
Abstract
The GM2 gangliosidoses are a group of lysosomal storage diseases caused by defects in the genes coding for the enzyme hexosaminidase or the GM2 activator protein. Four Jacob sheep from the same farm were examined over a 3-year period for a progressive neurologic disease. Two lambs were 6-month-old intact males and 2 were 8-month-old females. Clinical findings included ataxia in all 4 limbs, proprioceptive deficits, and cortical blindness. At necropsy, the nervous system appeared grossly normal. Histologically, most neurons within the brain, spinal cord, and peripheral ganglia were enlarged, and the cytoplasm was distended by foamy to granular material that stained positively with Luxol fast blue and Sudan black B stains. Other neuropathologic findings included widespread astrocytosis, microgliosis, and scattered spheroids. Electron microscopy revealed membranous cytoplasmic bodies within the cytoplasm of neurons. Biochemical and molecular genetic studies confirmed the diagnosis of GM2 gangliosidosis. This form of GM2 gangliosidosis in Jacob sheep is very similar to human Tay-Sachs disease and is potentially a useful animal model.
The GM2 gangliosidoses are a group of autosomal recessive lysosomal storage diseases caused by defects in the genes coding for the enzyme hexosaminidase (Hex), resulting in accumulation of GM2 ganglioside within lysosomes and eventual cellular dysfunction. 3,16,20,38 Hex is composed of 2 subunits, α and β, which combine to form 2 major isoenzymes, Hex A (αβ) and Hex B (ββ). Tay-Sachs disease (TSD) is caused by mutations in the gene coding for the α subunit resulting in Hex A deficiency. Mutations in the β subunit gene result in Sandhoff’s disease (SD), in which both Hex A and Hex B are deficient. A lack of Hex A results in the B variant of GM2 gangliosidosis, whereas the production of an inactive form of Hex A results in the B1 variant of the disease. Hex function is also reliant on the GM2 activator protein, and lack of this protein results in the AB variant form of gangliosidosis. GM2 gangliosides are most abundant in the nervous system, and the clinical picture of patients with the GM2 gangliosidoses is dominated by a progressive loss of neurologic function. Most patients are affected in infancy (infantile form), but disease can develop later (juvenile and adult-onset forms). Heterogeneity of the disease in regard to severity of clinical signs and age of onset can be attributed to a variety of mutations as well as poorly defined modifying genes and epigenetic factors. 13
Effective therapy for TSD and other GM2 gangliosidoses has remained elusive, and the development of reliable animal models will likely hasten the development of more promising treatment modalities. 27 Transgenic mice with targeted disruption of the HEXA gene have been developed to serve as a model of TSD. 6,24,31,34,46 Reports of spontaneous GM2 gangliosidosis in animals have included the cat, 4,7,8,21,22,48 dog, 2,9,18,32,47 Yorkshire pig, 25,28 Muntjak deer, 12 and American flamingo. 49 Here we detail the pathology of GM2 gangliosidosis in Jacob sheep, which is potentially a new model of TSD.
Materials and Methods
Four lambs from the same farm were presented to the Texas A&M University Veterinary Teaching Hospital (VTH) over a 3-year period. Lamb Nos. 1 and 2 were both 6-month-old intact males and were presented simultaneously. Lamb No. 3 was an 8-month-old female presented 2 years after lamb Nos. 1 and 2. Lamb No. 4 was another 8-month-old female presented 1 year after lamb No. 3. All 4 animals received clinical evaluations and 3 lambs were euthanatized given their poor prognosis. Lamb No. 2 died under anesthesia during a myelogram procedure. Serum biochemical analysis and complete blood counts were not performed on any of the lambs. To compare the degree of myelination and gliosis with a normal control, cerebral tissue from an 8-month-old, castrated male, mixed-breed lamb was used.
The animals were necropsied, and tissue samples were fixed in 10% neutral buffered formalin and processed routinely for histopathologic examination. Tissues examined included brain (cerebrum, hippocampus, thalamus, cerebellum, midbrain, pons, medulla oblongata), spinal cord (cervical, thoracic, lumbosacral), trigeminal ganglia, sciatic nerve, spinal nerves, dorsal root ganglia, lung, heart, liver, kidney, spleen, gastrointestinal tract (esophagus, abomasum, rumen, reticulum, omasum, duodenum, jejunum, ileum, colon), urinary bladder, trachea, bone, bone marrow, skeletal muscle, lymph node, thymus, adrenal gland, thyroid gland, gonads, and uterus. Sections were stained with hematoxylin and eosin (HE), and samples of brain and spinal cord were also stained with Luxol fast blue, Sudan black B, Alcian blue, toluidine blue, Weil’s, and periodic acid–Schiff (PAS) stains. Formalin fixed liver from 1 case was cryostat sectioned and stained with PAS. Transmission electron microscopy was performed on formalin-fixed brain and liver tissue according to standard protocols. Ultrathin sections were cut, poststained with uranyl acetate and lead citrate, and examined with an electron microscope. Attempts were made to examine formalin-fixed cerebrum using the Golgi method. Immunohistochemistry was performed using antibodies against glial fibrillary acidic protein (GFAP) (DakoCytomation, Carpenteria, CA) and microglia (Iba1) (Biocare Medical, Concord, CA) with Gill’s hematoxylin counterstain. Both antibodies are rabbit polyclonals and were used at dilutions of 1:1000 and 1:100, respectively.
The results of biochemical and molecular genetic analyses to confirm and characterize this disease have been published. 36 A brief summary of the results is included in the following section.
Results
Lamb Nos. 1 and 2 were presented to the VTH with suspected occipital condylar dysplasia, a disorder that has been described in the Jacob sheep breed. Lamb No. 1 had a 2-month history of ataxia in all 4 limbs, lack of weight gain, and decreased frequency of ambulation. On examination, this lamb had an upright posture and stood on its toes with the rear feet turned out. The lamb’s gait was very stiff, and the flexor tendons in the front limbs appeared contracted. The lamb resisted manipulation of the neck, had a slight intermittent head tilt to the right, and had proprioceptive deficits in all 4 limbs with the rear limbs more severely affected than the front limbs. Cerebrospinal fluid analysis was unremarkable. A grade III/VI systolic murmur was auscultated on the left side. Lamb No. 2 had a history of ataxia in the rear limbs of unreported duration that was considered less severe than that exhibited by lamb No. 1. Neurologic examination revealed ataxia and proprioceptive deficits in all 4 limbs with the rear limbs most severely affected. The lamb also had valgus tarsal and carpal deformities, most severe on the left side. A grade IV/VI left-sided systolic murmur was auscultated. Spinal radiographs and myelograms of lamb Nos. 1 and 2 were within normal limits. The first clinical sign exhibited by lamb No. 3 was weakness, which was followed by ataxia. The age of onset of the clinical signs was not reported. On examination, the lamb walked hesitatingly and exhibited sinking of the pasterns on tiring. The lamb exhibited posterior paresis and pollakiuria with urine dribbling. Lamb No. 4 developed laxity of the front fetlocks with sinking of the pasterns at 1 month of age, followed by weakness and ataxia in the rear limbs at 5 months. By 8 months, the lamb was frequently recumbent and showed evidence of blindness. At presentation, the lamb stood with splayed front limbs and had proprioceptive deficits, including knuckling of the fetlocks. Evaluation of the cranial nerves revealed an intact pupillary light reflex and an absent menace reflex.
At necropsy, the nervous system tissues of all 4 animals were grossly normal. The brains of lamb No. 3 and lamb No. 4 weighed 78.5 g and 88 g, respectively; the brain weights of lamb Nos. 1 and 2 were not recorded. Numerous 0.5-mm uroliths were in the urinary bladder of lamb No. 1, and similar 3- to 5-mm uroliths were in the urinary bladder of lamb No. 2. Whereas lamb Nos. 1 and 2 appeared to be in good body condition, lamb No. 3 appeared small for its age with reduced body fat stores and serous atrophy of fat. Lamb No. 4 was in fair body condition with reduced body fat stores and numerous nematodes, consistent with Haemonchus contortus, in the abomasum. The hearts of lamb Nos. 1 and 2 were grossly normal, and a cause for the murmurs could not be determined.
Histologic changes in the nervous system were severe in all cases and most severe in lamb No. 4. The majority of neurons in the brain and spinal cord were enlarged with a round to ovoid shape and eccentric nuclei. The cytoplasm of these neurons was distended by foamy to granular material (Fig. 1). In occasional neurons, the cytoplasmic material appeared as multiple distinct, round, 6- to 12-μm, clear vacuoles with a central, round, 3- to 5-μm eosinophilic center (Fig. 2). The cytoplasm was often diffusely affected, but in some cells the cytoplasmic material was only in the periphery of the cell or distending only 1 side of the neuron. The intraneuronal material stained positively with Luxol fast blue stain (Fig. 3), was weakly positive with Sudan black B stain (Fig. 4), and was negative with PAS, Alcian blue, and toluidine blue stains. Myelin staining with Luxol fast blue and Weil’s stains was decreased in the cerebral white matter compared with control tissue (Figs. 5 and 6). In the cerebrum, reactive astrocytosis was evident in both gray and white matter and was most conspicuous on HE staining within the superficial subpial areas and along the gray–white matter junction. The astrocytosis was highlighted with GFAP immunostaining (Figs. 7 and 8), and immunohistochemical staining for microglia demonstrated moderate microgliosis in both gray and white matter (Figs. 9 and 10). Swollen axonal segments (spheroids), ranging in diameter from 10 to 30 μm, were common in the white matter and the deeper lamina of the gray matter. Electron microscopy revealed membranous cytoplasmic bodies (MCBs) within neurons (Fig. 11). Staining of a frozen liver section with PAS revealed a small amount of PAS-positive material within the cytoplasm of hepatocytes. Storage material was not evident in the liver with electron microscopy. Golgi staining to demonstrate meganeurites was unsuccessful.
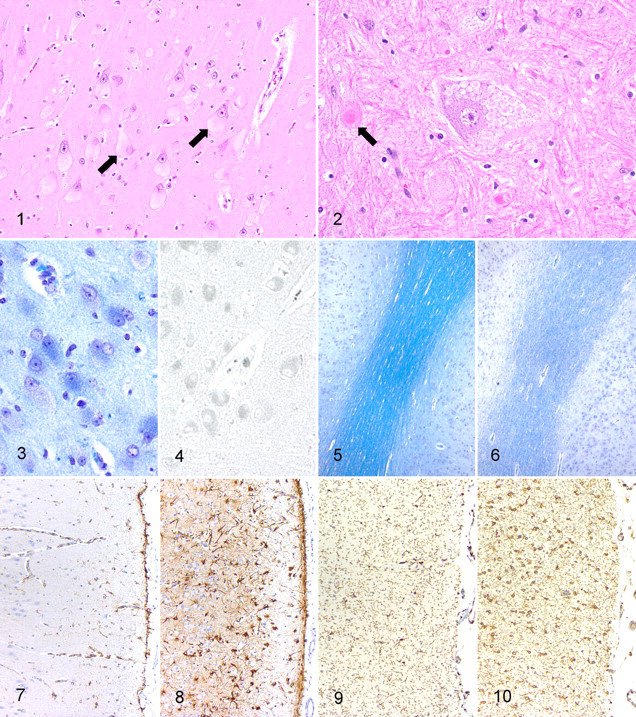
Cerebrum; lamb No. 1. Neurons throughout the cerebral cortex are distended by intracytoplasmic storage material (arrows). HE.
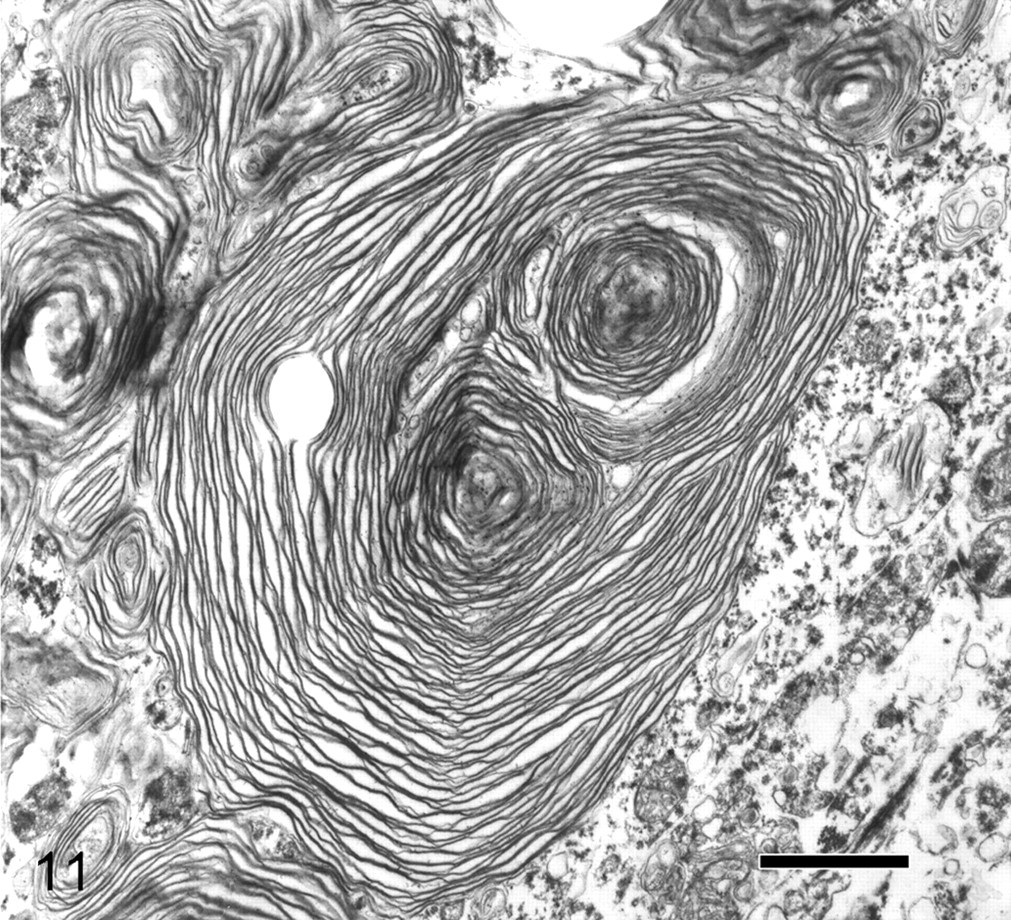
Brain; lamb. The cytoplasm of a neuron contains a membranous cytoplasmic body. Bar = 1 μm. Uranyl acetate and lead citrate.
In the cerebellum, Purkinje cells were markedly distended with cytoplasmic material, whereas neurons in the molecular and granular cell layers were less consistently affected (Fig. 12). Purkinje cell dendrites were often thickened. Spheroids were common in the white matter and were prominent in both the white matter and the granular layer in lamb No. 4. Swollen neurons, astrocytosis, microgliosis, and scattered spheroids were evident in the hippocampus, thalamus, midbrain, pons, medulla oblongata, and cervical, thoracic, and lumbosacral spinal cord. Minimal multifocal axonal degeneration, characterized by swollen myelin sheaths and occasional intramyelinic macrophages, was seen in the spinal white matter of lamb No. 4.
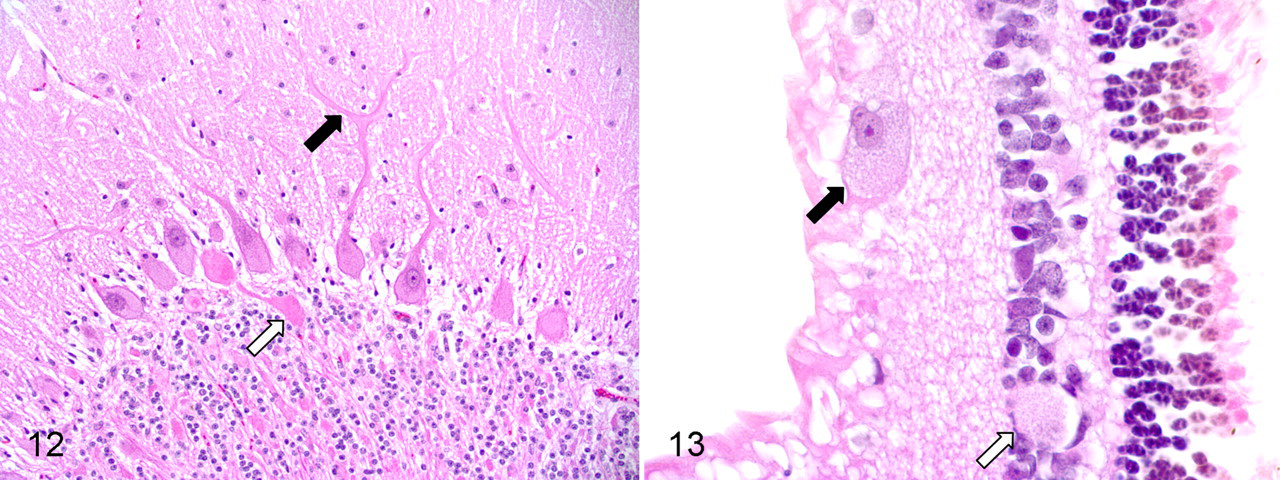
Cerebellum; lamb No. 4. Purkinje cell dendrites are thickened (black arrow). A spheroid is evident in the granular cell layer (white arrow). HE.
Neurons in the peripheral nervous system were also widely affected. Affected neurons were evident in the trigeminal ganglia, spinal dorsal root ganglia, and autonomic ganglia in the adrenal gland, epicardium, urinary bladder, and every part of the gastrointestinal tract examined. In the retina, ganglion cells were consistently and severely affected, and scattered inner nuclear cell layer neurons were also involved (Fig. 13). Peripheral nerves were normal, including the sciatic nerve, spinal nerves, and autonomic nerves within viscera. Significant findings were not evident in non–nervous system tissues.
A complete description of the biochemistry and molecular genetics of the disease is provided by Torres et al. 36 Briefly, β-hexosaminidase activity was reduced in the affected sheep to 29% of that in the control brain and 6% of that in the control liver. Determination of Hex A activity in the liver of an affected animal disclosed a marked deficiency, and this was confirmed by cellulose acetate electrophoresis of hexosaminidases A and B. Brain lipid analysis disclosed a 3-fold elevation in lipid-bound sialic acid, which by thin layer chromatography consisted almost entirely of GM2 ganglioside. The cDNA of Hex A was cloned and sequenced, revealing a single base change in a conserved region within the cDNA of the affected sheep that was not present in the cDNA of normal sheep. Specifically, a homozygous recessive missense (G to C transition) mutation was demonstrated at nucleotide position 1330 (C1330 → C), which resulted in an amino acid change from Gly444 to Arg. Transfection of the mutated sheep Hex A cDNA into COS cells and into TSD human skin fibroblasts confirmed the deficiency of Hex A activity. Analysis of blood spots on filter paper by restriction enzyme analysis then permitted genotyping for the identification of carriers of the mutation in more than 100 Jacob sheep from 3 different flocks. An autosomal recessive pattern of inheritance was established.
Discussion
Gangliosides play a number of important roles in brain development and function, including receptor-mediated cell signaling, ion channel modulation, and dendritogenesis. 30,43 They are found in the neuronal plasmalemma, where they colocalize with other glycosphingolipids and cholesterol in specialized areas of the membrane known as lipid domains or rafts. 42 The degradation of gangliosides occurs via the endosomal–lysosomal system, and deficiencies in hydrolases such as hexosaminidase result in intracellular ganglioside accumulation in the form of MCBs. 42 Gangliosides also accumulate within the axon hillock to produce noticeable enlargements called “meganeurites,” and new ectopic dendrites develop on the surface of meganeurites and the adjacent neuronal cell membrane. 26,43 Although MCBs cause the enlargement of neuronal cell bodies and the formation of meganeurites, the spheroids that develop in TSD are caused by the accumulation of organelles (mitochondria, membrane-bound vesicles, dense bodies), similar to those observed in primary neuroaxonal dystrophies. 41 The cause of the decreased myelin observed with the gangliosidoses and other lysosomal storage diseases is uncertain; it likely has multiple causes. Evidence points to effects on oligodendrocyte growth and function as well as the secondary effects of altered neuroaxonal development. 10,11,37
Neuronal death in TSD and SD, both in human patients and in their corresponding mouse models, appears to be the result of apoptosis. 15,17 In murine models, studies suggest that apoptosis is triggered by an alteration in endoplasmic reticulum calcium levels resulting in an unfolded protein response. 23,36 Inflammation clearly occurs during disease development, although the role this inflammation plays in the pathogenesis is still unclear. In SD mice, it has been shown that activated microglia are present prior to the onset of neurodegeneration. 40 Microglia in TSD and SD mice show enhanced MHC class II expression, evidence of nitric oxide formation, and elevations in inflammatory cytokines such as tumor necrosis factor, interleukin-1β, transforming growth factor-β1, and macrophage inflammatory protein-1α. 17,45 The astrocytosis that develops in the gangliosidoses appears to involve the lipid-signaling molecule sphingosine-1-phosphate (SIP), as SD mice with targeted deletion of the SIP gene develop milder disease than mice with the functional gene. 44 The results of a more recent study suggest that the combined downregulation of phosphorylated Akt and upregulation of phosphorylated extracellular, signal-regulated kinase (ERK) may be responsible for the proliferation of astrocytes in SD mice. 19
The clinical and pathologic findings of GM2 gangliosidosis in Jacob sheep are very similar but not identical to those described in human TSD. These sheep were euthanatized or died fairly early in the course of their disease, so some of the differences could be the result of the relatively limited duration of the ovine disease. Infantile, late infantile, juvenile, and adult-onset forms of TSD have been recognized. In the infantile form, affected infants are normal at birth and begin developing progressive psychomotor dysfunction at around 4 to 6 months of age. 29,33,39 Clinical signs include motor incoordination leading to decreased motor activity, blindness, an exaggerated extensor response to sound, and eventual seizures and obtundation. The sheep reported here exhibited progressive clinical signs that were similar to but milder than those described in humans. The most consistent findings were ataxia, proprioceptive deficits, and a decreased level of voluntary motor activity. Blindness was evident in 1 animal.
In the first 12 to 14 months of the human disease, gross pathologic findings include brain atrophy, widening of the sulci, and mildly dilated ventricles. 33,38,39 Between 15 and 24 months, the brain often increases in weight with enlargement of the cerebrum and atrophy of the cerebellum and brain stem. Brains are firm in consistency, are rubbery, and lack a clear distinction between cerebral gray and white matter. The brains of the sheep were considered grossly normal; however, the histologic findings were very similar to those described in human TSD patients. As in descriptions of the human disease, the majority of neurons throughout the central and peripheral nervous systems were affected, and the histologic, histochemical, and ultrastructural appearance of the storage material resembled that described in the human disease. 1,14,29,33,39 Decreased myelin staining is well recognized in human patients with infantile forms of the disease and was demonstrated in the sheep brains with Luxol fast blue and Weil’s myelin stains. Other findings common to both human and ovine cases include astrocytosis, microgliosis, and the presence of spheroids. Meganeurites could not be demonstrated in ovine cases with Golgi staining, and this was attributed to prolonged immersion of the tissue in formalin (7 years) prior to attempting the procedure. Changes that are included in descriptions of the human disease that were not observed in the sheep include the presence of fat-laden macrophages surrounding blood vessels, cystic degeneration of white matter, neuronophagia, and substantial neuronal loss in the cerebral cortex and cerebellum. Storage material can be demonstrated ultrastructurally in the liver of human TSD patients. 14,39 A small amount of PAS-positive material that could represent storage material was seen on a frozen liver section, but storage material could not be unequivocally demonstrated in the livers of the sheep. As with the Golgi staining, the lack of ultrastructural evidence of storage material in the liver might be explained by prolonged formalin fixation of the tissue prior to attempting the procedure.
An accurate animal model of GM2 gangliosidosis would be useful for gaining a better understanding of the pathogenesis and for the development of effective therapies. Although Hex B–deficient mice develop clinical signs that mimic SD at an early age, mice lacking Hex A do not develop clinical disease that closely resembles TSD. 6,24,31,34,46 Hex A–deficient mice exhibit the biochemical features of the disease at a young age but fail to develop clinical signs until at least 11 to 12 months of age, suggesting that murine ganglioside metabolism differs from that of humans. Hex B–deficient mice exhibit widespread neuronal ganglioside storage, but Hex A–deficient mice have storage material only in limited neuronal populations. Some of the key pathologic features of TSD and SD, including meganeurites, ectopic dendritogenesis, decreased myelin staining, astrocytosis, and microgliosis, have not been described in these mouse models. Gene therapy studies using Hex A deficient mice appear to show promise, 5 and therapeutic research using a species with spontaneous disease that more closely resembles the human disease would be very useful. Study of this model might also shed light on the pathogenesis of GM2 gangliosidosis.
Footnotes
Acknowledgements
We thank Fred and Joan Horak of St. Jude’s Farm, Lucas, Texas, for their excellent cooperation and assistance. We also thank Sarah Jones, Dr. Andy Ambrus, and Dr. Ross Payne for assistance with histochemical stains, immunhistochemistry, and electron microscopy, respectively, and acknowledge Dr. Steven Walkley for consultations on Golgi staining.
The authors declared that they had no conflicts of interest with respect to their authorship or the publication of this article.
The authors declared that they received no financial support for their research and/or authorship of this article.