
Abstract
Building, curating, and maintaining a compound collection is an expensive operation, beyond the scope of most academic organizations. Here we describe the selection criteria used to compile the LifeArc diversity set from commercial suppliers and the process we undertook to generate our representative LifeArc index set. The aim was to avoid a “junk in, junk out” screen collection to increase chemical tractability going forward, while maximizing diversity. Using historical LifeArc screening data, we demonstrate that the index set was predictive of ligandability and that progressable hits could be identified by mining associated clusters within our larger diversity set. Indeed, a higher percentage of index-derived hit clusters were found to have been progressed into hit-to-lead programs, reflecting better drug-likeness. In practice, the library has been shared widely with academic groups and used routinely within LifeArc to assess the ligandability of novel targets. Its small size is well suited to meet the needs of medium-throughput screening in labs with either limited automation, limited precious or expensive reagents, or complex cellular assays. The strategy of screening a small set in combination with rapid hit analog follow-up has demonstrated the utility of finding active clusters for potential development against challenging targets.
Introduction
The early drug discovery space was once the domain of large pharmaceutical companies. Over recent years, there has been an increase in participation from academic and not-for-profit drug discovery centers or translation centers. This has been driven by a number of factors, which have been discussed in depth elsewhere.1,2 However, what has become clear is that initiatives fostering collaboration between academic disease focused groups, translation centers, and pharma companies are needed to generate tools and platforms to probe disease biology and uncover novel therapeutic interventions. 3 Larger translational centers, such as ourselves, LifeArc (formerly MRC Technology), have evolved to catalyze and support academic translational research from target validation through to lead optimization, offering scientific expertise and capability alongside active project management. An area where we have invested expertise and resources is compound library design and collation, initially to support our own high-throughput screening (HTS) efforts, but increasingly to use as a mechanism to reach out to academic groups to seed early drug discovery efforts. To set the context of our current activities in this area, it is worth considering how the field has evolved.
HTS technologies have now become widely adopted within the academic and not-for-profit communities to search for probes and starting points for early-stage drug discovery.4,5 While the goals of these centers are nuanced (probe delivery vs drug discovery), there are more than 100 academic screening facilities and 14 medicinal chemistry facilities registered with the Society of Laboratory Automation and Screening (www.SLAS.org/resources/information). Typically, these facilities will have the capacity to screen more than 100,000 compounds against targets or assay systems of interest. Critical to success is not the capacity to screen large numbers, but the quality of the library screened and the ligandability of the target. Library design, collation, curation, and management is a significant investment, in addition to the automation required for the screening facility. To offset this burden, joint compound purchases or library sharing is often employed between groups. At LifeArc we have jointly purchased kinase, protein–protein interaction (PPI), and ion channel focused sets with other UK academic and not-for-profit drug discovery groups and shared our full diversity collection with the Euroscreenport and Leiden University screening facilities in Europe, exemplifying the need for collaboration in this early discovery phase.
In addition to the established facilities and translational centers highlighted above, many university-based academic research groups now have access to some automation within their host institution, and with limited funding can support the consumable burden of screening 10,000–20,000 compounds. For novel target-based screens, this provides an assessment of the ligandability of the target, that is, the ability of small drug-like compounds to bind and thus the appropriateness of investing in significant further screening of larger diversity sets or other chemical space. This moderate scale of screening operation also opens up the possibility of screening more complex biology—whole-organism or disease-relevant phenotypic assays. Access to sets of compounds can be limited, so to support such academic endeavors, we set out to compile a set of around 10,000 compounds to be made freely available to academic groups. These compounds represent both our diversity collection and beyond that, to the wider, commercially available compound landscape. Such a compound set not only allows an academic group to evaluate their assays and identify potential hit matter for follow-up studies, but also initiates an early collaboration with our own center, which can lead to a fuller joint drug discovery program, if appropriate. This paper describes in detail the process we undertook to create the representative index set and its subsequent deployment. We will show that probes or chemical starting points can be found from such a small screen.
A combination of screening capacity, storage capabilities, and budget had previously directed us to scale the full LifeArc screening collection at around 150,000 compounds. This consists of target class-directed (e.g., kinase sets) and modality-focused (e.g., fragment screening) libraries, alongside a diversity screening deck of around 100,000 compounds. The selection of the diversity collection has been a cyclical build, reviewing the commercial landscape and selecting compounds complementary to the existing sets every 3–4 years ( Fig. 1 ). This regular selection window has the benefit of allowing us to build a collection that reflects the latest commercial space while letting the commercial compound landscape develop significantly in novelty between each application of the selection methodologies,6–8 as described in the following sections.
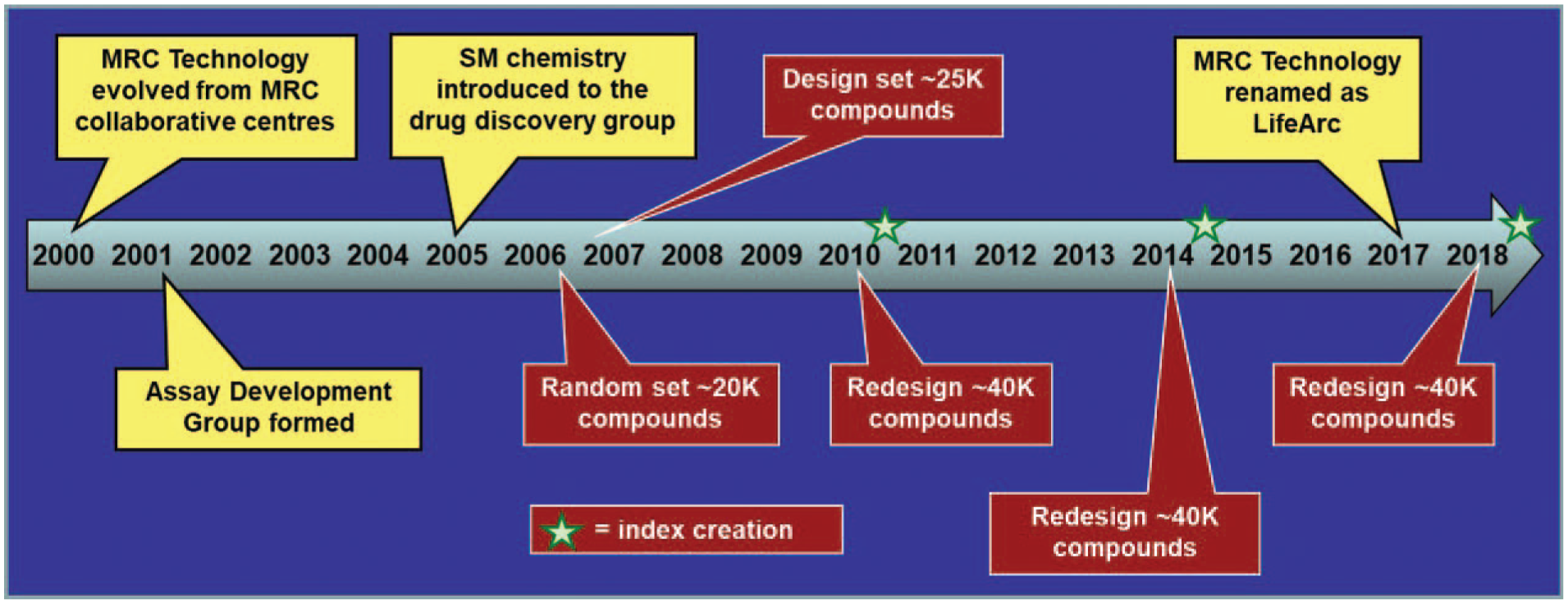
Timeline for building the diversity collection. The diversity set comprises the largest proportion of the LifeArc library collection. It is periodically updated to capture novel regions of commercially available chemical space, as indicated in the timeline.
The first iteration of the index set (~9000 compounds) was built following the 2010 collection enhancement; a subsequent rebuild following the 2014 diversity collection development increased this set to around 12,000 compounds. Once the latest 2018 round of diversity enhancement is completed, we will repeat the index build to ensure that it is representative of the collection as detailed herein.
Fundamental to any consideration around the development and use of the LifeArc index set is an understanding of how we developed the full diversity collection. The initial diversity developments between 2006 and 2010 were intended to represent as much of the commercial landscape as possible while minimizing the risk of missing potential structures of interest. The landscape of chemical space was sampled where feasible with small clusters of similar compounds rather than singletons; thus, if a compound substructure had the potential for activity at a target, then the hope was that more than one member of the series should be identified, enabling an early assessment of any structure–activity relationship (SAR). The next step of screen follow-up was then to explore that region of chemical space further through sampling commercial space and purchasing compounds. Subsequent diversity redesigns built further on this same principle. Therefore, when it came to design of the index set, the intention was to reference as many of the clusters as possible; any index set hit could then be followed up with initial mining of associated clusters within the diversity set, with this then leading to the wider commercial space as the second round of follow-up (or in parallel, if appropriate).
Library Design
As introduced above, the largest proportion of the LifeArc compound collection is composed of diversity sets that are periodically updated to capture novel regions of commercially available chemical space. This also helps ensure that screening stocks are fresh and boosts the chances that hit compounds and analogs are immediately available for fast follow-up. When selecting compounds to add to the collection, there are a number of desirable criteria: To ensure the efficient exploration of chemical space, selected compounds should be different from what is already present in the LifeArc collection and different from each other, that is, diverse. Computationally, it is relatively simple to ensure diversity based on structural similarity calculations. A more challenging objective is to ensure the selection of high-quality compounds, those that have at least a reasonable chance of progressing from hit to lead. The emphasis on quality is in part to reduce false positives in HTS, but also to avoid the frequently encountered situation where resources are wasted on hits that have poor prospects and are subsequently eliminated at follow-up triage or after attempts to resolve compound issues. The qualities that contribute to this are hard to define, but often involve some combination of the following: physicochemical property profiles that cause solubility or permeability problems, functional group liabilities that cause toxicity or compound instability, and synthetic complexities that limit the number and type of analogs that can be made, which can also slow down the data generation that drives medicinal chemistry optimization.
Computational approaches attempting to deal with compound quality have been improving over time, from simple reactive group filters 9 and individual property guidelines, 10 to more sophisticated approaches that identify features associated with various nefarious characteristics11–13 or composite scores that relate how combinations of properties and features impact prospects for development.14–16 There is rising concern that unbridled application of such filters could lead to dismissal of compounds capable of delivering legitimate hits, and it is rightly noted that a significant proportion of approved drugs do not conform to many of the guidelines and filters. 17 Closer inspection finds that in many cases, the exceptions have been specifically designed around particular mechanisms of action and modes of administration, for example, reactive anticancer drugs, greasy topical antibiotics, or intravenous peptide mimetics. Such specialist chemotypes are not suited to include in a diversity set for widespread HTS, but are justifiably included in other subsets in the LifeArc compound collection.
Diversity Set Selection
Great care is required to minimize the rejection of reasonable compounds while maximizing the removal of poor-quality compounds. For example, substructure flags for undesirable features are not necessarily comprehensive and vary in their severity depending on context. Furthermore, the accuracy in calculation of some key physicochemical properties, such as LogD, can be questionable. Finally, because diversity-based algorithms select compounds most dissimilar to anything else, often these are chemical oddities with less desirable features that rarely occur and so tend not to be captured by substructure flags and can be problematic to follow up due to a lack of analogs. Consideration of these factors is evident at several points in the selection procedure outlined in Figure 2 . In step 3, we cautiously applied hard property filters to remove only the most extreme examples, relying on definitive properties such as molecular weight and elemental composition. These were refined in step 5, also incorporating calculable properties such as LogD and QED, 15 for which acceptable cutoffs were determined by manual inspection. Medicinal chemistry expertise was also employed in step 4, where up to 100 examples of molecules hit by each substructure flag12,18,19 were inspected to decide if we would be happy to outright reject or accept such molecules. In several cases, we decided to refine the substructure queries to separate out the most interesting from the most offensive.
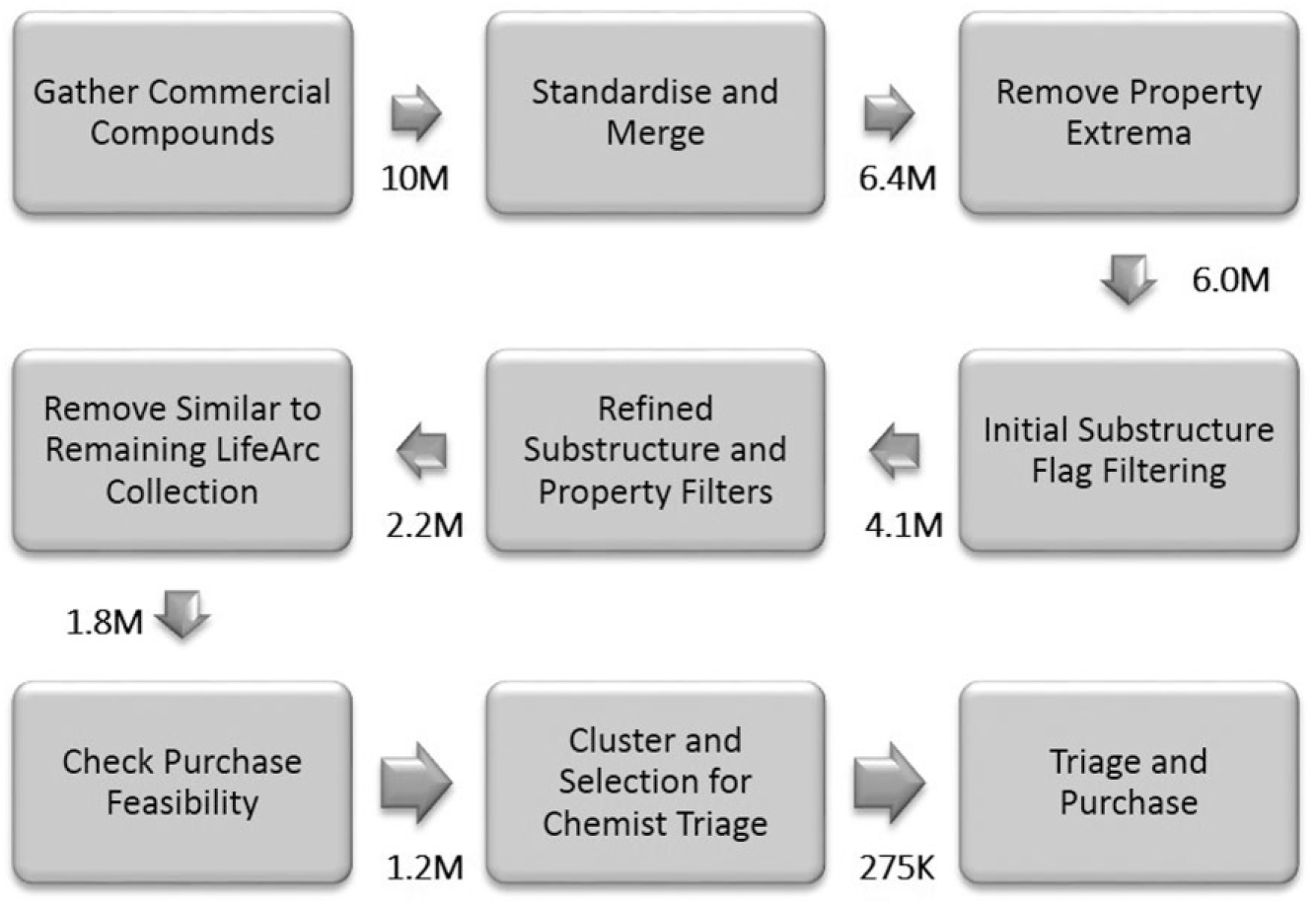
Flowchart providing an overview of the diversity set selection procedure.
We wanted to take advantage of the extensive amount of medicinal chemistry expertise at LifeArc to try to ensure that any compounds added to the collection are free from fundamental liabilities and have reasonable chances of being progressable. With this in mind, we decided to take a crowdsourcing approach such that only compounds independently approved by two chemists would be added to the collection. To facilitate this, the shortlisted compounds must be presented in manageable numbers of chemically meaningful groups. This can be achieved by clustering that aims to group similar compounds together into the same cluster while placing significantly different compounds into different clusters. A diverse selection of compounds can thus be obtained by selecting a suitable number of representative compounds from each cluster. It is important to strike the right balance between diversity and density when sampling chemical space; maximizing diversity, for example, by having just a single representative from each cluster, is prone to missing a series of actives simply by virtue of having the wrong group at a particular position on an otherwise suitable scaffold. Furthermore, the output from screening a set of singletons is much less encouraging and harder to follow up. Considering the typical degree of variation observed around commercial scaffolds (e.g., two to three points of variation with two to four fundamentally different types of group at each) and earlier work relating cluster size to the probability of discovering actives, 20 it was decided that 4–12 representatives per cluster is desirable. In order to get chemically meaningful clusters in a feasible time frame, a two-stage procedure was developed. First, the compounds were roughly clustered using ECFP_4 fingerprints with a sphere exclusion approach (radius 0.5), rejecting clusters of size less than 4. Second, the compounds in each cluster were subjected to a more sophisticated maximum common substructure (MCS) perception procedure, requiring that the MCS should match at least 75% of compounds in the cluster and contain at least nine atoms to ensure chemically meaningful clusters. The MCS was then used to align the cluster members for ease of viewing during the triage step, as described below. Selection of cluster representatives was based on pharmacophore feature counts, keeping the most attractive (highest QED) example for each combination of features, up to a maximum of 12 representatives per cluster.
A total of 275,000 molecules contained within 39,000 clusters of size 4–12 were available for triage by medicinal chemists. Clear verbal and written guidance was provided and can be summarized here as follows: “We are looking for hits, not drugs. We want to avoid nonprogressable compounds entering the library—minor liabilities can be tolerated, but multiple/inescapable liabilities should be avoided. Ultimately, is the compound worthy of a chance to be a screening hit—one that you would be willing to work on?” Compounds were presented via a webport in grids of structures aligned by their MCS, with a manageable 100 clusters per subset, allocated to each chemist randomly. Compounds or entire clusters can be rejected with a single click. Each compound is viewed by two chemists, keeping only those that are passed by both. A total of 14 chemists volunteered, each processing 13 subsets of 100 clusters, one in common with each other chemist. More than 70,000 unique molecules were examined and an agreed pass verdict of 62% (44,000 molecules) was obtained, of which 33,600 were purchased to form the 2015 diversity set, replacing the 41,667 molecules in the pre-2010 diversity set. An analysis of the scaffolds 21 in these sets found that there were 28,103 different scaffolds in the 2015 set compared with 15,041 in the pre-2010 set, meaning that although the new diversity set contains 20% fewer molecules, there are 90% more scaffolds. The diversity of the set is further analyzed using nearest-neighbor similarity distributions, as calculated by pairwise comparison of each molecule in the 2015 diversity set by ECFP_4 fingerprint Tanimoto similarity, as shown in Figure 3a . The results confirm the efficiency of this set in that it contains no trivial analogs (NN1sim > 0.9), no singletons (NN1sim < 0.4), and no compounds with too many analogs (NN50 < 0.5). Around 40% of the compounds have at least 5 reasonable analogs (NN5sim 0.5–0.7) and less than 10% have as many as 10 related analogs (NN10sim > 0.5) immediately available within the set to help define SAR, while many more analogs may be accessible commercially. The physicochemical property distributions are shown in Figure 3b , indicating a reasonable lead-like profile.
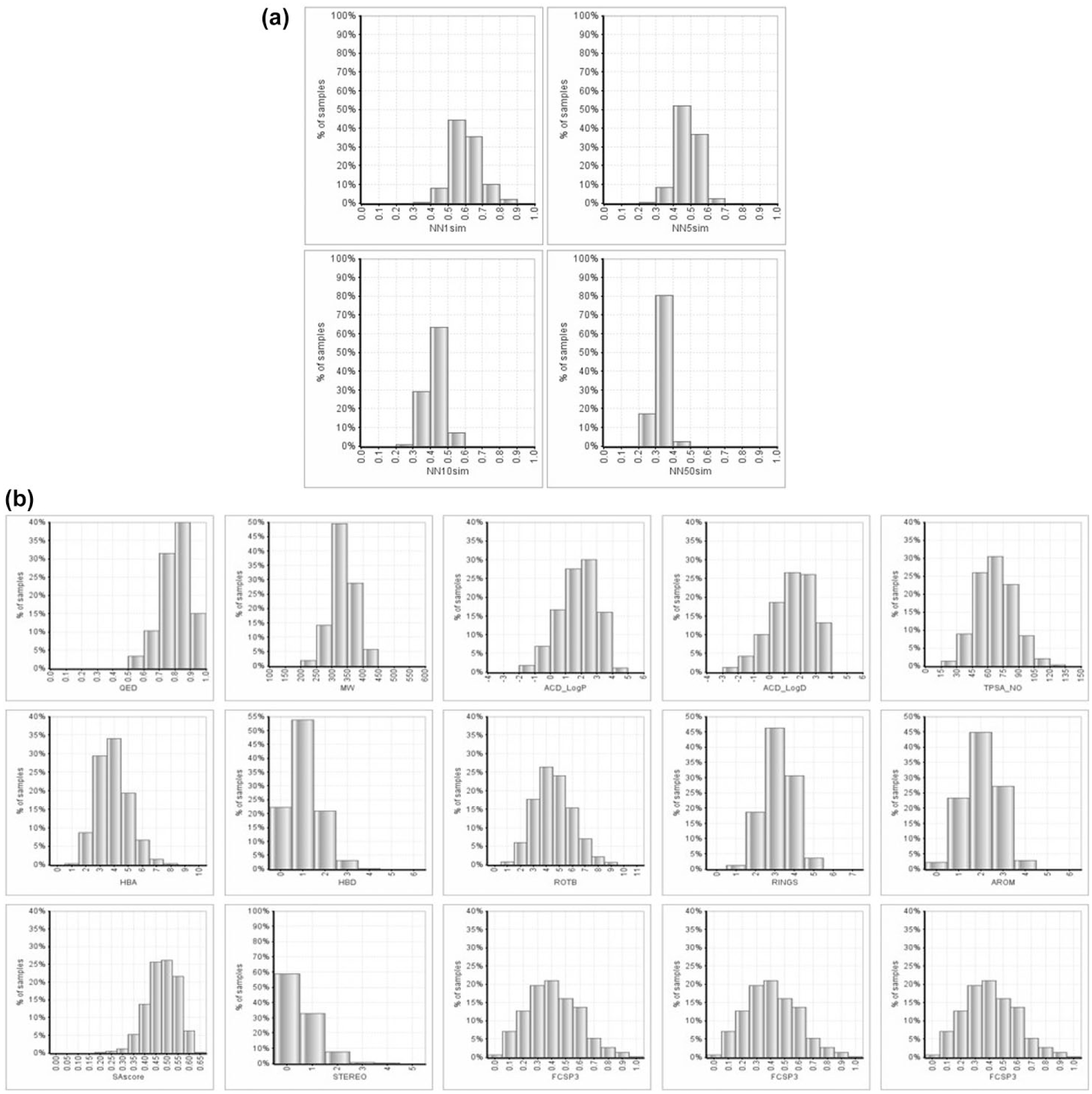
(
Index Set Selection
To ensure that the index set continues to be representative of the wider diversity collection, 2462 of the least attractive compounds representing the pre-2010 diversity set were removed and replaced by 3840 compounds selected from the 2015 diversity set. The primary aim was to select compounds that maximize the coverage of the 2015 diversity set. This can be achieved by selecting a single compound (highest QED) from each master cluster and selecting from the largest master clusters first, such that there are more analogs available for rapid follow-up of any index set hits. We also implemented additional constraints to ensure that the selection is diverse and complementary to the remaining index set; specifically, each selected molecule has a unique Murcko scaffold and similarity <0.5 with respect to any other compound in the index set, including the newly added ones.
In summary, the selection procedure has resulted in a diverse lead-like representative index set in accordance with the latest developability metrics, with the added assurance that for the latest index set members (drawn from 2015 diversity sampling), each compound has been independently approved by two medicinal chemists as suitable for hit follow-up. Furthermore, the decision to focus sampling on reasonably sized (4–12) clusters with a logical MCS for the index build provides a more rational potential for exploration of SAR from initial index set screening with subsequent full diversity cluster sampling and promotes faster progression from hits to lead series. An illustration of this is provided in Figure 4 , where a master cluster of molecules aligned by their MCS is shown in a grid to easily see what is active (red border), what is inactive (blue border), and what other commercially available compounds were not acquired (grey border), thus simplifying the visualization of screening results and selection of analogs for follow-up.
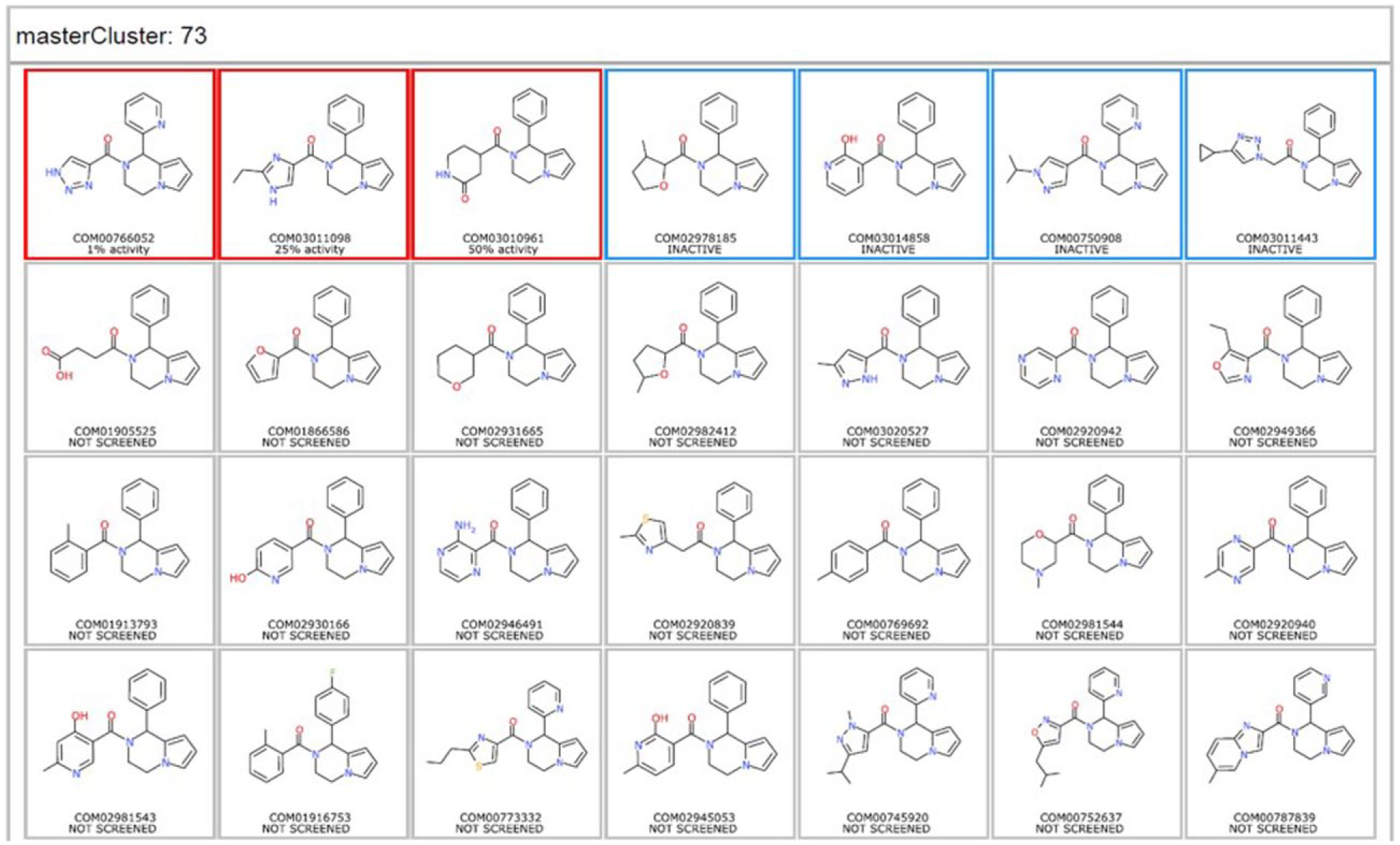
Illustration of how the clustering and selection procedure can impact visualization of the screening results and hit follow-up. A master cluster of molecules aligned by their MCS is shown in a grid, with the original index hit shown in the top left corner and other analogs available in the collection along the top row, with activities reported after hit follow-up. Other commercially available compounds not in the collection are shown in rows 2–4.
Sharing the Library
As part of our extensive project scouting activities, academics from around the globe are encouraged to submit proposals to access and screen the various sets within our library collection. These proposals are reviewed by a LifeArc panel of senior scientists regularly throughout the year and assessed on novelty, screening capacity, demonstrated assay robustness, and a clear strategy to confirm hits in orthogonal assays. An important consideration here is to avoid duplication, with either other groups or internal projects, and this is the main reason for any refusal to share the index set. For well-worked targets for which we do not believe the index set would be suitable for screening, we can offer alternative library sets for screening, for example, target-focused libraries. If assays have not been demonstrated robust in a previous screening campaign, we offer smaller sets to validate the screen performance before embarking on the index set. Once a proposal has been approved a work plan is agreed and the libraries are shared under a material transfer agreement (MTA). Each screen will be custom-made, but typically follows the generic strategy of a primary screen, single-point hit reconfirmation with a counter-screen to confirm active and not interfering with the assay technology, followed by dose–response confirmation in the primary assay. To simplify logistics, our library is offered in a single format—20 µL 1 mM stocks in DMSO plated in 384-SBS standard polypropylene plates, 320 compounds per plate, with columns 1–2 and 23–24 containing DMSO-only controls. Follow-up resupply from our 10 mM master stocks is provided for dose–response and orthogonal confirmation assays, along with any related analogs as per the cluster sampling described previously to identify any hit SAR. Library plating, cherry-picking, and the shipping of compounds are managed by SPECS (Zoetermeer, Netherlands), which curates our collection. At this point, we perform liquid chromatography–mass spectrometry (LC/MS) on the hit samples to confirm compound identity and purity. Throughout the screening campaign and follow-up confirmation stages, we provide computational chemistry support and feedback on the activities of the hits found, including identification of commercially available analogs for purchase.
Progression of compounds from screening is not solely dependent on their biological activity. Chemoinformatic analysis of HTS data is applied to identify active clusters within the screen set and determine which properties and features can be modified to optimize both the biological activity and the physicochemical property profile. Consequently, having a range of actives within a cluster provides a more informative SAR and improved prospects for development, compared with singleton actives or clusters where SAR is flat. Any physicochemical property or substructure flags are assessed alongside internal and external historical activity data from other screens to better understand whether nonspecific mechanisms of action are likely, so as to avoid wasting time and money following them up. Having confirmed active clusters demonstrates our ability to identify start points, though further testing in orthogonal and biophysical assays will be required to validate their mechanism of action.
During the main process of screening and hit follow-up, all compounds are supplied structure blinded; structures of confirmed actives (agreed on in the work plan) and any related compounds screened are shared once the robustness of the results has been ensured. If the project is of joint interest, then a collaborative project to develop the series identified may follow; otherwise, the host institution is free to develop any structural series independently, without further reach-through from LifeArc. Finally, we also collect internally and collate the data from all screens of our compound libraries (in-house and collaborative screening) to provide a data set that we can then use in future iterations of library and index set design. Following these processes, we have supported more than 100 screens with academic labs, of which the index set comprises about a quarter. Those collaborations that have completed screening and hit follow-up are summarized in Table 1 , from which it can be seen that the library has been widely shared across the world in both established screening groups and individual labs. A broad range of targets and phenotypic screens have been tested, with all but one screen generating hits that could be followed up. In this instance, the assay employed had a low DMSO tolerance, and consequently a low screening concentration (1 µM) had been used. The purpose of screening the index set for some groups was to validate their screening strategy ahead of larger screen campaigns, for example, the phenotypic screening of worm larvae and adult motility by the London School of Tropical Medicine 22 and validation of a miniaturized 384-format mass spectrometry assay platform for de-ubiquitinating enzymes at Dundee University (unpublished). For others, the index set was included to add additional diversity to planned large screen campaigns, for example, the screening of biased ligands against apelin 23 or the screening of malarial kinase pfCLKs (unpublished) in groups with access to HTS facilities. Notably, assay formats not usually considered amenable for HTS, for example, those involving radioactivity or cellular imaging, have been employed with success using this small screen set, providing tool compounds for further follow-up studies.
Completed Index Screens with Academic Collaborators.
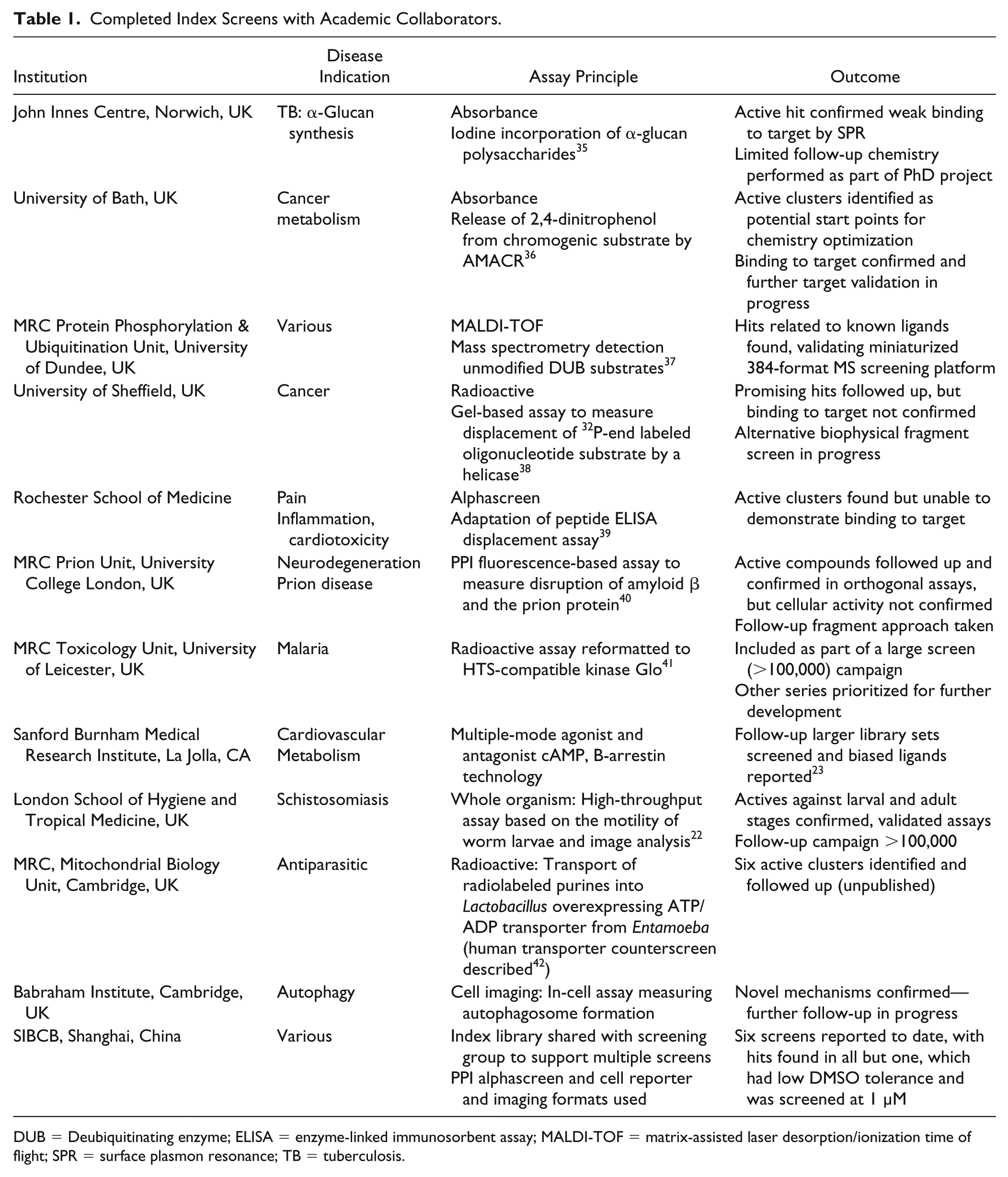
DUB = Deubiquitinating enzyme; ELISA = enzyme-linked immunosorbent assay; MALDI-TOF = matrix-assisted laser desorption/ionization time of flight; SPR = surface plasmon resonance; TB = tuberculosis.
Demonstrating Ligandability
As screening large collections can be expensive, it has been argued that using a smaller set of compounds should be predictive of potential screening success and indicate whether a target could be ligandable. Thus, to demonstrate whether screening our index set would be predictive of the outcome of screening our full diversity set, we performed a retrospective analysis of our historical screening data. As described earlier, the index set is a snapshot in time of our collection, which we regularly renew and replace, so HTS campaigns prior to 2010 or after 2014 will have different representatives of the index set present, dependent on the plates screened at the time. HTS campaigns for which at least 20% of the index set compounds existed within the larger data set were selected for analysis. Eighteen target-based screens were investigated: 9 cell-based (receptor or ion channel) and 9 enzyme or PPI targets. First, we compared hit rates from full screens versus the hit rate of the subset of index compounds present in the screening data. The hit rate from single-point screening reflects the screen noise and is expected to be evenly distributed across plates, unless there is an underlying plate trend or change in conditions through the screen. As the index set compounds are distributed throughout the library, and not on one plate, we did not expect to see a significant difference and none was observed (data not shown). Next, we compared confirmed hit rates. Here, compounds have confirmed activity in the dose–response mode and passed technology counterscreens, or any other screen-specific selectivity criteria. As such, they are potential true hits and a measure of the ligandability of the target. One of the 18 full high-throughput screens failed to find any confirmed hits with activity of interest, and several protein target-based screens had very low hit rates, indicative of poor ligandability. Confirmed hits from the index set were found in 14/18 screens. As shown in Figure 5a , hit rates do correlate, thus validating the selection of the index compounds as representative of the active chemical space and a good predictor of ligandability from diverse chemical library screening. The average confirmed hit rate from the index set is lower than that seen in the diversity set; this is attributed to the presence of active clusters of related compounds in the larger diversity set.
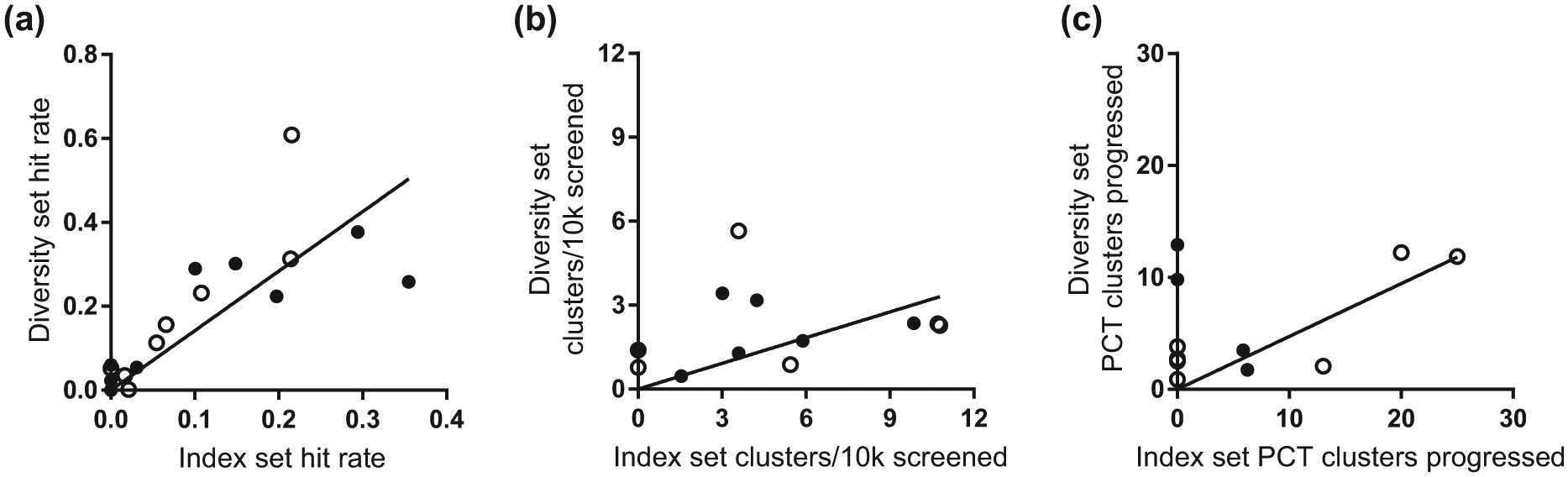
Retrospective analysis of index compounds. HTS campaigns for which at least 20% of the index set compounds existed within the larger data set were selected for analysis. Eighteen target-based screens were investigated: 9 cell-based (receptor or ion channel; solid circle) and 9 enzyme or PPI (open circle) targets. The linear regression correlation plots shown were calculated against the whole data set. (
Finding Progressable Hits
Screening a limited number of compounds inevitably reduces the number of hits found, but this can be rapidly expanded by testing related analogs. As such, our strategy for the index set was to ensure that analogs were available from our collection or easily commercially sourced. Taking this approach, we hoped to identify active clusters for ligandable targets. Analysis of the historical screen data suggested that this was feasible. A total of 13 out of the 18 full high-throughput screens produced active clusters that were progressed; of these, the index set identified at least one cluster in 9 cases. This may be an underestimate of the likely success rate for two reasons. First, hits were selected on stringent hit cutoffs applied to large screening data sets, such that weakly active compounds within the cluster were missed. This can be mitigated by screening a smaller set by applying more inclusive statistical cutoffs, for example, less than 3 standard deviations from the mean positive control. Using this approach, weak active hits may identify more active analogs in a cluster after hit expansion. Second, as discussed above, the index set is a snapshot of the library at a given time, and for some older screens, as few as 2000 compounds were present, meaning that targets with intrinsically low ligandability (low hit rate) would not necessarily be expected to yield hits within such a small sample of compounds. Nonetheless, normalizing the data to account for the relative numbers of compounds screened shows an enrichment of the number of clusters identified per 10,000 screened from the index set compared with the HTS data ( Fig. 5b ).
To examine further whether there might be any difference in the progressability of hits originating in the index set compared with those originating in the diversity set, we identified which clusters were developed by subsequent chemistry follow-up. Compounds with dose–response activity were clustered (sphere exclusion with radius 0.5 using ECFP_4 fingerprints with Tanimoto coefficient) in each of the studies to provide an indicator of the number of different chemotypes in the hits available for follow-up. Lists of all other (not present in the diversity or index set) compounds with dose–response data were compiled for each study, representing follow-up compounds originating from purchase and/or synthesis. Each hit cluster is classified as having progressed if any of the compounds in that cluster have a similarity (ECFP_4 fingerprints with Tanimoto coefficient) value greater than 0.5 to any of the follow-up compounds. The number of hit clusters containing an index set compound tends to be a small proportion of the total number of hit clusters, reflecting the fact that fewer index set compounds were screened. Nevertheless, all 13 studies generated hit clusters containing an index set representative, although one study did not contain any clusters without index set representatives, meaning that there were no clusters originating purely from the diversity set with which to compare. Of the 12 studies where hit clusters were available for both the index and diversity sets, all but one was found to have follow-up compounds. The percentage of clusters from the index and diversity sets that were followed up are shown in Figure 5c . It can be seen that a higher percentage of clusters containing index set compounds were progressed, although in six cases the only clusters that were progressed originated purely from diversity set compounds. There can be many reasons why some clusters are followed up rather than others, depending on their relative merits; potency, selectivity, or other factors relating to the desired biological profile are considered to determine if there is meaningful SAR in a cluster. Having data on a range of analogs tends to be more informative and encouraging, so it is not surprising to see that larger clusters have improved chances of being followed up, with particularly poor prospects for singletons, as shown in Figure 6a . These results support the rationale for choosing reasonably sized clusters rather than singletons when building the diversity set and selecting index set compounds that represent larger clusters from the diversity set. Compound quality is the other main factor affecting progressability, and it is likely that hits with poor property profiles and/or problematic chemical features will tend to be deprioritized from follow-up. Figure 6b shows the distribution of QED scores and occurrence of substructure flags (indicators of compound quality) for hit clusters, according to whether or not they are represented in the index set, and whether or not they were progressed. The results show that substructure flags occur in a greater proportion of lower-quality (low-QED) compounds and that flags are present more often in compounds that were not progressed (32% overall) compared with those that were progressed (26% overall). Furthermore, QED scores tend to be higher for compounds that were progressed than for those that were not. It is interesting to note that while the proportion of substructure flagged compounds is lower overall in the index set (20% compared with 33% in the diversity set), a substantial proportion of the index set hits that were progressed contain a substructure flag. Investigation of this surprising result revealed that all 18 compounds were flagged for containing a nitro group and were otherwise a perfectly reasonable set of compounds, as may be expected from their respectable QED scores (all greater than 0.5). This illustrates the value of employing medicinal chemistry expertise rather than hard computational filters to provide a more fine-grained distinction between less desirable groups that might be replaceable (e.g., nitro) and other features that are more likely to be problematic in a hit discovery setting (e.g., toxoflavin).
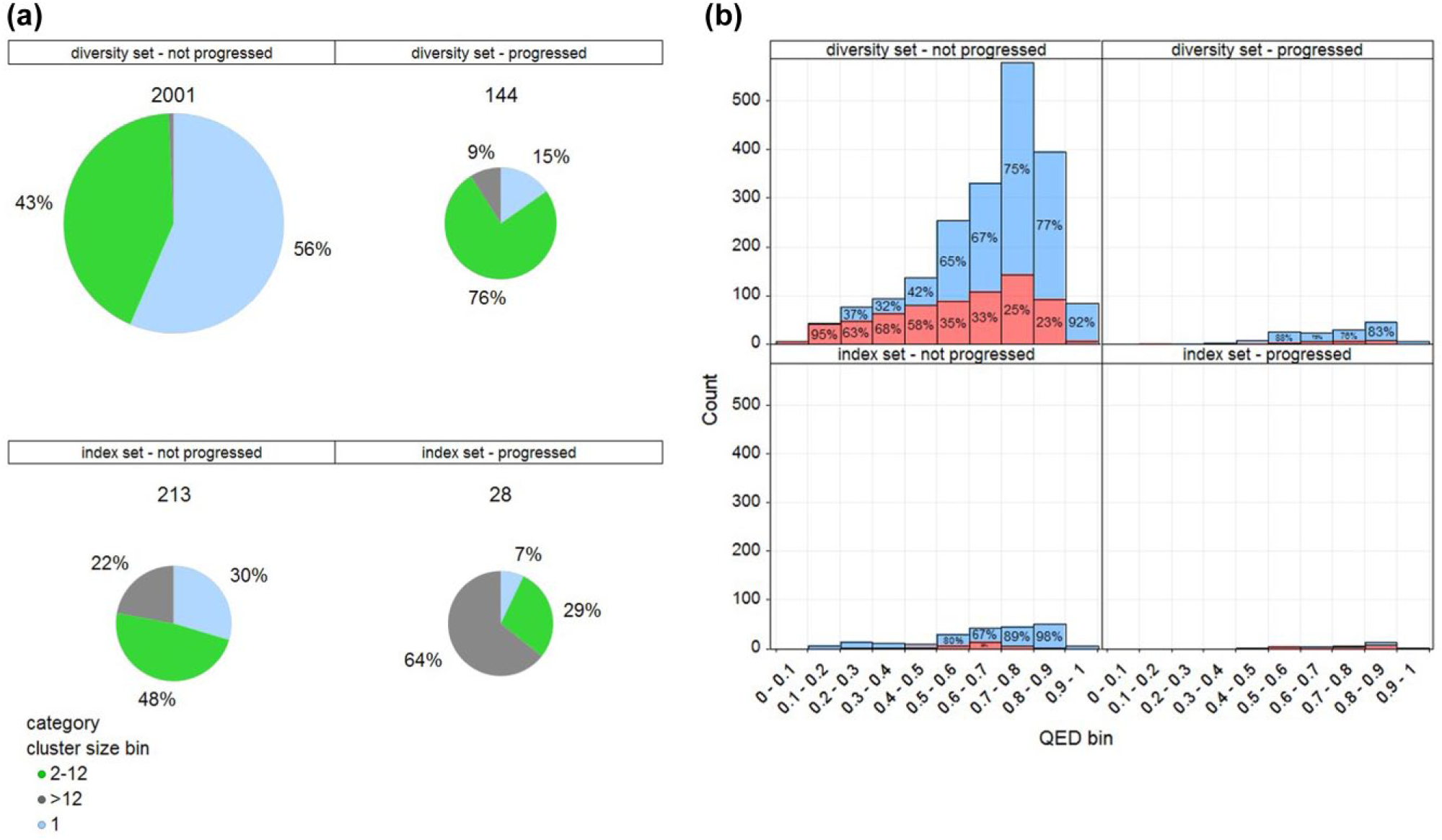
Assessment of hit quality. (
Frequent Hitter Analysis
Only two index set compounds have hit in more than three screens; these are shown in Figure 7 . Given the diversity of targets inhibited by these compounds, it is unlikely that they are acting by genuine mechanisms capable of optimization. It is interesting to note that each compound has a perfectly reasonable property profile, with a high QED score. Furthermore, although both compounds were present in the 2010 index set, compound A was later found to have a substructure flag (pan-assay interference compounds [PAINS], class B, pyrrole_A(118)) and was subsequently removed. However, compound B has no such flags, although reviewing the chemistry identifies that this scaffold is closely related to toxoflavin, which is capable of redox behavior that can result in misleading nonspecific inhibition.24,25 These observations emphasize the importance of not relying solely on computational methods when building compound collections and validate our approach of using a combination of chemistry expertise and data-driven curation.
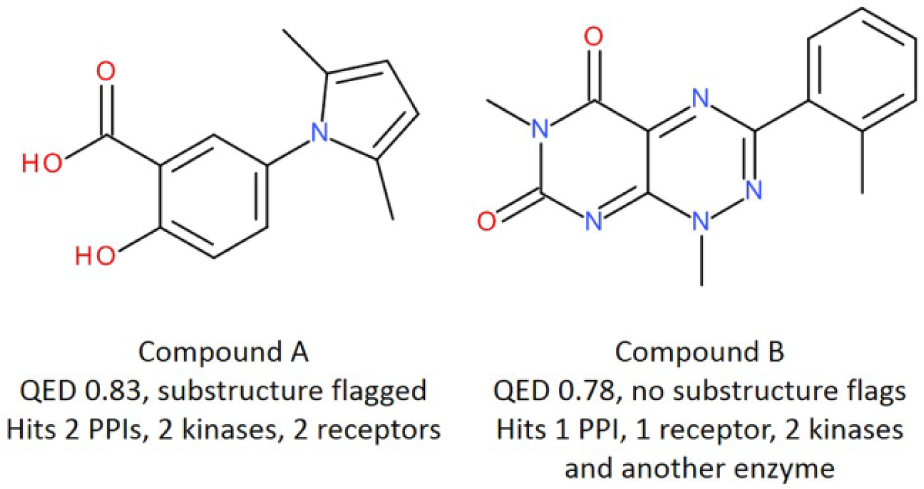
Frequent hitter exemplars identified for removal from the collection. Screening data compiled at LifeArc identified only two compounds from the index set that were identified as hits in three or more screens against diverse targets or screening technologies.
Even though there are very few individual compounds that hit multiple screens, variability in hit cutoffs can lead to important patterns being missed. We sought to learn more about the properties and features that contribute toward promiscuous behavior by examining the number of screens hit per cluster, that is, where different representatives in a series of compounds are found to hit unrelated targets when considering index representatives and their diversity set analogs. While the majority of clusters hit only a single target, the 14 “hottest” clusters hit four or five targets. Only three of these clusters are substructure flagged, two of which are related to compound A in Figure 7 in that all their members contain the benzoic acid pyrrole substructure, adding further support to the problematic nature of these chemotypes, 26 whose representative has been removed from our index set. While there are some structural commonalities between the remaining clusters, these are not sufficient to propose new substructure flags. Similarly, although correlation analysis and modeling approaches (naïve Bayesian classifiers and recursive partitioning trees) did not provide novel insights, many of the descriptors associated with hot clusters were related to higher aromatic content, consistent with the literature.27–29 For example, while 86% of molecules in the hot clusters had a fraction of SP3 carbon atoms (FCSP3) less than 0.4, this was true for only 77% of molecules in clusters hitting a single target.
Screening the Index Set
The index set has been screened broadly across targets and cell-based screens, both internally and through library sharing with the academic community. In total, 33 screens have been completed and their hit follow-ups (including analog testing in dose–response mode) reported. The data are summarized in Table 2 . These have been loosely categorized into five screen categories by target. In total, 476 compounds, almost 5% of the index library, have been reported as confirmed actives in one or more screens. After hit expansion, the majority are found to be singleton hits, with approximately a third falling into active clusters that demonstrate the potential for further optimization.
Index Set Performance LifeArc and Collaborator Screens.
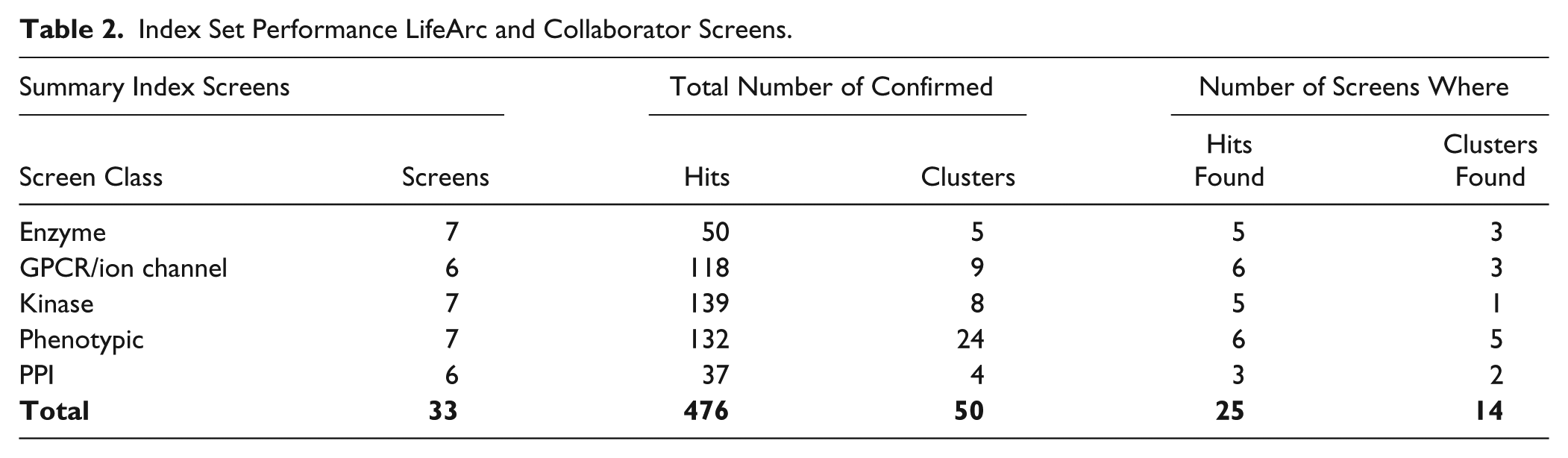
Overall, the number of screens reporting hits found (25/33) and active clusters (14/25) is less than that predicted from our historical data. This can be attributed to a higher proportion of PPI screens and kinase screens, which were not represented in the historical set. The low ligandability of PPI targets reflects their challenging nature and for which “beyond rule of 5” collections (natural products, macrocycles, or peptidomimetics) 30 may be more suitable. Hits for kinases were found, but these tended to be weak actives (>10 µM) that did not fall into clusters. Although kinases are generally regarded as highly ligandable targets, the diversity set is not designed to meet the specific requirements of the ATP pocket, for which we have a kinase-focused set with suitable hinge binding motifs. The index set does, however, have the potential to identify allosteric sites, and therefore we include screening of the index set alongside our kinase library to gauge this potential.
The index set has been most productive in cell-based screens. Several G protein-coupled receptor (GPCR) screens have been conducted from which agonists, antagonists, and allosteric modulators have been found. The hits found have provided chemical starting points from which chemistry programs have been initiated and progressed. Success has also been found targeting two-pore potassium channels (K2P) (unpublished data), a target class for which there is a dearth of ligands in the literature. K2P channels carry background (or leak) potassium current in a variety of cell types and primarily act to maintain resting membrane potential and have been implicated in a variety of pathophysiological conditions, including migraines and pain. Assays are challenging and K2Ps are thought to be a difficult target class to modulate with small molecules. Recently, we initiated a number of collaborative phenotypic screening projects in disease-relevant models in neuroscience. Each assay employs different readouts—high-content imaging or reporter-based different cells or different time points from which common or novel mechanisms, aggregate clearance, or autophagy may emerge. The first of these screens have been completed and hit follow-up is in progress. Similarly, in the anti-infective arena, both target-based and phenotypic assays have been initiated to investigate antimicrobial resistance using whole-cell reporter-based systems and phenotypic biofilm formation assays. These screens are in progress and will be read out in the future.
Future Prospects for Library Design
We continue to revise the composition of our index set to ensure that it is of the highest quality, partly to reflect the changing nature of our compound collection and what is available commercially, but also in response to the increasing level of characterization of our compounds. For example, many index set compounds have been screened tens of times and never been found to be active, so it could be argued that they might be “dark chemical matter” and better replaced by chemotypes that have some precedence for biological activity. However, it is rightly noted that if such chemotypes are discovered as hits in the future, then their prospects may be enhanced by the cleanliness of their activity profile. 31 We already have a set of compounds with known biological activities and ADMET (absorption, distribution, metabolism, excretion, and toxicity) data that are often used for phenotypic screening, and while there may be some utility in incorporating some of these into our index set, there is also benefit to be gained from incorporating novel chemotypes into the index set for characterization. Macrocycles are an example of an interesting area of chemical space where we have recently expanded our collection and whose representatives are to be integrated into the index set. Around 6% of approved drugs are macrocycles, and although they occupy “beyond rule of 5” space, 30% are orally bioavailable. 32 The predominant indications of these are oncology and infection, but other macrocycles are in clinical development against a broad range of target classes, including PPIs, polymerases, proteases, and kinases, which fits well with our areas of therapeutic focus and with the diversity of collaborative projects where we share our index set. Macrocycles have traditionally been regarded as challenging, both from a synthetic perspective and in terms of balancing their physicochemical property profile to permit cell penetrance and solubility. However, data from recent studies 33 have allowed us to carefully build a set of macrocycles enriched in chemotypes that should be cell permeable and where a reasonable range of analogs are available to explore SAR. Another aspect that is influencing the design of our next-generation index set is the results generated on our fragment set, which rivals the index set in having been screened against over 30 diverse targets. The lower complexity of fragments tends to provide much higher hit rates than typical drug-like libraries. This amplifies differences among fragment hit frequencies, revealing which pharmacophoric building blocks stand out as privileged binders 34 and which are found to never offer productive interactions of sufficient strength to be detected. We are using this information to direct the selection of new diversity and index set compounds toward those bearing the more productive pharmacophores. It is hoped that this will boost hit rates by avoiding the selection of representatives containing features that do not tend to confer productive binding interactions. The index set concept is also something we think could be useful to apply to our fragment set—selecting a smaller subset of fragments experimentally confirmed as capable of interacting with a wide range of different target types and whose behavior (solubility, technology interference, etc.) has been well characterized. Such a fragment set is particularly valuable for probing target ligandability when throughput is limited by the sheer number of targets. Focusing on a smaller set of fragments also enables resource-intensive, information-rich crystal-soaking experiments to be employed as a primary screen to expedite the follow-up of hits from fragment screening, akin to how the index set enables information-rich, lower-throughput assays.
Summary
The index set provides a useful pre-HTS tool to gauge the ligandability of unknown targets and assess screen quality. At LifeArc, we routinely run this as part of our hit identification strategy on novel targets. Where feasible, this is also supplemented by fragment-based biophysical approaches. Screening the index set alone is not a perfect substitute for screening a large set of compounds. However, using it in conjunction with hit analog follow-up, enhanced by the cluster selection process connecting our diversity set to the index set, provides a good chance of identifying a tool compound. This could be invaluable for validating the target or provide a useful reference compound for subsequent screens. This is particularly useful for academics with limited budgets, materials, or access to full HTS facilities. At LifeArc, this potential has been exemplified for ion channels for which ligands had not previously been described.
The LifeArc index set helps us build confidence in a target and foster further collaborative studies with the academic community to help translation. Important in this process is management of the library, by keeping it up to date to ensure that commercially available analogs are available for follow-up studies. Through our network of academic collaborators, we are building a knowledge base of compound activities against specific targets, pathways, and phenotypic assays. Through sharing knowledge gained from screens, we are better able to advise collaborators on the best routes forward for translation of their target. Through feedback, we can inform the design of the next set and continue to evolve the LifeArc library.
Footnotes
Declaration of Conflicting Interests
The authors declared no potential conflicts of interest with respect to the research, authorship, and/or publication of this article.
Funding
The authors received no financial support for the research, authorship, and/or publication of this article.