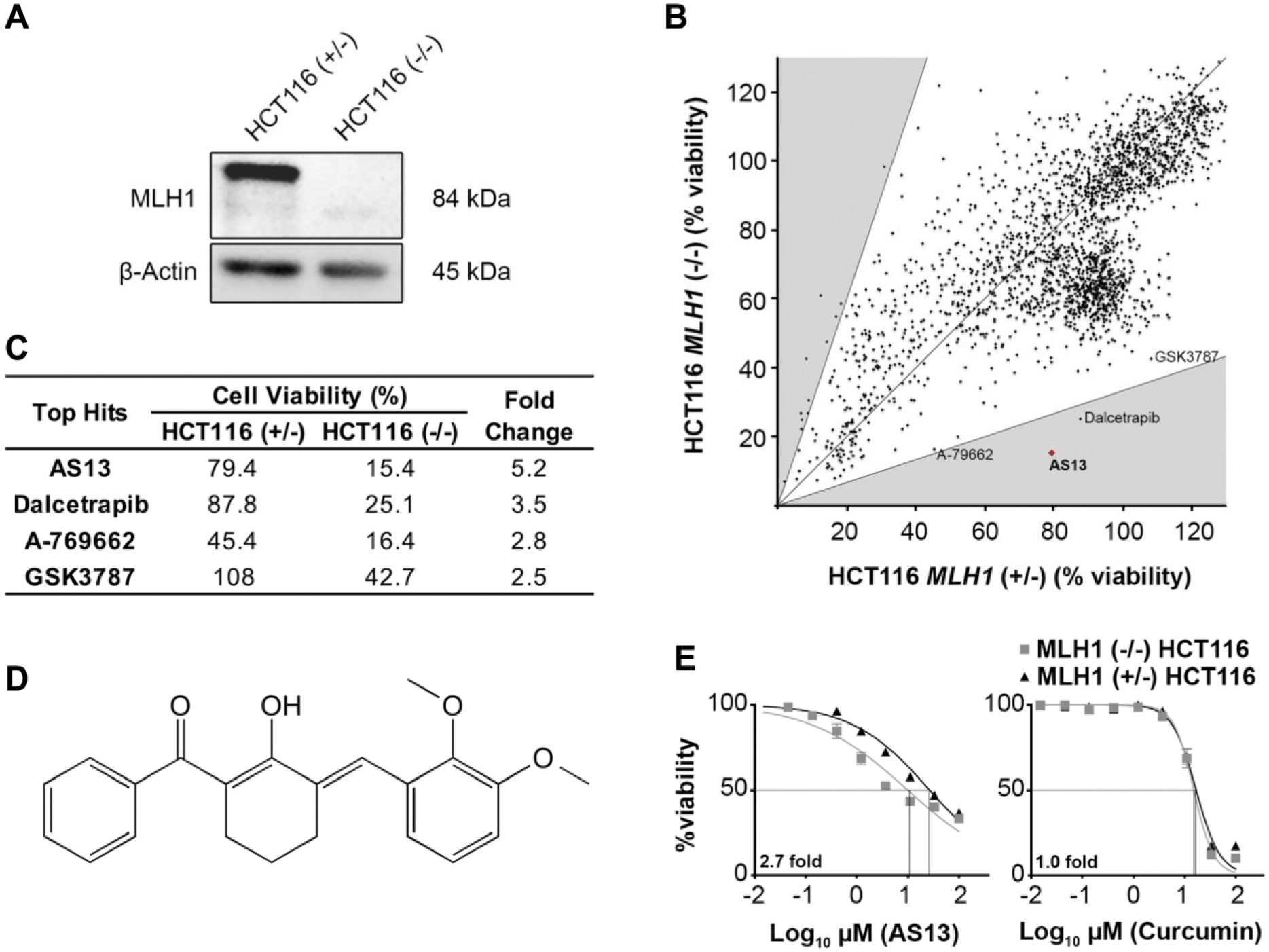
Identification of AS13 selectivity toward HCT116 MLH1 (–/–) cells. (
With the observed preliminary selectivity of AS13, we next performed a secondary dose–response screen (72 h) of all the derivatives in the AS series (AS1–AS32) including AS13, to determine the IC50 on the isogenic lines HCT116 MLH1 (–/–) and (+/–) ( Suppl. Table S1a,b ). We found that AS13 was the only analogue that conveyed a 2.7-fold specificity toward the HCT116 MLH1 (–/–) compared with the corrected counterpart with IC50 values of 9.54 ± 1.98 µM and 25.8 ± 3.01 µM, respectively ( Suppl. Table S1a ). This notable difference in selectivity is shown in Figure 1e in contrast to curcumin, the parent compound used for the synthesis of this diarylpentanoid series. Broadly, curcumin is nonselective toward MLH1 deficiency.
SAR Analysis of 2-Benzoyl-6-(2,3-Benzylidene)-Cyclohexenol Selectivity toward MLH1 Deficiency
The interpretation of SAR is often confounded by solubility, protein binding, and membrane permeability, which guide the understanding of substituent groups that have inhibitory effects. Based on the results obtained for 32 analogues of AS1 (from which AS13 is derived), the SAR conferring AS13 with selectivity toward HCT116 MLH1 (–/–) was investigated. As shown in Supplemental Table S1a , it was observed that analogues with methoxy, heteroatom, or hydroxyl groups at the para- position of the aryl (Ar) substituent (AS4, AS7, and AS9) had low activity in both isogenic cell lines HCT116 MLH1 (+/–) and (–/–), with the analogues with a hydroxyl group having the most inhibition on cell viability (IC50 >100, ~88, and ~55 µM, respectively) ( Suppl. Table S1a ). On the other hand, substitutions at the ortho- and meta- positions were observed to confer greater activity than para- substitution. These substitutions are heteroatom (IC50 ~41, ~37, and ~40 µM; AS5, AS6, and AS30) and hydroxyl (IC50 ~37 µM; AS8) groups, except for methoxy substitutions (>100 and ~55 µM; AS2 and AS3). The greater biological activity of meta-substituted diarylpentanoid analogues in the present study was in concordance with the reported higher anti-inflammatory and anticancer properties of similarly meta-substituted curcuminoids.22,26 Interestingly, dual additions and permutations of heteroatoms and hydroxyl and methoxy groups at the meta- (C3 or C5) and para- (C4) positions (AS10, AS15, AS17, AS18, AS19, and AS20) showed increased activity (IC50 16–62 µM) in both isogenic pairs compared with their singly substituted counterpart (AS3 and AS4, AS6 and AS7, AS8 and AS9). In particular, we observed an increase in cell viability inhibition with the addition of two hydroxyl groups (HCT116 isogenic pairs: 19.4 ± 2.35 µM; AS10) compared with the addition of methoxy groups (29.1 ± 4.33 µM; AS15). Although the observed difference is minimal, the data may suggest the potential of hydroxyl functional groups having greater biological activity than halogens and methoxy moieties. This tested series of analogues also included several stereoisomers, namely, AS13 (2,3-dimethoxy), AS14 (2,5-dimethoxy), and AS15 (3,4-dimethoxy). Unlike AS15, both AS13 and AS14 have methoxy groups in ortho- (C2) and meta- (C3 or C5) positions. In HCT116 MLH1 (+/–), AS13, AS14, and AS15 exhibited relatively similar IC50 values of 25.8 ± 3.01, 34.2 ± 5.37, and 30.0 ± 6.68 µM, respectively ( Suppl. Table S1a ). Interestingly, in HCT116 MLH1 (–/–), AS13 exhibited a greater inhibitory effect (IC50 9.53 ± 1.98 µM) while the activity of AS14 (IC50 35.9 ± 2.26 µM) and AS15 (IC50 27.7 ± 3.64 µM) remained similar to that of its counterpart cell line. This suggests that dual substitution at C2 and C3 positions, exemplified by AS13, could likely be a key requirement for selectivity toward MLH1-deficient cells without changing its activity in MLH1-proficient cells.
AS13 Selectively Targets MLH1 Mutant Cells
As the HCT116 isogenic pair reflect a single genetic variation, in particular MLH1, we investigated the specificity of AS13 against different genetic pools of MLH1-deficient and -proficient lines of different cancers. The deficient lines include DU145, a prostate cancer cell line that harbors a hemizygous splice mutation between exon 1 and exon 2 of the MLH1 gene, resulting in early truncation, 24 and KM12, a CRC cell line with hypermethylation of the MLH1 gene, resulting in loss of protein expression. 27 Cell lines with MLH1 proficiency include HT29 (colorectal), PC3 (prostate), and MCF7 (breast), as well as noncancer counterparts, NP460 (nasopharyngeal epithelial) and NPC252 (fibroblast). We then standardized our treatment to 72 h using an MTT-based colorimetric assay. As shown in Figure 2a , basal levels of MLH1 protein were only detected in the proficient lines in contrast to those known to be deficient. Analysis of AS13 treatment on HCT116, KM12, and DU145 MLH1-deficient lines gave IC50 values of 18.9 ± 3.61, 17.3 ± 1.92, and 19.3 ± 4.17 µM, respectively, while the proficient lines (HT29, PC3, and MCF7) and the two normal phenotypic cell types (NP460 and NPC252) gave an average IC50 of >83 µM ( Fig. 2b ).
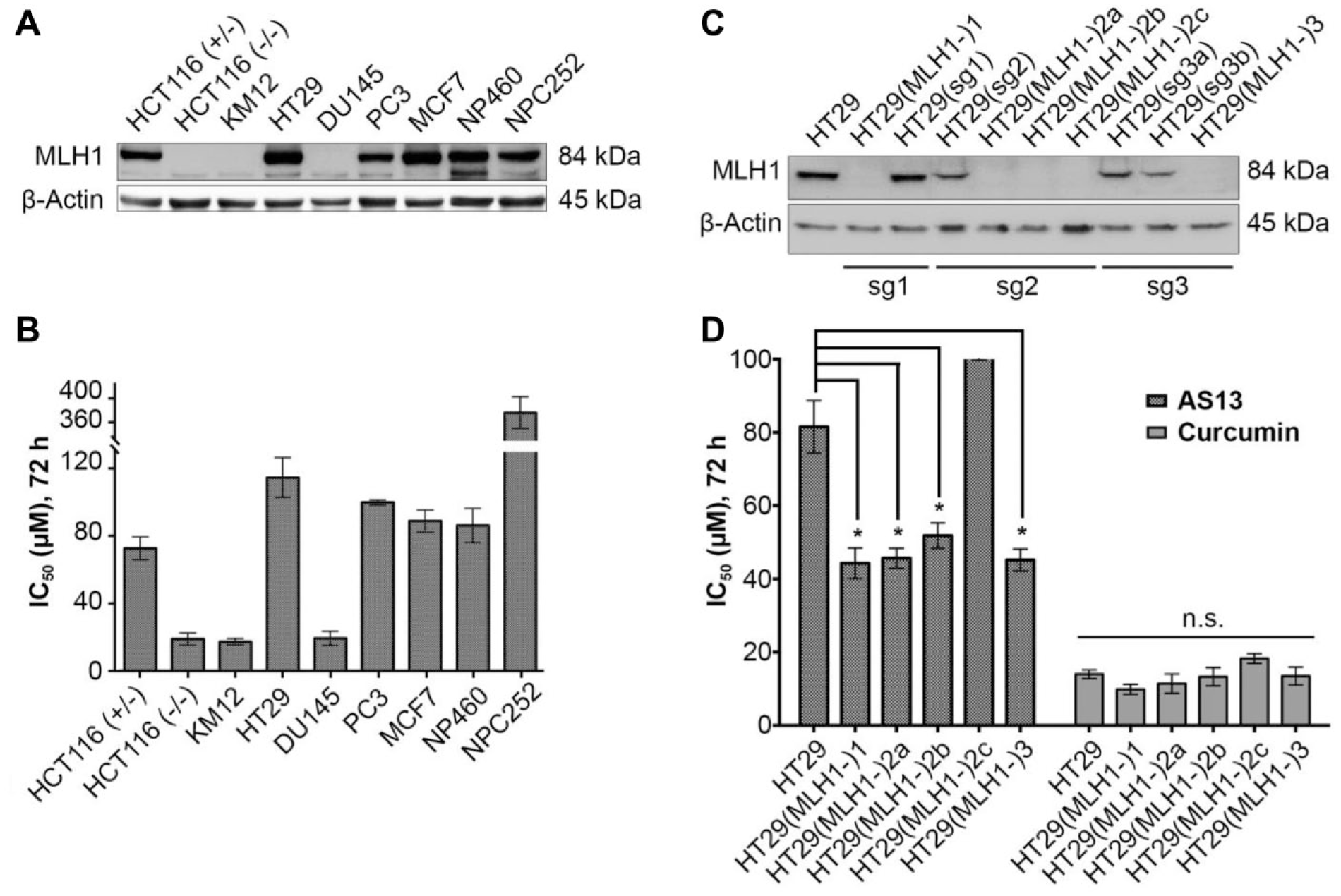
AS13 is sensitive to MLH1 deficiency. (
To further support our observation, we generated an MLH1 isogenic line from HT29 cells using the CRISPR-Cas9 technique. Compared with the HCT116 isogenic, which is a knock-in of MLH1 25 , the HT29 isogenic contains a knockout of MLH1. As shown in Figure 2c , loss of MLH1 protein was found in five out of nine MLH1-edited HT29 lines, encoded as HT29(MLH1-)1, HT29(MLH1-)2a, HT29(MLH1-)2b, HT29(MLH1-)2c, and HT29(MLH1-)3, respectively. Among the generated HT29 MLH1 knockout, HT29(MLH1-)2b has a homozygous deletion of CTGC (c.949_952del) resulting in a truncated MLH1 protein ( Suppl. Fig. S1a,b ). Interestingly, both HT29(MLH1-)2c and HT29(MLH1-)2a contain different mutations in each MLH1 allele. HT29(MLH1-)2c has a homozygous insertion of C and T at the same area (c.952_953insC and c.952_953insT), while HT29(MLH1-)2a has a common deletion of A and G (c.953del and c.955del) with multiple nonsynonymous mutations; both lead to a truncated protein. The chromatograms of the HT29 MLH1 (–/–) knockout isogenic are included in Supplemental Figure S1a,b . Analysis of AS13 treatment on these HT29 MLH1 knockout lines showed a decline in IC50 (average IC50 ~45 µM; p < 0.05) except HT29(MLH1-)2c compared with HT29 MLH1 (+/+) parental lines (IC50 81.5 ± 7.16 µM) ( Fig. 2d ). Of note, curcumin, which was shown to have no selectivity in an earlier screen, consistently exhibited no preference to either the knockout lines or the parental HT29 line (average IC50 ~13 µM). Collectively, the data thus far suggest selectivity of AS13 to MLH1-defective cells.
AS13 Induces Cytostasis in HCT116 MLH1 (–/–) but Is Cytotoxic in HT29 MLH1 Knockouts
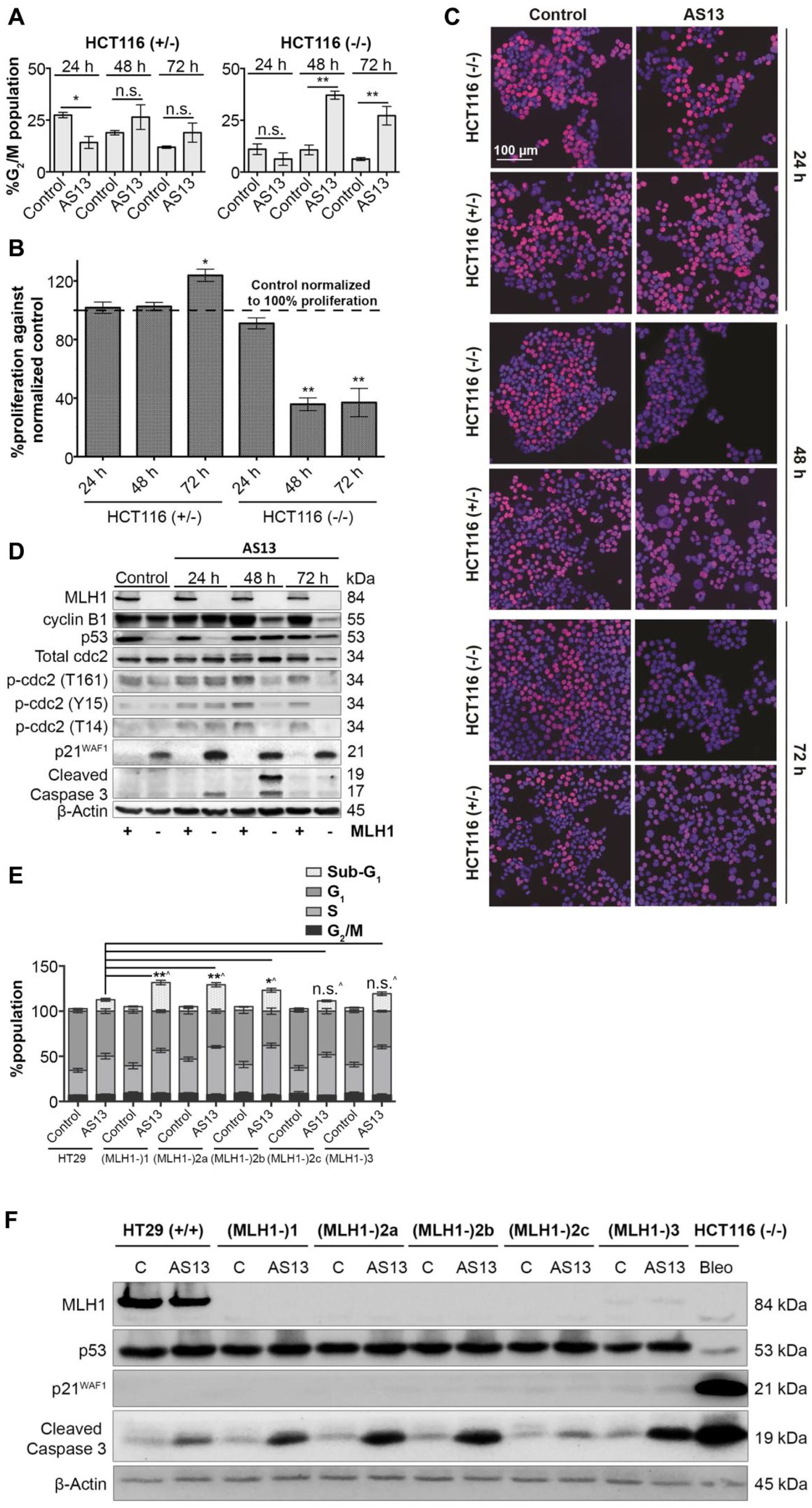
With the observed selectivity, we next sought to understand differences in cell cycle progression and proliferation upon AS13 treatment in both HCT116 and HT29 MLH1 isogenic lines. For these studies, cells were cultured in the presence of AS13 (10 µM for HCT116; 50 µM for HT29) over 24, 48, and 72 h. The test concentrations were selected based on the IC50 value of AS13 at 72 h. As shown in
Figure 3a
, AS13-treated HCT116 MLH1 (–/–) (48 h) had a notable increase of cell population in the G2/M phase compared with the respective vehicle control (10.6%–37.1%; p <0.01), and this increased with 72 h treatment (6.2%–27.8%; p <0.01). Notably, no statistically significant differences were observed between AS13-treated cells and vehicle control in the G1, S, and G2/M phases of HCT116 MLH1 (+/–) cells (
Effect of AS13 on cell cycle and proliferation in HCT116 and HT29 MLH1 isogenic lines. (
As AS13 was halting HCT116 MLH1 (–/–) cells at the G2/M phase from the cell cycle, we evaluated the overall proliferation of cells upon AS13 treatment using the Click-iT EdU cell proliferation assay. We quantified the amount of incorporated EdU in newly synthesized DNA, as a measurement of proliferation. Supporting our cell cycle analysis, we observed that proliferating cells were notably reduced, ~65% from 48 h in HCT116 MLH1 (–/–) compared with the control ( Fig. 3b,c ). On the other hand, quantification of the amount of incorporated EdU in HCT116 MLH1 (+/–) remained unchanged from 24 and 48 h. However, there was a 23% increase in MLH1 (+/–) cell proliferation observed at 72 h. Thus, with these data we hypothesized that in HCT116 MLH1 (+/–) cells, prolonged AS13 treatment increases the activity of the MLH1-dependent DNA repair mechanism. Consequently, HCT116 MLH1 (+/–) cells are able to repair AS13-induced DNA lesions and further proliferate, and hence the increased incorporation of EdU into the DNA.
Western blot analysis of G2/M-associated proteins revealed that treatment with AS13 (10 µM) in HCT116 MLH1 (–/–) gradually decreased protein levels of both cyclin B1 and total cdc2, including its activating (T161) phosphorylated form ( Fig. 3d ), supporting the observation of cell cycle arrest at G2/M. However, the protein level of inhibitory (T14 and Y15) phosphorylated cdc2 was found to also decrease. Of note, there was an induction in p53 that was accompanied by an increase in p21WAF1, further suggesting a p53-dependent upregulation of p21WAF1 and cell cycle arrest. On the other hand, in HCT116 MLH1 (+/–), AS13 treatment broadly did not induce observable changes in cdc2 or its phosphorylated levels, suggesting no cell cycle arrest as supported by the EdU proliferation assay.
As a consequence of cell cycle arrest, cell death was observed selectively in HCT116 MLH1 (–/–) whereby the sub-G1 population of HCT116 MLH1 (–/–) (23.6 ± 0.70%, 72 h) was greater than that of HCT116 (+/–) (11.9 ± 0.73%, 72 h; p < 0.01) ( Suppl. Fig. S2a ). This is also evident in immunoblotting analysis as the level of cleaved caspase 3, an apoptosis marker, is upregulated upon AS13 treatment from 24 to 48 h in HCT116 MLH1 (–/–) but not in HCT116 MLH1 (+/–) ( Fig. 3d ). There was a decrease of cleaved caspase 3 expression observed from 48 h to 72 h in HCT116 MLH1 (–/–), which could partly be due to the degradation of unstable cleaved caspase 3 by 72 h, 28 while at the same time shifting toward cytostatic survival mode.
Furthermore, we also examined the cell cycle progression of HT29 isogenic lines. Unlike HCT116 isogenic lines, no significant changes in cell cycle were observed in AS13 (50 µM, 72 h)-treated HT29 isogenic lines, indicated by comparable cell populations in G1, S, and G2/M phases ( Fig. 3e ). Interestingly, we noted that AS13 treatments increased the sub-G1 population much greater in HT29 MLH1 knockout lines (20%–32%; p < 0.01), with the exception of HT29(MLH1-)2c and HT29(MLH1-)3, when compared with the parental HT29 MLH1 (+/+) (12.6 ± 1.58%), indicating cell death upon AS13 treatments. This observation was reinforced by the biochemical analysis whereby cleaved caspase 3 was upregulated upon AS13 treatment in HT29 MLH1 (–/–) cells compared with HT29 MLH1 (+/+), with the exception of HT29(MLH1-)2c ( Fig. 3f ). In addition, we found that AS13 treatment did not reduce cell proliferation ( Suppl. Fig. S2b,c ), supporting the cell cycle analysis. Our data thus far suggest that AS13 promotes cytotoxicity in HT29 MLH1 (–/–), with the exception for HT29(MLH1-)2c, while it induces cytostasis in HCT116 MLH1 (–/–). This observed disparity is likely due to the p53 status in these cell lines where HT29 harbors a mutant p53 29 that is unable to induce p53/p21-mediated cell cycle arrest ( Fig. 3f ) and confers cytotoxicity instead. Regarding HT29 MLH1 knockouts, HT29(MLH1-)2c was not sensitive to AS13 ( Fig. 2d ) including menadione, a POLG inhibitor ( Suppl. Fig. S2d ), partly due to the presence of other unexpected genetic alterations that could have occurred during the gene editing process. 30
AS13 Increases the Amount of 8-Oxoguanine Lesions by ROS in HT29 MLH1 Knockout Cells
Given the selectivity of AS13 for MLH1 deficiency, and that MLH1 is responsible for the repair of mismatch DNA lesions, we hypothesized that AS13 induces excessive unrepaired DNA lesions due to malfunctional MLH1, which eventually compromises cell survival. To prove our hypothesis, we first evaluated the accumulation of 8-oxoG, the most common DNA oxidized base, which has also been reported to correlate with POLG, 8-oxoguanine glycosylase (OGG1), or mitochondria-related kinases/MLH1 synthetic lethality effects.16,17 As AS13 induced cytotoxicity in HT29 MLH1 knockouts, we assessed the level of 8-oxoG in DNA extracted from HT29 cells treated with AS13 and control using a competitive ELISA method. Hydrogen peroxide, a known inducer of oxidative stress including modification of guanine to 8-oxoG, 31 was used as a positive control to validate the assay. As shown in Figure 4a , hydrogen peroxide (100 µM) elevated the 8-oxoG concentration by 1.4-fold (6.07 ± 0.56 ng/mL; p < 0.05) and 2.5-fold (12.1 ± 1.90 ng/mL; p < 0.01) in HT29 (control: 4.24 ± 0.074 ng/mL) and HT29(MLH1-)2b (control: 4.87 ± 0.637 ng/mL), respectively. The difference in 8-oxoG concentration is expected as HT29 is proficient in MLH1 and thus repairs 8-oxoG. Similarly, AS13 (50 µM) and menadione (12.5 µM), a POLG inhibitor, elicit similar observations of increasing 8-oxoG levels by 1.8-fold (8.93 ± 1.28 ng/mL; p < 0.05) and 1.6-fold (7.79 ± 2.06 ng/mL; p = 0.14) in the deficient MLH1 line, HT29(MLH1-)2b, while having minimal increase in levels of HT29 (from 4.24 to 5.74 ng/mL and from 4.24 to 3.97 ng/mL, respectively). This is consistent with the earlier observation of AS13 selectivity in MLH1-deficient cells. To exclude the possibility of bias, we treated both HT29 MLH1 (+/+) and HT29(MLH1-)2b with 5-FU (10 µM), a chemotherapy known to inhibit DNA synthesis and that has not been reported to have selective elevation of 8-oxoG levels in the MLH1 isogenic. Indeed, we observed that 5-FU treatment did not induce much elevation of the 8-oxoG level compared with the endogenous level in both HT29 and HT29(MLH1-)2b.
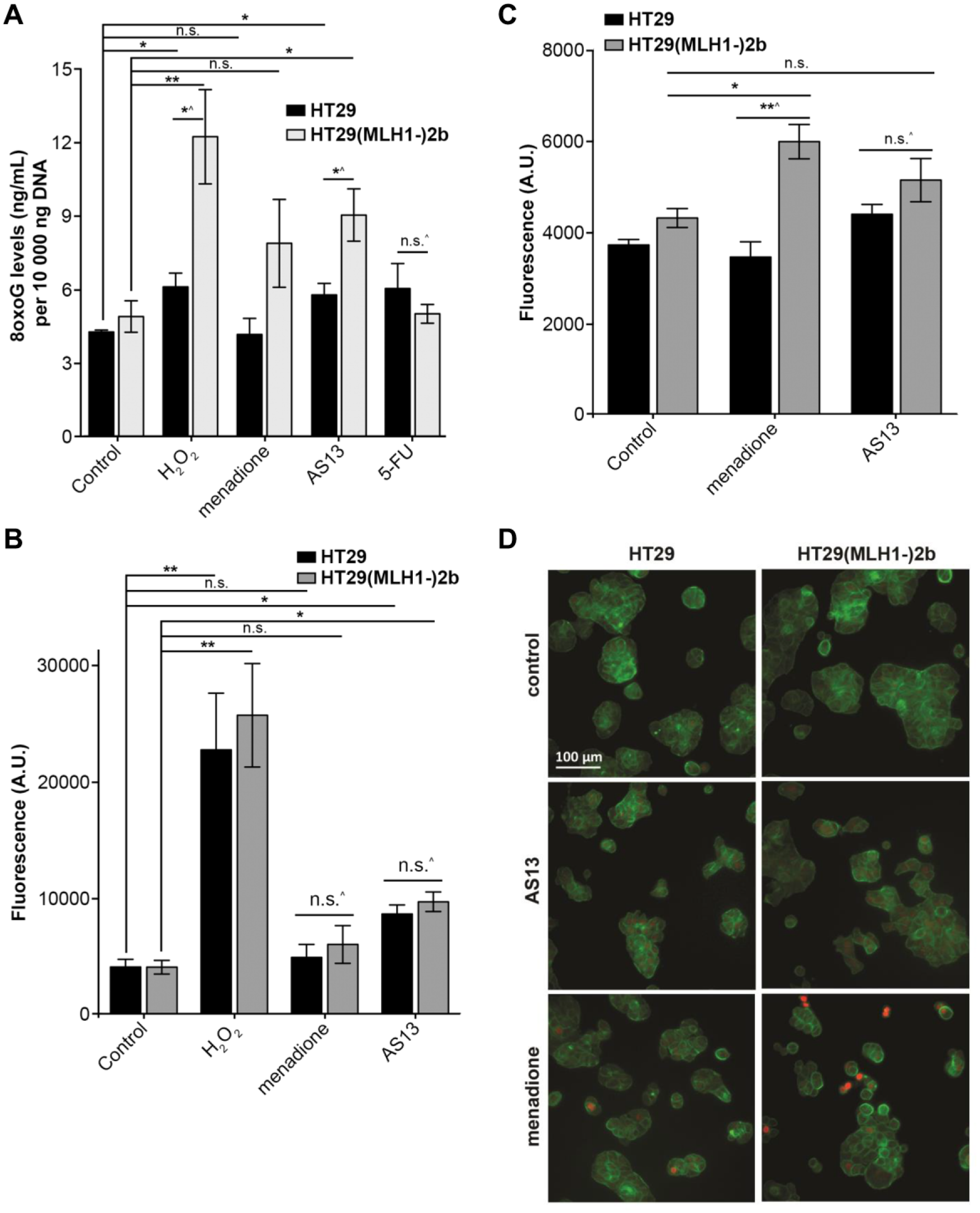
The selectivity of AS13 toward MLH1-deficient cells is associated with an increase in oxidative damage. (
Next, we evaluated if the observed increase in 8-oxoG is a result of elevated reactive oxidation species (ROS) in the cells. We measured levels of ROS in HT29 and HT29(MLH1-)2b after AS13 treatment. Hydrogen peroxide (200 µM) was used as a control to elicit ROS in both HT29 MLH1 (+/+) and HT29(MLH1-)2b, with a geometric mean of fluorescence signal (A.U.) of 22,698 ± 4857 and 25,659 ± 4433, respectively ( Fig. 4b ). Increased ROS was observed in both isogenics upon AS13 treatment, regardless of MLH1 status (HT29: A.U. 4080–8622; p < 0.05; HT29(MLH1-)2b: A.U. 4059–9666; p <0.05). This indicated that AS13 induced ROS in both HT29 and HT29(MLH1-)2b, but because only HT29 can repair the damage, a lower number of 8-oxoG lesions resulted ( Fig. 4a ).
Concurrently, we also assessed if AS13 induced the formation of DNA DSBs in the synthetic lethal relation with MLH1 deficiency.16,18 The reported mechanism for this is outlined in Supplemental Figure S3 , where intracellular ROS causes the formation of 8-oxoG lesions leading to MUTYH being recruited. The inhibition of POLG down this pathway can lead to the endonuclease, APE, accumulating unrepaired single-stranded breaks (SSBs), which ultimately leads to DSBs. We monitored this DSB formation by the detection of γ-H2A.X (S139), which is specifically phosphorylated at S139 in response to DSBs. 32 Menadione was found to elicit a higher intensity of γ-H2A.X in HT29(MLH1-)2b (A.U. 5991 ± 375) compared with HT29 MLH1 (+/+) (A.U. 3455 ± 335; p < 0.01), which is expected as a POLG inhibitor ( Fig. 4c ). Interestingly, AS13 did not induce significant changes in γ-H2A.X fluorescence intensity compared with vehicle control in both HT29 MLH1 (+/+) (A.U. 3722–4395) and HT29(MLH1-)2b (A.U. 4315–5146) cells. An immunofluorescence visual representation is shown in Figure 4d , and cells with DSBs can be seen with red spots in the nucleus. Interestingly, increased concentrations of AS13 (75 and 100 µM) also yielded nonsignificant changes in γ-H2A.X fluorescence intensity for both HT29 MLH1 (+/+) and HT29(MLH1-)2b, as shown in Supplemental Figure S4a . Overall, AS13 selectively increased 8-oxoG levels specific to MLH1 deficiency by inducing ROS.
Discussion
In this study, we identified AS13, a novel compound that confers selectivity to MLH1 deficiency. AS13 is a diarylpentanoid analogue, synthesized based on a curcumin backbone. Curcumin is the active ingredient of turmeric and has been widely studied for various bioactivities, including antiproliferative and anti-inflammation. However, its poor stability and limited bioavailability diminish its usefulness for clinic utility. 33 As a consequence, attention has been focused in the area of derivative synthesis, with the intention to improve the stability and bioavailability of curcumin, including our current study. We have synthesized a series of diarylpentanoids, which are 5-carbon spacer series of curcumin that are known for its metabolic stability at physiological pH. 22 This diarylpentanoid series omitted the ethylene group from the basic curcumin structure while including a cyclic ring with the keto-enol functional group, with the aim to reduce electron delocalization and hence increase its stability from degradation.
Diarylpentanoids have gained increasing attention due to their potential anti-inflammatory, antiproliferative, and antiangiogenic benefits. For example, Ohori et al. 34 has reported that symmetrical diarylpentanoids with 3,5-methoxy substitutions inhibit growth by downregulating β-catenin, K-ras, cyclin D1, c-Myc, and ErbB-2 in a panel of different cancer cell lines, with as low as one-eighth the concentration at which curcumin normally has an effect. Furthermore, from our series of 32 diarylpentanoids, we were able to deduce a potential SAR that likely contributes to the observed selectivity of AS13 in an MLH1-deficient background. Based on our analysis, we noted that concurrent substitution at C2 and C3 positions of the diarylpentanoid analogue may grant a preference and inhibitory effect toward the MLH1 mutant. However, more analogues with both C2 and C3 substitutions, such as dual hydroxyl groups (hydroxyl version of AS13), or rearrangement of methoxy groups at different substitution positions (C2 and C4, and C3 and C5) is needed to further strengthen our observation here.
MMR proteins play critical roles in the repair of DNA lesions during DNA replication as well as restoring oxidatively damaged DNA. 35 Thus, it is perhaps not surprising that HCT116 MLH1 (–/–) and HT29 MLH1 knockouts are more sensitive to AS13 exposure, as observed in our study, where lesions induced by AS13 may not be optimally repaired in the absence of MLH1. Of note, although MLH1 deficiency is not the only genetic variable within the cancer cell line panel tested, AS13 can still exhibit its selectivity toward MLH1-deficient cells such as HCT116, KM12, and DU145. In particular, the observed sensitivity of AS13 in MLH1 mutant is encouraging, as all the cancer cell lines tested are MSH2 functional ( Suppl. Fig. S4b ). To further extend the understanding of AS13 on other MMR genes, the viability of LoVo (MSH2-deficient) and DLD-1 (MSH6-deficient) lines was determined through dose escalation treatment. We observed that DLD-1 was not sensitive to AS13, having an IC50 value greater than 100 µM ( Suppl. Fig. S4c ). Interestingly, LoVo confers marginal sensitivity toward AS13 (IC50 ~67 µM) when compared with HT29 (MSH2 proficient; IC50 86.88 µM) and HT29(MLH1-)2b (IC50 53.02 µM). However, the potency of AS13 toward MSH2 deficiency cannot be fully concluded without validation using MSH2 isogenic cell lines such as HAP1 MSH2 knockouts (Horizon Discovery UK), which is not addressed in this study. Our results also indicate that the p53 status of cell lines potentially influences the potency of AS13. We observed that AS13 is more effective toward MLH1 deficiency with p53 wild-type as shown in HCT116 MLH1 (–/–) (IC50 9.54±1.98 µM) compared with HT29 MLH1 (–/–), a p53 mutant line (IC50 ~45 µM). A plausible explanation for this difference could be the p53-mediated upregulation of MUTYH, an enzyme critical for modulating cell death (or repair) through DNA damage, 36 conferring further sensitivity in HCT116 MLH1 (–/–). It has been shown that the decrease in overall cell viability by cumulative oxidative lesions or POLG/OGG1 inhibition ( Suppl. Fig. S3iii ) is dependent on increasing MUTYH activity.16,36 In support, Martin et al. showed that POLG inhibition in combination with MUTYH silencing in p53 wild-type HCT116 MLH1 (–/–) cells rescues the treated cells from cellular death. 16
Here, we provide evidence that AS13 can trigger MLH1-selective effects through an oxidative stress-mediated mechanism and hence cytotoxicity in MLH1-deficient cells. AS13 elicits genetic instability by generating ROS in both MLH1-proficient and -deficient cells. Specifically, an MLH1-deficient background is permissive to an increased level of ROS and hence can lead to suboptimal repair or oxidatively damaged DNA, which eventually is incompatible with viability. There are several oncology drugs currently used in the clinics that are thought to provoke their therapeutic effects via oxidative damage, 37 including menadione, motexafin gadolinium, and β-lapachone. However, caution is recommended as such induced excessive oxidative damage could potentially promote cancer progression.17,38 Nonetheless, compounds with a modest level of ROS and oxidative DNA damage such as AS13 identified from our currently study could potentially provide a viable therapeutic window. Although AS13 is low in potency with modest selectivity, the opportunistic discovery of AS13 warrants further drug lead optimization and development with the aim to improve the potency as well as the selectivity of MLH1 deficiency and could provide better therapeutic development for patients with MMR-deficient tumors.
Supplemental Material
Supplementary_Material_by_Song,_et_al – Supplemental material for Novel 2-Benzoyl-6-(2,3-Dimethoxybenzylidene)-Cyclohexenol Confers Selectivity toward Human MLH1 Defective Cancer Cells through Synthetic Lethality
Supplemental material, Supplementary_Material_by_Song,_et_al for Novel 2-Benzoyl-6-(2,3-Dimethoxybenzylidene)-Cyclohexenol Confers Selectivity toward Human MLH1 Defective Cancer Cells through Synthetic Lethality by Dedrick Soon Seng Song, Sze Wei Leong, Kwok Wen Ng, Faridah Abas, Khozirah Shaari, Chee Onn Leong, Felicia Fei-Lei Chung, Chun Wai Mai, Ling Wei Hii, Pei Jean Tan and Vyomesh Patel in SLAS Discovery
Footnotes
Acknowledgements
We would like to thank Professor Dr Teo Soo Hwang and Dr Lee Hong Boon for initiating the screening programme for synthetic lethality; Professor Ashok Venkitaraman from the University of Cambridge for the initial discussion on synthetic lethality; Professor Nordin Lajis, who initiated the synthesis of the dicarbonyl diarylpentanoid system; and Mariani Masjidan for assistance in the analysis of the chemical structures of the compounds used in this study. We are also grateful to Li-Sia Heng, Kah-Syhuan Lee, Farid Nazer, Beng Fye Lau, Bryan Lye, and Norazwana Samat for help with data analysis.
Supplemental material is available online with this article.
Authors’ Note
Kwok Wen Ng completed this work as part of his employment at Cancer Research Malaysia. He is currently affiliated with the Centre of Foundation Studies, Universiti Teknologi MARA, Persiaran Cybersouth, Selangor, Malaysia. Felicia Fei-Lei Chung is currently affiliated with Mechanisms of Carcinogenesis (MCA), Epigenetics Group (EGE), International Agency for Research on Cancer, World Health Organization, Lyon, France.
Declaration of Conflicting Interests
The authors declared the following potential conflicts of interest with respect to the research, authorship, and/or publication of this article: Mr. Song and Drs. Ng, Tan, and Patel are employees of Cancer Research Malaysia and completed this work within the scope of their employment.
Funding
The authors disclosed receipt of the following financial support for the research, authorship, and/or publication of this article: This study was supported by Cancer Research Malaysia internal funds. We would like to express our gratitude to all the donors of Cancer Research Malaysia, a nonprofit organization. We are committed to an understanding of cancer prevention, diagnosis, and treatment through a fundamental research program. The authors also thank the Ministry of Education (MOE) of Malaysia for financial support under the project ERGS/1/11/STG/UPM/01/24 for compound synthesis.
References
Supplementary Material
Please find the following supplemental material available below.
For Open Access articles published under a Creative Commons License, all supplemental material carries the same license as the article it is associated with.
For non-Open Access articles published, all supplemental material carries a non-exclusive license, and permission requests for re-use of supplemental material or any part of supplemental material shall be sent directly to the copyright owner as specified in the copyright notice associated with the article.