
Abstract
Synthetic lethality (SL) is a promising therapeutic concept that relies on the indirect targeting of vulnerabilities acquired through genetic mutations. Ovarian and endometrial cancers frequently exhibit mutations in the breast cancer (BRCA), phosphatase and tensin homolog (PTEN), AT-rich interactive domain-containing protein 1A (ARID1A) and TP53 genes, as well as DNA repair pathway deficiencies. Poly(ADP-ribose) polymerase inhibitors (PARPis) have demonstrated remarkable efficacy in various cancers with BRCA mutations and homologous recombination deficiency. In addition to PARPi, there has been an expansion of drugs exploiting the selective vulnerability of cancer cells via SL, such as WEE1 kinase and Ataxia Telangiectasia and Rad3-related protein (ATR). WEE1 inhibitors have shown encouraging results in combination with chemotherapy, increasing the objective response rate in patients with platinum-resistant TP53-mutated ovarian cancer. ATR inhibitors are currently being evaluated in ARID1A-mutated tumours, with preliminary results confirming their therapeutic potential.
The physiological role of each synthetic lethality target in the context of gynaecological cancers. This figure highlights the specific molecules involved and the consequences of inhibiting each gene, demonstrating how synthetic lethality strategies exploit these vulnerabilities for therapeutic intervention. Created in BioRender by Lebeau (2025; https://BioRender.com/g66j127).
Introduction
Gynaecological cancers are characterised by a significant genetic diversity. High-throughput sequencing techniques, such as those employed by The Cancer Genome Atlas (TCGA) project, have enabled detailed mapping of the molecular alterations in these tumours. Understanding the prevalence of these mutations across the different gynaecological tumours allows more rational and precise use of targeted therapies.
Alterations in genes such as AT-rich interactive domain-containing protein 1A (ARID11), phosphatase and tensin homolog (PTEN) and BRCA1/2 are common. For example, ARID1A mutations occur in approximately 45% of ovarian endometrioid and clear cell carcinomas and 25% of endometrial cancers, while BRCA1/2 mutations are frequently found in high-grade serous ovarian cancers (HGSOCs).1–3 These mutations often disrupt DNA repair mechanisms, leading to genomic instability. Synthetic lethality (SL) offers an opportunity to exploit these vulnerabilities, with strategies such as poly(ADP-ribose) polymerase inhibitors (PARPis) in BRCA-mutated cancers 4 or Ataxia Telangiectasia and Rad3-related protein (ATR) inhibitors in ARID1A-deficient tumours. 5
These advances have led to the identification of specific mutations that can be exploited for the development of targeted therapies, allowing for treatments based on precision medicine. 6 An increased understanding of the genomic profiles of gynaecological cancers has facilitated the optimisation of therapeutic strategies. Among the mutations associated with tumourigenesis, there are ‘gain-of-function’ mutations leading to oncogenic protein overexpression that can be easily targeted, as well as ‘loss-of-function’ mutations in tumour suppressor genes that disrupt cell growth regulation. Targeting these later mutations is more challenging, as they do not involve oncogenic protein inhibition, but require the exploitation of vulnerabilities by identifying SL. 7 In this review, we describe SL, which has emerged as a promising therapeutic approach for cancers associated with loss-of-function mutations. We also discuss interesting targets in ovarian and endometrial cancers, along with completed and ongoing clinical studies.
SL overview
SL is a biological concept describing a relationship between two genes, where the alteration of both genes leads to cell death, while alteration in only one of the genes is tolerable. By targeting a gene that is synthetically lethal with a mutated or deregulated oncogene, SL-based therapies can selectively eliminate cancer cells while sparing normal cells, thereby reducing off-target toxicities. Research on SL has led to the identification of a wide array of SL mechanisms that vary in complexity and contextual dependency conditions. These mechanisms can be classified into four main categories (Figure 1).

Representation of the four categories of synthetic lethality. (a) Standard synthetic lethality, where the mutation of a gene, such as BRCA1, renders the cell vulnerable to PARP inhibition. (b) Synthetic dosage lethality, exemplified by the interaction between Cyclin E1 (CCNE1) and the Wee1 inhibitor, which affects cell cycle control. (c) Conditional synthetic lethality, influenced by environmental factors such as human papillomavirus (HPV), oxidative stress or hypoxia, as seen in HPV-positive cells treated with a PARPi. (d) Complex synthetic lethality, which incorporates polygenic interactions and environmental factors, increases the complexity of the tumour response.
Standard SL
At the cellular level, functional redundancy ensures that compensatory mechanisms can be activated in cases of an inactive or mutated protein. 8 SL is an approach involving the interaction between two or more genes. The alteration (mutation or inhibition) of one of these genes can be viable for the cell, while an expression anomaly of both genes leads to cell death9,10 (Figure 1(a)).
This concept is now widely studied in the development of cancer treatments, particularly in relation to mutations frequently observed in tumour cells.11,12 The use of PARPi in patients with BRCA-mutated ovarian cancer has paved the way for SL as an effective therapeutic strategy.13–15
Synthetic dosage lethality
Alterations in gene copy number and the epigenetic regulation of some genes are common tumour abnormalities, leading to gene and protein overexpression. Various therapeutic strategies can therefore be considered, including inhibition of the overexpressed proteins or the synthetic dosage lethality (SDL) approach. This latter strategy is based on gene overexpression inducing cellular stress that can only be tolerated if other genes remain functional. 10 When mutations cause a loss of function, or in the case of pharmacological inhibition of these proteins, tumour cells apoptosis can be induced. SDL is particularly relevant in cases of oncogene deregulation, where direct inhibition is often too toxic for normal cells due to their essential roles in cell cycle, survival, differentiation, apoptosis, proliferation and metabolism (Figure 1(b)). For example, the oncogene c-myc, which regulates approximately 15% of all genes under normal conditions, is frequently overexpressed in more than 50% of cancers. Targeting c-myc appears to be a relevant strategy in oncology; however, its involvement in many crucial cellular functions represents a therapeutic challenge. 16 Various strategies to target c-myc have been tested, including SDL. Inhibition of cyclin-dependent kinase 1 (CDK1) by the small molecule purvalanol A induces downregulation of the surviving in myc-driven cells, leading to specific apoptosis of c-myc overexpressing cells.17,18 Moreover, c-myc induces replication stress and DNA damage through excessive replication-fork firing, making c-myc overexpressing tumours more sensitive to checkpoint kinase 1 (CHK1) inhibition and consequently, CHK1 inhibition leads to massive cell death in c-myc overexpressing cancer cells.19–21
The overexpression of protein CCNE1, which is frequently observed in gynaecological cancers due to high copy number amplification, exhibits SDL when combined with the protein Wee1 inhibitor.
Conditional SL
Beyond the genetic contribution, the environment, tumour heterogeneity and external conditions can significantly influence SL. 22 The recent concept of conditional SL refers to a specific lethal phenotype that also depends on internal factors (such as hypoxia and high reactive oxygen species (ROS) production) and/or external factors (such as the use of agents inducing DNA anomalies). These factors can explain the heterogeneity of treatment responses (Figure 1(c)).
Hypoxia is a recognised hallmark of solid tumours, arising from the rapid proliferation of cancer cells and the development of aberrant, inefficient vasculature. This results in a heterogeneous oxygen landscape within the tumour, with regions of moderate (1%–2%) and areas of severe hypoxia or anoxia (<0.01%). Both acute and chronic hypoxia have been shown to suppress homologous recombination repair (HRR) by downregulating key effectors such as RAD51, BRCA1 and BRCA2.23–25 This hypoxia-induced homologous recombination deficiency (HRD) sensitises tumour cells to PARPi, leading to SL under conditions of severe hypoxia (<0.5%). 26
Additionally, SL is essentially influenced by the tumour microenvironment in cervical and vulvar cancers. Indeed, tumorigenesis results in most cases from the integration of the HPV genome into the host genome, rather than being caused by specific oncogenic mutations in DNA repair pathways or cell cycle regulation. HPV-positive tumours are associated with several intrinsic characteristics: low tumour hypoxia, high T cells density in the tumour microenvironment 27 and a p53 wild-type status. 28 Additionally, the E6 and E7 viral oncoproteins expressed in tumour cells interact with over 20 proteins (including CHEK2, CLK2, ERCC3) involved in DNA repair mechanisms. 29 The virus exploits these DNA repair pathways to facilitate its own replication, ultimately compromising the host cell and leading to genomic abnormalities accumulation. The use of PARPi in HPV-positive tumour cells exploits conditional SL, leading to the accumulation of unrepaired DNA damage and ultimately causing cell death (NCT01281852, NCT737664, NCT03476798, NCT03644342). 30
Complex SL
SL associated with genetic mutations often exhibits incomplete penetrance, suggesting the influence of additional genetic (polygenic) and environmental factors. 31 Indeed, the concept of complex SL reflects the biological redundancy inherent in cancer cells. Unlike a binary relationship between two genes, complex SL is context-dependent, influenced by genetic and environmental factors, and contributes to the variability in therapeutic responses. Tumour cells have redundant pathways and compensatory systems that can be modulated in response to mutations. The lethal effect of a partnership (mutations/inhibitions) depends on the presence of these compensatory systems, which are modulated by additional genetic mutations, epigenetic regulation and environmental factors such as metabolism, hypoxia and inflammation. Indeed, it has become apparent that the penetrance of pairwise genetic interactions differs significantly across different cancer types (Figure 1(d)). The interplay between genetic mutations and underlying epigenetic modifications creates a network of disturbances that differs from one cancer to another, leading to varied therapeutic outcomes, as seen in the frequent combination of mutations in KRAS/PIK3CA 32 in colorectal cancer, BRAF/PTEN in melanoma 33 or ARID1A/PIK3CA in clear cell ovarian carcinoma. 34
While certain pairwise genetic interactions, such as those between BRCA genes and PARPi, have shown therapeutic promise, resistance mechanisms have revealed that other factors, such as mutations in DNA repair proteins such as TP53BP1 or poly(ADP-ribose) glycohydrolase (PARG), can alter treatment efficacy. For example, HRD can be bypassed by loss-of-function mutations in DNA repair proteins such as TP53BP1, which restore HRR and confer resistance to PARPi.35,36
Unlike cumulative toxicity resulting from multiple non-specific inhibitions, complex SL relies on a functional, non-additive and often selective dependence of cells harbouring the mutated partner. Moreover, combinatorial studies (via CRISPR or RNAi) have shown that only a minority of SL interactions are consistently reproduced across multiple cell lines, underscoring their complex nature. 37
Mechanisms and targets of SL in gynaecological cancers
DNA repair
Among the hallmarks of cancer described by Hanahan and Weinberg,38,39 which define the biological characteristics enabling normal cells to transform into tumour cells, genomic instability is a key factor. It typically arises from defects in DNA repair mechanisms, leading to the accumulation of mutations that promotes malignant transformation. Cells are subjected to various forms of DNA damage, which can arise from both intrinsic (such as replication errors and cellular metabolism generating ROS) and extrinsic factors (such as radiation, viral infections and environmental exposure). 40 These DNA lesions are resolved by various DNA repair mechanisms, thereby maintaining genomic integrity. Each DNA repair mechanism targets specific types of damage. Base Excision Repair (BER) addresses damage caused byoxidised or alkylated bases, 41 while Nucleotide Excision Repair (NER) manages more extensive lesions, such as those induced by UV radiation or chemical agents. 42 For double-strand breaks (DSBs), often caused by ionising agents or replication errors, two repair mechanisms may be involved: HRR or non-homologous end joining (NHEJ). 43
Homologous recombination repair
DSBs are repaired primarily through HRR, initiated by ATM kinase, which phosphorylates proteins like BRCA1, thereby stabilising the damaged DNA and recruiting repair complexes. At the 3′ extremities, BRCA2 and PALB2 facilitate the recruitment of RAD51, which forms a nucleoprotein filament. This filament searches for a homologous sequence in the sister chromatid to enable strand pairing and invasion of the double helix, allowing for DNA synthesis to faithfully repair the break.
The HRR pathway plays a crucial role in gynaecological cancers. 44 According to TCGA, HRD is found in approximately 50% of epithelial ovarian,2,45,46 50% of non-endometrioid endometrial and 12% of endometrioid endometrial cancers (Figure 2). 47
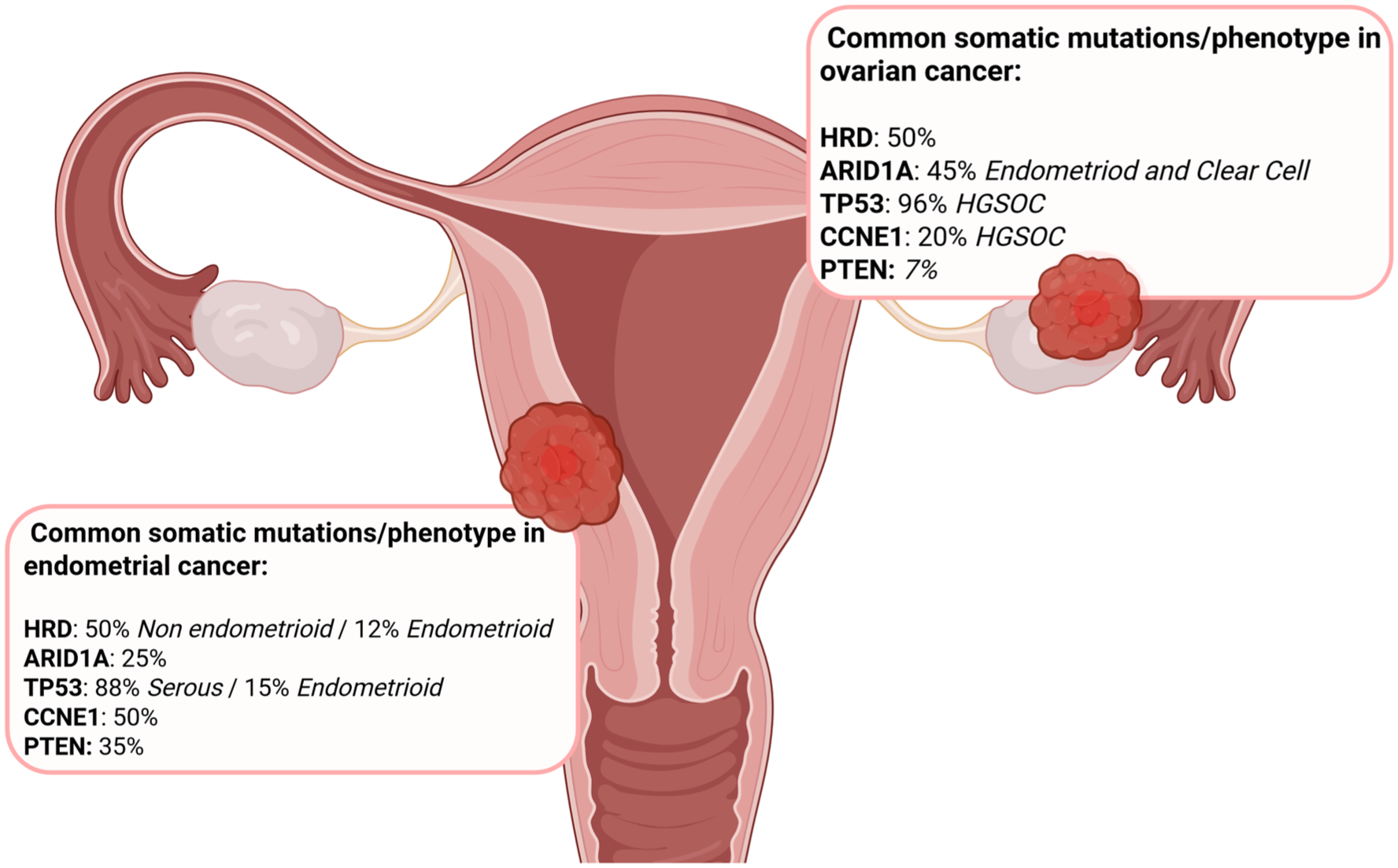
Overview of the most common mutations identified in ovarian and endometrial cancers, classified by tumour site.
HRD may result from mutations in key proteins including BRCA1, BRCA2, RAD51, RAD52, PALB2 and ATM. In the case of an HRD-positive tumour, DSBs accumulate, leading to increased genomic instability. Alternative repair pathways, such as NHEJ, take over, allowing the cell to survive but at the cost of increased errors and genomic instability. 48 PARP is involved in repairing single-strand breaks (SSBs), which must be corrected rapidly to prevent their progression to DSBs during DNA replication. In response to SSB, PARP binds to the damaged site and adds ADP-ribose chains (PARylation) to itself and nearby proteins. This post-translational modification serves both as a recruitment signal for DNA repair factors and as a means to decondense chromatin, thereby enhancing accessibility to the damaged site. This step is essential for initiating repair pathways such as BER, the primary mechanism for resolving SSBs. In the presence of pharmacological PARP inhibition, SSBs persist. During DNA replication, the progression of the replication fork requires the opening of the DNA helix and strand separation. When the replication machinery encounters an unrepaired SSB, it becomes interrupted. Continued helicase and polymerase activity prior to the damage site generates torsional stress, which can cause collapse of the replication fork and physical breakage of the opposing strand – thereby converting a single-strand lesion into a DSB. These secondary DSBs are normally repaired by HRR, a high-fidelity process relying on a homologous DNA template. In HR-deficient cells, such as those with BRCA1 or BRCA2 mutations, repair cannot proceed effectively, resulting in genomic instability and cell death.
Inhibition of PARP results in the accumulation of SSBs, which evolve into DSBs that cannot be effectively repaired in HRD-positive tumour cells, ultimately leading to cell death. PARP inhibition exploits this HRD-related vulnerability by amplifying DNA damage beyond a tolerable threshold, thereby inducing apoptosis.
However, ovarian cancers inevitably develop resistance to PARPi during or after maintenance therapy. 49 This resistance is due to secondary mutations restoring BRCA function,50,51 activation of pro-survival signalling pathways (such as RAS and PI3K/AKT),52,53 and stabilisation of replication forks. 54 Treatment with PARPi increases activation of ATR, which plays a crucial role in maintaining genome integrity by stabilising replication forks and activating cell cycle checkpoints at the S and G2/M phases. 55 Selective pharmacological ATR inhibitors have been developed, and their use in combination with PARPi could potentially overcome resistance and enhance DSBs accumulation.56–58
AT-rich interactive domain-containing protein 1A (ARID1A)
ARID1A gene mutations are common in gynaecological tumours, particularly in endometrioid and clear cell ovarian cancers (approximately 45%), as well as in endometrial cancers (approximately 25%) (Figure 2).3,59–61 Notably, the majority of ARID1A mutations are of the ‘loss-of-function’ type.
ARID1A (also known as BAF250a) is a key subunit of the SWI/SNF chromatin remodelling complex, involved in DNA repair, cell cycle regulation and epigenetic control of signalling pathways. 62
This complex is recruited to sites of DNA DSBs through interactions with ATR. 63 ARID1A facilitates chromatin decompaction at these sites, enhancing DNA accessibility and allowing the efficient recruitment of HRR proteins, such as RAD51. 64 In addition, ARID1A recruits and stabilizes topoisomerase II alpha (TOP2A), an enzyme essential for resolving DNA supercoiling by introducing transient DNA breaks. Loss of ARID1A impairs both chromatin decompaction and TOP2A function, resulting in limited access for repair proteins and increased DNA damage.65,66 ARID1A also directly regulates p21 (CDKN1A), a cyclin-dependent kinase inhibitor (CKI) that governs the G1/S checkpoint in response to genotoxic stress temporarily halting cell cycle progression to allow repair. 67 Epigenetically, ARID1A represses the expression of enhancer of zeste homolog 2 (EZH2) polycomb repressive complex 2 (PRC2) subunit, which silences tumour suppressor genes such as PTEN through histone methylation. 68
Loss-of-function mutations in ARID1A impair chromatin remodelling, compromising HRR by reducing RAD51 access to DSBs and destabilising TOP2A, thereby increasing DNA supercoiling and replication stress. The inability to upregulate p21 prevents G1/S checkpoint arrest, allowing the propagation of damaged DNA. Increased EZH2 activity silences tumour suppressors such as PTEN, thereby activating the PI3K/Akt signalling pathway. These alterations lead to genomic instability and make cells highly dependent on ATR, offering therapeutic opportunities via SL with ATR, PARP or EZH2 inhibitors. 69 Indeed, pharmacological inhibition of ATR induces replication fork collapse and accumulation of DSBs, triggering SL. 5 Similarly, as ARID1A-deficient cells display HRD, they are sensitive to PARPi, which block SSB repair and lead to toxic DSB accumulation. Moreover, EZH2 inhibitors such as tazemetostat may restore tumour suppressor gene expression (e.g. PTEN), downregulate PI3K/Akt signalling and suppress tumour cell proliferation.68,70 Molecular profiling reveals the frequent co-occurrence of ARID1A and PTEN/PI3K mutations, particularly in ovarian and endometrial cancers.34,71
Cell cycle checkpoints and regulation
The mitotic cell cycle is an essential process for the proliferation of normal cells, occurring in four phases: G1 (cell growth), S (DNA synthesis), G2 (mitosis preparation) and M phase (mitosis). This mechanism is regulated by the G1/S and G2/M checkpoints, which ensure genomic integrity before progression to the next phase. Various proteins are involved in cell cycle regulation, including the regulatory proteins p53 and Wee1 kinase, CHK1/CHK2, as well as cyclins and CDKs. Before DNA replication, ATM and ATR proteins are recruited to the sites of DNA lesions. They activate CHK1/2 and p53, which inhibits the CCNE1/CDK2 activity complex through negative regulation of p21.72,73 Tumour cells frequently hijack these cell cycle checkpoints to continue proliferating despite accumulating DNA damage, which in turn enhances their invasiveness and immunosilencing.
TP53/Wee1
The p53 protein is a transcription factor involved in various cellular functions, primarily surveillance of genomic integrity. Under normal conditions, cells contain low levels of p53, which is rapidly degraded by the MDM2 complex. 74 In response to cellular stress stimuli (such as DNA damage, nutrient deprivation or oncogene activation), p53 levels increase due to protein stabilisation: signals from ATM, ATR, CHK1 and CHK2 inhibit MDM2 and stabilise p53 through acetylation. In cases of replication errors or DNA damage, p53 induces the expression of p21 protein, which inhibits CDKs to block cell cycle progression at the G1/S transition. If DNA damage is irreparable, p53 activates pro-apoptotic genes (BAX, PUMA, Caspases) and represses anti-apoptotic genes (Bcl-2). 75
The TP53 gene is the most frequently mutated gene in ovarian cancers, with approximately 96% of HGSOCs carrying mutations.2,76 In endometrial cancer, its prevalence varies by histological subtypes: 88% in serous compared to 15% in endometrioid cancers1,77 (Figure 2). These are typically loss-of-function mutations.
The Wee1 kinase regulates the cell cycle by preventing progression from G2 to mitosis. When p53 is mutated, the G1/S checkpoint becomes ineffective, making the cell dependent on the G2/M checkpoint, which is regulated by Wee1 kinase. 78 This dependency can be therapeutically exploited. By targeting Wee1, CDK1 and CDK2 activity is inhibited, forcing cells blocked in G2 due to DNA damage to progress into mitosis, leading to mitotic catastrophe and apoptosis of tumour cells. The loss of p53 function contributes to genomic instability, resulting in DNA damage accumulation. This damage is detected by the ATR and ATM proteins, which activate DNA repair mechanisms. In p53-mutated cells, inhibition of the ATM/CHK2 and ATR/CHK1 pathways inactivates the repair of accumulating DNA damage, leading to SL.79–82
Cyclin E1
CCNE1 is a protein essential for cell cycle progression during the S phase. Entry and progression through the cell cycle depend on cyclin activity associated with CDKs, which are specific to each phase. CCNE1 forms a complex with CDK2, inducing phosphorylation of the retinoblastoma protein (Rb) and the release of the transcription factor E2F, which is essential for the transcription of genes involved in DNA synthesis. 83
The CCNE1 gene copy number is significantly increased in approximately 20% of HGSOC 84 and 50% of serous endometrial carcinomas (Figure 2). This amplification is associated with resistance to platinum-based therapies and reduced survival outcomes.85–87 In ovarian cancer, CCNE1 overexpression is an early event in tumorigenesis occurring before the emergence of serous tubal intraepithelial carcinoma. 84
CCNE1 overexpression is strongly associated with p53 mutation, leading to cell cycle dysregulation. In p53 wild-type cells, CCNE1 overexpression induces p53 expression, which allows for cell cycle arrest to facilitate DNA repair. 88 In contrast, when p53 is mutated, CCNE1 activity persists, resulting in genomic instability. 89 CCNE1 amplification and overexpression, associated with p53 ‘loss of function’, allow tumour cells to bypass the G1/S checkpoint despite DNA damage. The G2/M checkpoint is crucial for preventing cell division. This dependency can be targeted by Wee1 and Protein Kinase Membrane Associated Tyrosine/Threonine 1 (PKMYT1; a protein that prevents premature entry into mitosis) inhibitors, inducing SL. 90 Currently, there are four ongoing phase I clinical trials evaluating PKMYT1 inhibitors.
Oncogenic signaling pathways
Phosphatase and tensin homolog
The PI3K/Akt/mTOR signaling pathway is involved in numerous pro-tumorigenic processes, such as proliferation, cell survival and cell motility. 91 PTEN functions as a tumour suppressor by negatively regulating this signaling pathway. By removing a phosphate group from phosphatidylinositol-3,4,5-triphosphate (PIP3), PTEN inactivates this pathway. In contrast, when PI3K is activated, it adds a phosphate group to phosphatidylinositol-3,4-bisphosphate (PIP2), catalyzing the conversion of PIP2 to PIP3, thus activating the signaling pathway. This mechanism functions as a switch (off/on), regulating the PI3K/Akt/mTOR pathway activation or inhibition.
PTEN loss-of-function mutations are observed in approximately 47% of endometrial cancers and 7% of ovarian cancers (Figure 2). 92
Loss of PTEN results in continuous pathway activation, driving tumour progression and resistance to conventional treatment. 93 In addition to its role as a PI3K/Akt/mTOR pathway inhibitor, PTEN can also be found in the nucleus, where it regulates the transcription of the RAD51 protein (a major player in HRR), as well as of cyclin D1 (involved in G1 phase progression of the cell cycle).94–96 Inhibitors targeting DNA repair mechanisms and the G2/M checkpoint may act synergistically to induce SL. The gain-of-function PIK3CA and KRAS mutations are commonly observed in ovarian and endometrial cancers.97,98 SL does not generally apply to these types of mutations, as they can be directly targeted with specific inhibitors, such as those developed for KRAS.
Clinical trials evaluating SL target in gynaecological cancers
The SL concept has found significant clinical application with the use of PARPi in tumours exhibiting HRD, revolutionising daily practice in breast, pancreas, prostate and ovarian cancers. There is a growing interest in evaluating new inhibitors for other loss-of-function mutation types. Currently, these agents are in the early stages of development, with studies primarily aiming to assess their toxicity and efficacy without specifically targeting particular mutations (Table 1).
Clinical trials evaluating a potential synthetic lethality target in ovarian and endometrial cancers.

5-y PFS, 5 years PFS; ARID1A, AT-rich interactive domain-containing protein 1A; ATR, Ataxia Telangiectasia and Rad3-related protein; BRCA, breast cancer; CCC, clear cell carcinoma; CCNE1, cyclin E1; CHK1/2, checkpoint kinases 1/2; CR, complete response; CT, chemotherapy; DCR, disease control rate; DoR, duration of response; EZH2, enhancer of zeste homolog 2; HGSOC, high-grade serous ovarian cancer; HR, hazard ratio; HRD, homologous recombination deficiency; HRRm, Homologous Recombination Repair gene mutated; ITT, intention-to-treat; mDoR, median DoR; MMRd, mismatch repair-deficient; MMRp, mismatch repair-proficient; mPFS, median PFS; MTD, maximum tolerated dose; NE, non evaluated; ORR, objective response rate; OS, overall survival; PARPi, poly(ADP-ribose) polymerase inhibitor; PFS, progression-free survival; PR, partial response; PSOC, platinum-sensitive recurrent ovarian cancer; QD, continuous daily; R, randomization; RP2D, recommended phase II dose; w/, with; w/o, without.
PARP inhibitors
PARP is a protein involved in the repair of SSBs. When inhibited, unrepaired SSBs can be converted into DSBs. In HRD-positive cells, DSBs accumulate, leading to genomic instability and ultimately cellular toxicity and death.
Historically, PARPi have been evaluated in the recurrent, platinum-sensitive setting. The phase II Study 19 and the phase III NOVA and ARIEL3 trials demonstrated the efficacy of olaparib, niraparib and rucaparib, respectively, as maintenance therapy in relapsed ovarian cancer. In these trials, recurrence rates in the intention-to-treat (ITT) population ranged from 42% to 64%, with a marked improvement in progression-free survival (PFS) for HRD-positive patients (hazard ratio (HR) 0.32–0.38) and the most pronounced benefit in BRCA-mutated patients (HR 0.18–0.27).99–101 The SOLO2 trial, focused exclusively on BRCA-mutated patients, confirmed a 70% reduction in the risk of relapse following PARPi maintenance. 102 In the treatment setting (rather than maintenance), the ARIEL4 trial assessed rucaparib versus chemotherapy in BRCA-mutated patients and showed clinical activity, albeit with a less pronounced benefit (HR 0.64). 103 This gradient of efficacy reflects the underlying biology: BRCA1/2 mutations result in complete loss of HR, making tumour cells highly dependent on alternative repair pathways such as BER, where PARP is essential. Conversely, HRD-positive tumours without BRCA mutations often exhibit partial or transient HRR defects, commonly due to loss of heterozygosity or epigenetic alterations. Some clones may regain HRR function through mechanisms such as demethylation of BRCA1, thereby diminishing PARPi sensitivity. 104
PARPi have also been evaluated in the first-line maintenance setting. The phase III SOLO1 and PRIMA trials assessed olaparib and niraparib, respectively, in newly diagnosed ovarian cancer. SOLO1 included only BRCA-mutated patients and demonstrated an impressive median PFS of 56.0 months versus 13.8 months in the placebo arm (HR 0.30; 95% CI: 0.23–0.41), with an overall survival (OS) HR of 0.55 despite 44.3% of patients in the control arm receiving a PARPi in later lines.105,106
In contrast, the PRIMA trial reported a 34% reduction in the risk of progression in the overall population and 49% among HRD-positive patients. No OS benefit was observed, likely due to the study being powered for PFS, the higher rate/percentage of patients receiving subsequent PARPi in the placebo arm and the inclusion of a higher-risk, more heterogeneous population (residual disease post-surgery, FIGO stage IV, use of neoadjuvant chemotherapy and partial response to platinum-based chemotherapy). 107 The higher proportion of patients achieving R0 resection, could also explain the improved efficacy in the SOLO1 trial. Achieving R0 during primary debulking reduces the likelihood of clonal resistance emergence, potentially influencing long-term outcomes. Moreover, the longer treatment duration in the PRIMA study (3 years) raises concerns regarding adherence, particularly given the need for dose modifications or interruptions. Although treatment duration does not significantly impact toxicity profiles, it may influence the efficacy of subsequent therapies. Clinical benefit from chemotherapy appears diminished in patients previously treated with first-line PARPi.
Combination strategies have also been investigated. The PAOLA-1 trial evaluated olaparib plus bevacizumab versus bevacizumab alone as first-line maintenance therapy. In the ITT population, the combination failed to improve OS (56.5 vs 51.6 months; HR 0.92; 95% CI: 0.76–1.12; p0.4118). However, a significant benefit was observed in HRD-positive patients, particularly those with BRCA mutations, with recurrence risk reduced by 38% and 40%, respectively. 108 The ICON7 trial previously showed that bevacizumab conferred benefit only in a high-risk subgroup for progression (FIGO stage III-IV disease following suboptimal debulking, or stage I–II disease with grade 3 or clear cell histology; HR 0.78), with no effect in the overall population (HR 0.99). 109 High-risk features were further defined using the KELIM score, a model based on the CA125 elimination rate. In PAOLA-1, high-risk HRD-positive patients exhibited an HR of 0.46, compared to 0.26 in the low-risk group – indicating potential therapeutic benefit even among patients with poor prognostic features. 110 The ATHENA-COMBO trial assessed the addition of the immune checkpoint inhibitor nivolumab to rucaparib. BRCA-mutated tumours often express neoantigens, attracting tumour-infiltrating lymphocytes and upregulating Program-Death Ligand 1 (PD-L1), suggesting potential synergy between PARPi and immune checkpoint inhibitors. Among 863 enrolled patients (44% HRD-positive), no benefit from nivolumab addition was observed. In the HRD-positive group, median PFS was 28.9 months (rucaparib + nivolumab) versus 31.4 months (rucaparib alone). Similar results were found in the ITT population (mPFS 15.0 vs 20.2 months), questioning the added value of immune checkpoint blockade in this setting. 111
Long-term analyses confirm the role of PARPi as maintenance therapy post-chemotherapy, demonstrating prolonged PFS and an extended chemotherapy-free interval. This interval allows patients to recover from prior cytotoxic treatments and better tolerate subsequent future regimens. However, maintenance treatment with PARPi can be accompanied by the development of resistance. In the SOLO-1, PAOLA-1 and PRIMA trials, 20%–45% of patients discontinued treatment due to disease progression. Treatment failure to PARPi is frequently associated with cross-resistance to platinum-based chemotherapy, largely because both therapeutic classes exploit similar mechanisms of cytotoxicity. The predominant mechanism of resistance to PARPi involves the restoration of HRR, most commonly through secondary reversion mutations in BRCA genes. Once HRR is reactivated, tumour cells regain the ability to efficiently repair DNA DSBs induced by both PARPi and platinum agents. In addition, the selective pressure exerted by PARPi treatment eliminates sensitive clones, promoting the expansion of resistant subclones. This HRR restoration contributes to broad chemoresistance and significantly complicates the management of recurrent disease. However, concerns persist regarding long-term toxicity, particularly myelodysplastic syndrome (MDS) and acute myeloid leukaemia (AML). These adverse events were observed in heavily pre-treated patients, with incidences of 3.7% in ARIEL3 and 8% in SOLO2.112,113 In these studies involving patients with relapsed ovarian cancer, cumulative exposure to multiple lines of cytotoxic chemotherapy likely contributed to the increased risk of therapy-related MDS. In contrast, follow-up data from first-line maintenance trials PRIMA, SOLO1 and PAOLA-1 have shown low incidences of MDS/AML (1.2%, 1.5% and 1.7%, respectively) comparable to those observed in the placebo arms.
In endometrial cancer, the role of HRD as a predictive biomarker remains insufficiently explored. To date, the DUO-E and RUBY Part II clinical trials have investigated the efficacy of combining anti-PD-1/PD-L1 therapy with PARPi.114,115 The DUO-E trial is a phase III, randomised, 1:1:1 study evaluating the combination of carboplatin/paclitaxel with durvalumab and olaparib in 718 patients with advanced (FIGO 2009 stages III–IV) or recurrent endometrial cancer. Three treatment arms were compared: a control group (carboplatin/paclitaxel plus placebo followed by placebo maintenance), a durvalumab group (carboplatin/paclitaxel plus durvalumab followed by durvalumab maintenance with placebo), and the durvalumab + olaparib arms (carboplatin/paclitaxel plus durvalumab followed by durvalumab and olaparib maintenance). While the DUO-E study concluded that the combination of durvalumab and olaparib provides clinical benefit irrespective of HRR gene-mutated (HRRm) status, notable differences in the degree of benefit were observed across molecular subgroups. In the ITT population, the combination significantly improved mPFS with a HR of 0.55 (95% CI: 0.43–0.69). This effect was even more pronounced in the HRRm group (HR 0.30 (95% CI: 0.15–0.58)). These results suggest a potentially predictive role for HRRm status, despite benefits being observed beyond this subgroup. Exploratory analyses also indicated improved PFS among patients who were either PD-L1 positive or HRRm positive when durvalumab alone was added to chemotherapy, whereas the benefit appeared more limited in TP53-mutated tumours. Notably, the addition of olaparib further enhanced PFS across several subgroups, including those with TP53-mutations and serous histology, supporting the hypothesis of a synergistic interaction between PARP inhibition and immune checkpoint blockade in specific molecular contexts. However, in the mismatch repair-deficient (MMRd) subgroup (around 30% of endometrial cancers) the addition of olaparib did not improve survival. MMRd tumours are typically characterised by high tumour mutational burden and PD-L1 expression, but also substantial T-cell infiltration, making them highly responsive to immune checkpoint inhibitors alone. The lack of added benefit from PARPi in this setting highlights that this population may already achieve maximal therapeutic effect with immunotherapy alone. These findings suggest that HRD testing may be most valuable in guiding treatment decisions within the MMRp subgroup, which accounts for approximately 70% of endometrial cancers.
In contrast, the RUBY Part II trial did not assess HRD status, limiting interpretation of its results in relation to this biomarker. The study included 291 patients with advanced (FIGO stage III/IV) or recurrent endometrial cancer randomised into two arms: one receiving maintenance treatment with dostarlimab combined with niraparib for 3 years (n = 192) versus a placebo arm (n = 99). Baseline characteristics were similar between the treatment arms, with notably 74% of patients identified as MMRp. In the overall population, the authors showed a mPFS improvement in the dostarlimab-niraparib arm compared with the control arm, 14.5 versus 8.3 months (HR 0.60; 95% CI, 0.43–0.82; p = 0.0007). Among the MMRp group, the mPFS was 14.3 and 8.3 months in the dostarlimab and the placebo arms, respectively (HR 0.63; 95% CI, 0.44–0.91; p = 0.006). 114 These findings underscore the heterogeneity of treatment responses in endometrial cancer and highlight the need for future studies to incorporate HRD testing. Improved molecular stratification could enable more precise patient selection, thereby maximising the therapeutic benefit of PARP inhibitors and immune checkpoint inhibitors in this setting.
PARPis have transformed the treatment of BRCA-mutant and HRD-associated cancers by exploiting the concept of SL. However, their clinical application is frequently limited by haematological toxicities: these led to dose reductions in 71% of patients in the PRIMA and 66% in the NOVA trial, with treatment discontinuation in some cases.116,117 Most clinically approved PARPi act as pan-inhibitors targeting PARP1, PARP2, PARP3, despite their distinct biological roles. PARP1 is the primay sensor of DNA SSBs and initiates repair through PARylation. In contrast, PARP2 is activated by 5′-phosphorylated DNA nicks (such as Okazaki fragments) and plays a structural role in facilitating DNA ligation by ligases 1 and 3. Catalytically inactive PARP2 compromises this ligation process, causing replication fork collapse, particularly detrimental to rapidly proliferating cells such as erythroblasts. This mechanism explains the haematological side effects observed with pan-PARPi. 118 To address this limitation, next-generation PARPi with enhanced selectivity for PARP1 have been developed. Among them, saruparib demonstrates over 500-fold greater selectivity for PARP1 compared to PARP2, with the goal of maintaining anti-tumour efficacy while reducing haematopoietic toxicity. 119 Preclinical studies have shown that saruparib has potent and durable anti-tumour activity in patient-derived BRCA1/2-mutant models of breast, ovarian and pancreatic cancers, alongside a favourable safety profile. Saruparib is currently under investigation in multiple phase I/II and III clinical trials, including PETRA and EvoPAR-Ovarian01 in ovarian cancer.In Part A of the phase I/II PETRA study (NCT04644068), saruparib was administered as monotherapy at doses ranging from 10 to 140 mg daily to 61 patients with advanced solid tumours (19.3% of ovarian cancers) harbouring BRCA1/2, PALB2 or RAD51C/D mutations. The treatment demonstrated a favorable safety profile with a low incidence of haematological and gastrointestinal adverse events, even among heavily pretreated patients (median of 3 prior lines of therapy, 45% with previous exposure to PARPi and/or platinum-based chemotherapy). The recommended dose was established at 60 mg daily, with a maximum tolerated dose of 90 mg. Pharmacodynamically, saruparib demonstrated a fold coverage (ratio of plasma concentration to effective concentration) of 31.7, markedly higher than other PARPi (niraparib 0.36, talazoparib 0.5, rucaparib 2.44, olaparib 2.44), indicating prolonged and selective PARP1 inhibition, which may underpin its improved efficacy and tolerability profile. The study is ongoing through multiple stages, investigating dose escalation of saruparib in combination with other agents (paclitaxel, carboplatin-paclitaxel, trastuzumab deruxtecan and datopotamab deruxtecan) in a total of 306 patients with breast, ovarian or prostate cancers. 120
The phase III, randomised, double-blind, placebo-controlled EvoPAR-Ovarian01 study, currently in preparation, will evaluate saruparib as second-line maintenance therapy in patients with platinum-sensitive relapsed ovarian cancer who progressed following PARPi maintenance after first-line carboplatin-paclitaxel chemotherapy combined with bevacizumab. This study plans to enrol 570 patients stratified into 3 cohorts based on BRCA and HRD status: BRCA-mutated, HRD-positive without BRCA mutation, and an exploratory/descriptive HRD-negative cohort.
ATR inhibitors
ATR inhibition, a key mechanism involved in the detection and repair of DNA DSBs, represents a promising therapeutic approach for targeting tumours with specific molecular vulnerabilities. Preclinical data have demonstrated increased ATR dependency in tumours with ARID1A loss-of-function mutations due to inherent defects in genomic stability. This creates a context of SL that can be therapeutically exploited. This dependency is particularly relevant in gynaecological malignancies, where ARID1A mutations are commonly observed, notably in clear cell ovarian and endometrial carcinomas.
In the NCI-9944 trial, the addition of berzosertib to gemcitabine in patients with platinum-resistant ovarian cancer did not significantly improve OS (HR 0.79 (90% CI: 0.52–1.2)). The absence of clinical benefit may be attributed to the absence of patient selection based on ARID1A mutational status. However, enhanced efficacy observed in subgroups with a platinum-free interval of less than 3 months, or with low replicative stress, suggests that clinical and biological stratification could improve treatment outcomes.121,122
As monotherapy, ATR inhibitors have shown limited efficacy in unselected populations. Conversely, their combination with PARPi, which target complementary DNA repair pathways, may enhance anti tumour activity. The phase II CAPRI study evaluated the combination of olaparib and ceralasertib in patients with platinum-sensitive relapse, reporting an objective response rate (ORR) of 48.5% and encouraging PFS, irrespective of genomic instability status. However, the absence of a control arm and a lack of molecular stratification by BRCA, HRD or ARID1A status limit the interpretation of these results and may conceal differential responses.123,124 In contrast, the ATARI study incorporated rigorous molecular and histological stratification, enabling a more refined assessment of ceralasertib efficacy, both as monotherapy and in combination. In cohort 1A, patients with clear cell ovarian or endometrial carcinoma with confirmed ARID1A loss received ceralasertib monotherapy, achieving an ORR of 14%. In cohort 2, patients with the same histology but without ARID1A loss were treated with the combination of ceralasertib and olaparib, also resulting in an ORR of 14%. In comparison, cohort 3, which included patients with other histological subtypes (endometrioid, carcinosarcoma, cervical) treated with the same combination therapy, demonstrated a higher ORR of 24%.125,126
EZH2 inhibitors
Tazemetostat, a selective EZH2 inhibitor, is currently under investigation in the first phase II clinical trial specifically targeting ARID1A-mutated solid tumours. In this single-arm study (NCT05023655) is evaluating tazemetostat as monotherapy, with ORR as the primary endpoint according to RECIST 1.1 criteria. 127
WEE1 kinase inhibitors
WEE1 kinase functions as a key regulator of the cell cycle, acting as a critical checkpoint that prevents premature mitotic entry in response to DNA damage or replicative stress. Tumours harbouring TP53 mutations and/or CCNE1 amplification, both major regulators of the cell cycle, exhibit increased dependency on WEE1 activity to avoid mitotic catastrophe. Inhibiting WEE1 with agents such as adavosertib or ZN-c3 forces damaged cells into unscheduled mitosis, leading to genomic instability and cell death. This SL approach is particularly promising in cancers characterised by high levels of replicative stress, such as those with TP53 mutations or CCNE1 amplification.
Several clinical trials have investigated the potential of WEE1 inhibitors in ovarian and endometrial cancers. In platinum-resistant TP53-mutated ovarian cancer, three studies have assessed adavosertib in combination with chemotherapy (carboplatin or gemcitabine), reporting encouraging ORRs ranging from 41% to 67%.128–130 Within the CCNE1-amplified subgroup, response rates were numerically higher, although statistical significance was not reached. 128
The multicentric phase II IGNITE study assessed the efficacy of adavosertib in women with recurrent platinum-resistant HGSOC based on CCNE1 gene amplification level (< or >8 copies). CCNE1 protein expression was evaluated by immunohistochemistry, and CCNE1 gene copy number was determined using fluorescence in situ hybridisation. Patients were stratified into two cohorts: cohort 1, characterised by CCNEE1 protein overexpression (H-score >50) with CCNE1 gene amplification (copy number >8; n = 21); and cohort 2, with CCNE1 protein overexpression but without gene amplification (n = 59). Among the enrolled patients, 83% had received two or more prior lines of chemotherapy. The ORR was 38% in the CCNE1-amplified cohort and 45% in the overexpressed but non-amplified cohort. Treatment-related adverse events occurred in 97% of patients (n = 78), with dose reductions required in 45% (n = 36), primarily due to neutropenia or diarrhoea. CCNE1 overexpression appears to be a reliable biomarker of response to adavosertib, independent of CCNE1 gene amplification. 131
In platinum-sensitive ovarian cancer, adavosertib also demonstrated clinical benefit, achieving an ORR of 74.6% compared with 69.4% in the placebo arm. Moreover, the study identified variable sensitivity to adavosertib according to TP53 mutation subtype, with hotspot and missense mutations conferring greater benefit than truncating variants. 132
In the population of patients with ovarian cancer who relapse following treatment with PARPi, therapeutic options become markedly limited. Several studies are currently investigating strategies to overcome PARPi resistance or to resensitise tumours to targeted therapies. Among these approaches, the EFFORT trial, a non-comparative phase II study, evaluated the efficacy of adavosertib alone (n = 39) or in combination with olaparib (n = 41) in 80 patients who relapsed after PARPi therapy. The majority of patients were platinum-resistant (64%) and heavily pretreated, with a median of four prior lines of therapy (range 1–11). The results demonstrated an ORR of 23% with adavosertib monotherapy and 29% with the combination, and mPFS of 5.5 and 6.8 months, respectively. Although a high incidence of grade 3/4 adverse events was observed (83%), most toxicities were manageable with treatment interruptions (88%) and/or dose reductions (71%).133,134
In endometrial cancer, particularly uterine serous carcinomas where TP53 mutations occur in nearly 90% of cases, 1 two phase II studies have shown antitumour activity with adavosertib monotherapy, with ORRs between 26% and 30%, and a 6-mPFS rate of 47.1%. 135 The ADAGIO study confirmed these findings, although toxicity was notable (grade ⩾3 adverse events reported in 68.8% of patients).136,137
Finally, ZN-c3, a next-generation selective WEE1 inhibitor, has shown promising preliminary efficacy and tolerability in phase I studies involving patients with solid tumours. 138 Multiple ongoing clinical trials are currently evaluating ZN-c3 in ovarian and endometrial cancers (NCT04158336, NCT05431582, NCT04516447).
Despite strong preclinical rationale supporting the inhibition of ATR, WEE1, and other key regulators of the DNA damage response (DDR) and cell cycle checkpoints, clinical trials to date have yielded only modest results. Tumour cells frequently activate compensatory signalling pathways to overcome the inhibition of a single CHK. For instance, ATR or WEE1 inhibition may lead to the upregulation of CHK1 or mTOR signalling, sustaining cell cycle progression and DNA repair despite therapeutic pressure. This functional redundancy diminishes the efficacy of monotherapy and suggests that combination strategies targeting multiple partners within the DDR network may be required. Furthermore, ATR, WEE1 and other inhibitors have been associated with considerable toxicities, particularly haematological (such as neutropenia and thrombocytopenia) and gastrointestinal side effects. These toxicities often necessitate dose reductions, treatment interruptions or discontinuations, limiting the delivery of optimal therapeutic intensity and negatively impacting clinical efficacy. To maximise clinical benefit, the identification and validation of robust predictive biomarkers that reflect tumour dependence on specific DDR pathways (like CCNE1 e.g.) is imperative.
CHK1/2 inhibitors
Checkpoint kinases play a pivotal role in cell cycle regulation by activating the G2/M and S-phase checkpoints. Upon DNA damage, CHK1/2 delay cell cycle progression, allowing time for DNA repair. Similar to WEE1 inhibitors, CHK1/2 inhibitors bypass this protective mechanism, forcing damaged cells to enter mitosis prematurely, ultimately leading to mitotic catastrophe and subsequent cell death.
CHK1/2 inhibitors are of particular interest when used in combination with DNA-damaging therapies such as chemotherapy or radiotherapy. By impairing the cell’s ability to repair DNA, these inhibitors potentiate the cytotoxic effects of such treatments. Additionally, SL can be achieved in tumours with deficiencies in ATM, ATR or p53 pathways.
Several CHK1 inhibitors have been evaluated in clinical trials, yielding variable results. AZD7762 was, initially tested as monotherapy and later in combination with gemcitabine, but its development was discontinued due to cardiotoxicity. 139 Prexasertib, another CHK1 inhibitor, was studied in patients with platinum-resistant or-refractory ovarian cancer, showing modest ORRs (6.1%–12.1%) across different cohorts. 140 A monocentric proof-of-concept study in patients with BRCA wild-type HGSOC mutations reported a partial response rate of 33%. 141 SRA737, another CHK1 inhibitor, was evaluated in a phase I/II trial, establishing a maximum tolerated dose of 1000 mg and a recommended dose of 800 mg. 142 The oral CHK1 inhibitor GDC-0575 failed to demonstrate clinical activity when administered as monotherapy. 143
While CHK1 inhibitors show therapeutic promise, their clinical development has been hampered by toxicity concerns and limited efficacy as single agents.
Discussion
SL offers a significant therapeutic advantage by enabling targeted treatment, particularly in cases of somatic mutations acquired within the tumour, without affecting healthy cells. Moreover, this approach allows targeting of loss-of-function mutations, particularly in tumour suppressor genes. By coupling mutation in these genes with the inhibition of a complementary gene, the resulting accumulation of mutations induces tumour cell apoptosis. This strategy neutralizes the advantage conferred by the loss-of-function mutation, turning the tumour cell’s weakness into a therapeutic opportunity.
Tumour sequencing is necessary to identify potential targets for SL. For example, mutations in BER genes, less studied in gynaecological tumours, could also be exploited for SL. The TCGA data analysis of breast, colon and uterine cancers revealed that PD-L1 expression is negatively correlated with the expression of BER genes (such as NTH1, XRCC1, POLB, LIG3). Alterations in this repair pathway may predict the effectiveness of anti-PD-1/PD-L1 immunotherapy. 144
The use of PARPis in HRD-positive cancers represents one of the most significant success stories in the field of SL. However, SL has shown more limited anti-tumour effects for other types of mutations. Emerging techniques evaluating genetic perturbation, such as CRISPR-Cas9 and RNA interference, along with high-throughput screening, have identified numerous synthetic lethal effects driven by genetic interactions, generally between a tumour driver gene and a target gene.10,145,146 This approach remains reductionist, focusing on binary interactions, whereas genetic interactions in cancer are often polygenic and involve multiple genes.
SL can also exhibit incomplete penetrance within tumours, partly due to intratumoural clonality. In many cases, certain tumour clones harbor the mutation that renders them vulnerable to SL strategies, while other do not, thereby limiting overall treatment efficacy. Furthermore, the influence of the tumour microenvironment, such as the presence of oncogenic viruses (e.g. HPV) or the local microbiome, can affect cellular responses to inhibitors targeting survival pathways, adding further heterogeneity. Indeed, although lethal effects have been identified, validation in different preclinical models is not always reproducible, suggesting that the presence of a mutation alone may not be sufficient to induce lethality. Tumour cells carrying similar mutations may respond differently depending on the tumour model and microenvironment. Additionally, the specific type of mutation also influences treatment response: for example, missense mutations, which result in a single amino acid substitution in the protein, can have different biological effects from truncating mutations that prematurely halt protein production. This diversity in mutations types (including point mutations, nonsense mutations and frameshifts) can influence the efficacy of SL-based treatments. 132 SL is based on the principle of pharmacologically inhibiting a partner gene of a loss-of-function mutation, thereby conferring a high degree of specificity in killing mutant while sparing normal cells. However, significant toxicities are frequently observed in clinical practice. These adverse effects predominantly impact rapidly proliferating tissues, such as the bone marrow and gastrointestinal mucosa, whose cells require efficient DNA repair mechanisms to resolve the physiological stress occurring during DNA replication. Although these normal cells do not carry the targeted mutations, sustained inhibition of key DDR effectors such as PARP, WEE1 or ATR impairs their repair capacity, leading to cytotoxicity. SL should not be considered a strictly binary phenomenon but rather a spectrum of cellular sensitivity, where non-mutated cells may still partially depend on the inhibited pathways. Furthermore, the inherent plasticity of cellular signalling networks and the activation of compensatory pathways can contribute both to therapeutic resistance in mutant tumour cells and to sensitivity in normal cells.
In conclusion, while the concept of SL remains a convincing and theoretically powerful approach in oncology, its clinical application requires in-depth research incorporating the tumour’s polygenic profile, the tumor microenvironment and the type of mutation.
Conclusion
In this review article, we have provided an overview of the most frequently observed loss-of-function mutations (HRD, TP53, ARID1A and PTEN) and gene amplifications (CCNE1) in ovarian and endometrial cancers, with particular emphasis on their biological roles in normal cells. These genetic alterations play a central role in tumour progression and represent key targets for SL-based strategies. Tumour cells acquire such mutations and develop diverse escape mechanisms, exploiting cellular processes to support their survival. For example, defects in DNA repair pathways enable tumour cells to sustain rapid proliferation and increase resistance to conventional treatments. Similarly, mutations in cell cycle proteins such as p53 enable the cell to bypass checkpoints and thereby ensure its survival. These mutations provide a selective advantage to tumour cells by activating compensatory pathways that support tumour viability.
Footnotes
Acknowledgements
The authors thank the teams from the Departments of Gynaecology, Oncology and Human Genetics at CHU de Liège for their valuable discussions and expertise, which sparked our interest in synthetic lethality and deepened our understanding of the topic. Their constructive feedback greatly contributed to the development of this manuscript.