
Abstract
Familial dysautonomia (FD) is an autonomic and sensory neuropathy caused by a mutation in the splice donor site of intron 20 of the ELP1 gene. Variable skipping of exon 20 leads to a tissue-specific reduction in the level of ELP1 protein. We have shown that the plant cytokinin kinetin is able to increase cellular ELP1 protein levels in vivo and in vitro through correction of ELP1 splicing. Studies in FD patients determined that kinetin is not a practical therapy due to low potency and rapid elimination. To identify molecules with improved potency and efficacy, we developed a cell-based luciferase splicing assay by inserting renilla (Rluc) and firefly (Fluc) luciferase reporters into our previously well-characterized ELP1 minigene construct. Evaluation of the Fluc/Rluc signal ratio enables a fast and accurate way to measure exon 20 inclusion. Further, we developed a secondary assay that measures ELP1 splicing in FD patient-derived fibroblasts. Here we demonstrate the quality and reproducibility of our screening method. Development and implementation of this screening platform has allowed us to efficiently screen for new compounds that robustly and specifically enhance ELP1 pre-mRNA splicing.
Introduction
Familial dysautonomia (FD; Riley–Day syndrome, hereditary sensory and autonomic neuropathy type III, MIM 223900) is a rare, congenital sensory and autonomic neuropathy caused by an mRNA splicing defect that results in reduced levels of the elongator complex protein 1 (ELP1), previously known as inhibitor of kappa B kinase complex-associated protein (IKAP). 1 Patients with FD have a complex neurological phenotype due to a striking progressive depletion of unmyelinated sensory and autonomic neurons with diminished pain and temperature perception, decreased or absent myotatic reflexes, and proprioceptive ataxia. In addition, lack of afferent baroreceptor signaling causes complete failure of blood pressure regulation and recurrent episodes of hypertension with tachycardia, skin flushing, diaphoresis, nausea, and vomiting, which together are referred to as “dysautonomic crises.”2–5
FD is caused by a single-nucleotide T-to-C change at position +6 of the 5′ splice site of intron 20 in the ELP1 gene (formerly known as IKBKAP) that leads to aberrant skipping of exon 20. Importantly, all FD patients possess at least one copy of the IVS20+6T>C mutation. A total of 99.5% of patients are homozygous for the major mutation, and the remaining are compound heterozygotes for the splicing mutation and missense mutations,2,6 so the development of a splice-modifying therapy would be beneficial for all patients. Despite the fact that FD is a recessive disease, homozygous mutant cells express both wild-type (WT) and mutant (MU) ELP1 mRNA, and are therefore capable of producing full-length functional ELP1 protein. 1 Thus, the mutation weakens but does not completely inactivate the 5′ splice site of exon 20. We have shown that the relative amount of MU and WT ELP1 transcripts varies between tissues. The lowest levels of WT ELP1 production are observed in tissues from the central and peripheral nervous system. 7 This is consistent with the selective degeneration of sensory and autonomic neurons observed in the disease; 2 therefore, FD results from ELP1 protein levels falling below tissue-specific thresholds. ELP1 is the scaffolding member of the human Elongator complex.8,9 Elongator is a six-subunit protein complex that is highly conserved in eukaryotes and participates in several cellular processes. First identified by its association with RNA polymerase, the Elongator complex is required in the nucleus for the efficient transcriptional elongation of a subset of genes through the acetylation of histone H3.8,10 Since its discovery, Elongator has also been implicated in a variety of other cellular functions: acetylation of α-tubulin and cytoskeleton organization,11,12 exocytosis, 13 zygotic paternal DNA methylation, 14 sensitivity to DNA-damaging agents, 15 and tRNA modification.16,17 So far, the cellular functions that have been most thoroughly investigated and linked to disease pathology in FD are transcriptional dysregulation caused by changes in transcriptional elongation, 18 translational impairment of codon-biased genes due to the role of the Elongator complex in tRNA modification, 19 and cytoskeleton organization, the disruption of which impacts neurite outgrowth and axonal branching.11,20,21
Our discovery that FD patients retain the capacity to make both WT ELP1 mRNA and protein offered an exciting, direct approach toward the development of therapies aimed at increasing levels of cellular ELP1 via splicing modification. With this goal in mind, we participated in the NINDS-sponsored Neurodegeneration Drug Screening Consortium, 22 during which we identified the plant cytokinin kinetin (6-furfurylaminopurine) as a potent splicing enhancer for ELP1 in both FD lymphoblast and fibroblast cell lines, resulting in nearly complete correction of the splicing defect.23,24 ELP1 protein levels in patient cells return to normal levels after 1 week in culture with kinetin. 23 Furthermore, we have shown that administration of kinetin modifies splicing in vivo in a transgenic mouse model of FD in which human FD-ELP1 is expressed from a transgene carrying the FD mutation. Kinetin improved human ELP1 mRNA splicing and significantly increased exon 20 inclusion in all tissues tested, including brain. Human ELP1 protein expression was also increased in treated animals, demonstrating that using splicing modulators to increase ELP1 protein levels is a feasible route to therapy. 25
The discovery of kinetin as a modulator of ELP1 splicing provides direct evidence of molecular efficacy. Despite this initial early promise, a small, short-term safety trial of kinetin in FD patients showed limited potency and efficacy on peripheral ELP1 splicing in humans, as well as some adverse events due to high dosing, highlighting the need for more potent and efficacious drug candidates.26,27
In 2012, our FD project was selected for funding by the NIH Blueprint Neurotherapeutics Network (https://neuroscienceblueprint.nih.gov/bpdrugs/). This program provides support for small-molecule drug discovery and development, from hit-to-lead chemistry through phase I clinical testing. A kinetin analog, 2-chloro-N-(furan-2-ylmethyl)-7H-purin-6-amine, also known as RECTAS, has been described in the literature to have a modest increase in potency over kinetin. 28 This provided evidence that an improvement in potency for this chemical series is possible and set the stage for our medicinal chemistry work. In the present study, we describe the development and validation of a cell-based reporter assay for a rapid and efficient measurement of ELP1 exon 20 splicing. The assay recapitulates the in vivo splicing pattern and provides a straightforward readout for the quantification of ELP1 exon 20 inclusion. The screening method has been optimized and validated to attain a high level of sensitivity and reproducibility and to achieve a moderately high throughput. This splicing assay enabled the identification of splicing modulators compounds (SMCs) with significantly increased potency and efficacy relative to those of kinetin, representing the first fundamental step in the development of a targeted treatment for FD.
Material and Methods
Rluc-FD-Fluc Minigene
Rluc-FD-Fluc plasmid was derived using the ELP1 FD minigene 23 containing the ELP1 genomic sequence spanning exons 19–21 inserted into spcDNA3.1/V5-His Topo (Invitrogen, Waltham, MA). Firefly luciferase coding sequence (Fluc) was cloned into the minigene from the pGL4.14[luc2/Hygro] vector and inserted at the 3′ end of ELP1 exon 21. Renilla luciferase coding sequence (Rluc) was cloned into the minigene from the pGL4.82[hRluc/Puro] vector and inserted in frame adjacent to the start of ELP1 exon 19.
Using site-directed mutagenesis, the ATG start codon in the firefly luciferase coding sequence was changed to CTG to inactivate independent translation. Rluc and Fluc coding sequences are in frame and expressed if exon 20 is included in the transcript; skipping exon 20 puts the Fluc coding sequence out of frame with the Rluc ATG start codon and prevents expression of Fluc.
Cell Culture
HEK-293T (ATCC, Manassas, VA) cells were cultured in Dulbecco’s modified Eagle’s medium (D-MEM, Gibco, Waltham, MA; cat. 11995-065) with 10% fetal bovine serum (FBS, Sigma, St. Louis, MO, cat. 12306C). Primary FD fibroblasts were purchased from Coriell Cell Repository and cultured in D-MEM with 10% FBS. Both media were supplemented with 1% penicillin/streptomycin (Corning, Corning, NY; cat. 30-009-CI). Cells were incubated at 37 °C with 5% CO2 in a humidified incubator.
Rluc-FD-Fluc Minigene Transfection
HEK-293T cells were seeded in six-well culture plates at 1.20 × 106 cells/well in D-MEM, 10% FBS without antibiotics, and incubated overnight to reach approximately 90% confluence.
Transfection was performed using FuGENE HD reagent (Promega, Madison, WI; cat. E2311) using the FuGENE-DNA ratio at 3.5:1. After 4 h of incubation at 37 °C, cells were plated at a density of 30,000 cells/well in a poly-
Preparation of Splicing Modulator Compound and Treatment of Cultured Cells
SMCs were manufactured by Albany Molecular Research Inc. (AMRI, Albany, NY). A total of 520 kinetin derivatives were synthesized and screened using our assay.
The same batch was split into two separate vials each containing about 1 mg of compound for dissolution into 100% DMSO to yield 40 mM stock solutions. Working solutions (10×) were prepared by dilution to 5% DMSO in phosphate-buffered saline (PBS). The final DMSO concentration in the treated or untreated cells was 0.5%. Kinetin and zeatin were purchased from Sigma (cat. K3253 and cat. Z0164).
Luciferase Assay
HEK-293T cells transfected with Rluc-FD-Fluc minigene were seeded in 96-well plates as described above and treated for 24 h with 10 μL of compounds prepared from 40 mM stocks as 10× solutions and added directly to the cell culture. After treatment the cells were washed once in PBS and lysed for 25 min at room temperature using 50 µL/well of passive lysis buffer (Promega, cat. E1941). Luciferase activity was measured in 20 μL of cell lysate using the Dual-Luciferase Reporter Assay reagents (Promega, cat. E1960) and the GloMax 96 Microplate Luminometer (Promega), following the manufacturer’s instructions and in a 96-well format. The integration time on the luminometer was set at 10 s. Test compounds were serially diluted in DMSO and PBS to generate a concentration–response curve over eight concentrations, with each point run in quadruplicate. The final concentration of DMSO in the media was kept at 0.5%. Cells cultured in the presence of 0.5% DMSO were used as a control and run in each plate in quadruplicate.
Splicing Analysis of ELP1 Transcripts
Semiquantitative RT-PCR was used to analyze ELP1 exon 20 splicing both in the cells transfected with the Rluc-FD-Fluc minigene and in human FD fibroblasts.
RNA was isolated from cells using QIAzol Reagent (Qiagen, Hilden, Germany; cat. 79306) following the manufacturer’s instructions. cDNA was generated using the Superscript III system (Invitrogen, cat. 18080093). cDNA produced from 500 ng of starting RNA was used to perform PCR in a 20 µL volume, using GoTaq green master mix (Promega, cat. MT123). Primers used for amplification were as follows: Rluc-FD-Fluc minigene primers P1 forward (CCTGAGCA GCAATCATGTGT) and P2 reverse (CTCGGCGTAGGTA ATGTCC), which amplify both the WT and MU transcripts generated from the plasmid and not from the endogenous ELP1 ( Fig. 1 ); ELP1 primers Ex19F forward (CCTGAG CAGCAATCATGTG) and Ex23R reverse (TACATGGTCT TCGTGACATC) were used to amplify WT and MU ELP1 transcripts in the FD fibroblast.
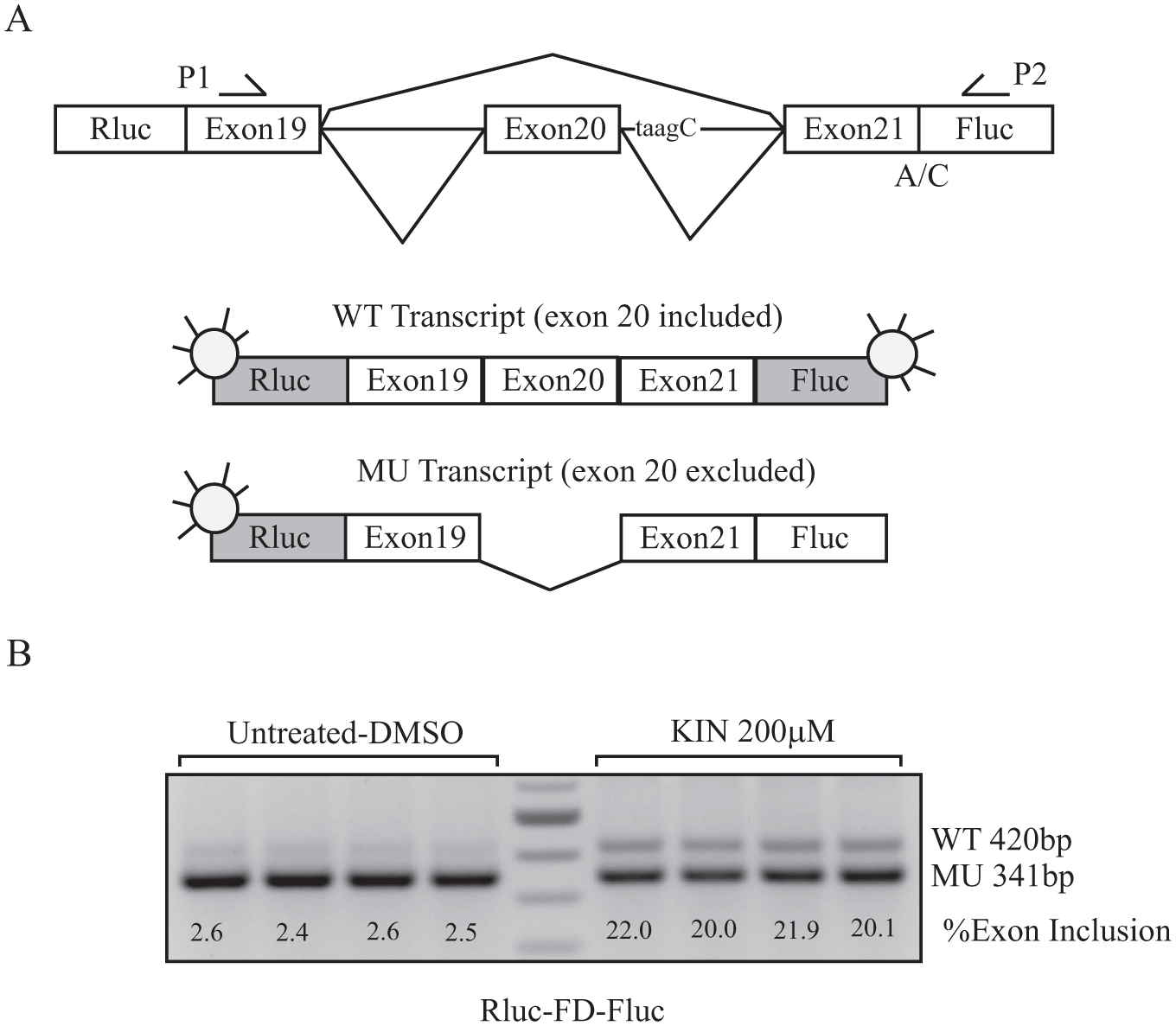
Rluc-FD-Fluc minigene. (
PCR was performed as follows: 32 cycles of (94 °C for 30 s, 58 °C for 30 s, 72 °C for 30 s), products were resolved on a 1.5% agarose gel and visualized by ethidium bromide staining.
WT/MU ratios were obtained using the integrated density value (IDV) for each band, quantified using Alpha 2000TM Image Analyzer and ImageJ software. The level of exon 20 inclusion was calculated as the relative density value of the WT band and expressed as a percentage, as previously described. 7 Validation of the semiquantitative RT-PCR method for the measurement of exon 20 inclusion has been previously demonstrated using RT-qPCR. 7
Firefly Inhibition Assay
Firefly luciferase (Sigma, cat. L9506) was prepared in 500 mM tris-acetate buffer, pH 7.5. The enzyme was diluted as 20 nM in passive lysis buffer one time (Promega, cat. E1941) and 0.1% bovine serum albumin (BSA). Compounds were prepared as described. Three different concentrations were tested for each SMC. After 30 min of incubation at room temperature, firefly activity was measured using luciferase assay reagent (Promega, cat. E1501) diluted three times in water before use with the GloMax 96 Microplate luminometer (Promega). The luminometer was set with an integration time of 10 s. The optimal firefly enzyme concentration and dilution of the luciferase assay reagent have been previously determined to maintain the assay in the linear range ( Suppl. Fig. S2 ).
Screening Cascade
SMCs were first assessed for firefly enzyme inhibition (counterscreen), and compounds that did not inhibit the enzyme (negatives) were advanced through the luciferase assay (primary assay). Compounds positive for firefly inhibition were evaluated using RT-PCR in Rluc-FD-Fluc-transfected HEK-293T cells as described. SMCs that exhibit appropriate activity in the primary assay were considered positive hits and were further characterized in FD fibroblasts using RT-PCR (secondary assay). Each experiment was performed in duplicate using separate compound preparations ( Suppl. Fig. S3 ).
Statistical Analysis
Statistical analysis was performed by ANOVA test with multiplicity correction (Dunnett’s) using GraphPad Prism software (GraphPad Software, La Jolla, CA). p > 0.05 was scored as not significant. *p ⩽ 0.05, **p ⩽ 0.01, ***p ⩽ 0.001, and ****p ⩽ 0.0001 were scored as significant.
Results
Primary Assay: Development of a Luciferase Assay for Rapid Assessment of ELP1 Splicing Correction
To screen newly synthesized kinetin derivatives and identify SMCs with improved efficacy and potency, we developed a luciferase-based splicing assay using an FD minigene construct containing the genomic sequence spanning exons 19–21 of the human ELP1 gene cloned in a pcDNA3.1/V5-His Topo backbone. The FD minigene carries the IVS20+6T>C FD mutation and faithfully models endogenous splicing, with the IVS20+6T>C mutation leading to nearly complete skipping of exon 20 in the transcripts. 23 The minigene splicing efficiency is typically assessed using RT-PCR, which is time-consuming and difficult to scale up for screening purposes. To develop a simple and reliable method to assess splicing, we designed a dual-reporter construct adding firefly luciferase (Fluc) downstream of exon 21 and renilla luciferase (Rluc) upstream of exon 19. An A-to-C mutation was inserted at the ATG start codon of the firefly reporter to inactivate independent translation. The Rluc-FD-Fluc construct has been sequenced, and both reporters were verified to be in frame.
Rluc is expressed each time a transcript is generated from the reporter plasmid, while Fluc is only expressed if exon 20 is included in the transcript, thereby keeping Fluc in frame ( Fig. 1A ). Renilla luciferase is used to normalize for total transcription, cell number, cell viability, and transcriptional off-target effects, whereas firefly luciferase activity is a measure of the level of exon 20 inclusion. Evaluation of the Fluc/Rluc ratio yields the percent exon 20 inclusion in the splicing assays. Similar in vivo (in cells) reporter constructs have been used for measuring increased exon inclusion in SMN2 mRNA.29–31 Splicing of the Rluc-FD-Fluc construct is shown in Figure 1B . In the untreated cells, the IVS20+6T>C mutation leads to nearly complete skipping of exon 20 in the context of the ELP1 minigene (mean of exon 20 inclusion: 2.5 ± 0.1% [n = 4]), whereas after kinetin treatment, exon 20 inclusion increases to 21 ± 1.1% (n = 4).
To confirm that the Fluc/Rluc ratio can be used as a rapid and quantitative measure of exon 20 inclusion, we performed three independent dose–response experiments in HEK-293T cells transfected with Rluc-FD-Fluc and treated with kinetin for 24 h using eight different concentrations, ranging from 0.1 to 400 μM. Next, we calculated the EC50 of kinetin using data from RT-PCR and luciferase expression and compared the results. We measured the ratio of firefly to renilla luciferase activity and plotted the ratio as normalized relative luciferase units (RLU). The three experiment replicates generated clear and reproducible dose–response curves ( Fig. 2A ). The mean EC50 of kinetin was 36.2 ± 3.6 μM (± SD) as determined using the luciferase assay. In order to confirm that the assay was accurately measuring splicing, we quantified exon 20 inclusion using RT-PCR and plotted the dose–response curve of the normalized percent exon inclusion ( Fig. 2B ). As a negative control, we treated cells with zeatin (6-(4-hydroxy-3-methylbut-2-enylamino)purine), a cytokinin that we have previously shown not to affect FD ELP1 splicing. 23 As expected, zeatin treatment did not change splicing of the minigene ( Fig. 2C , D ). The correlation between the luciferase and the RT-PCR assay results was evaluated by plotting the RLU values obtained from the luciferase primary assay against the normalized percent exon inclusion calculated by RT-PCR, yielding an R2 of 0.93 ( Fig. 2E ).
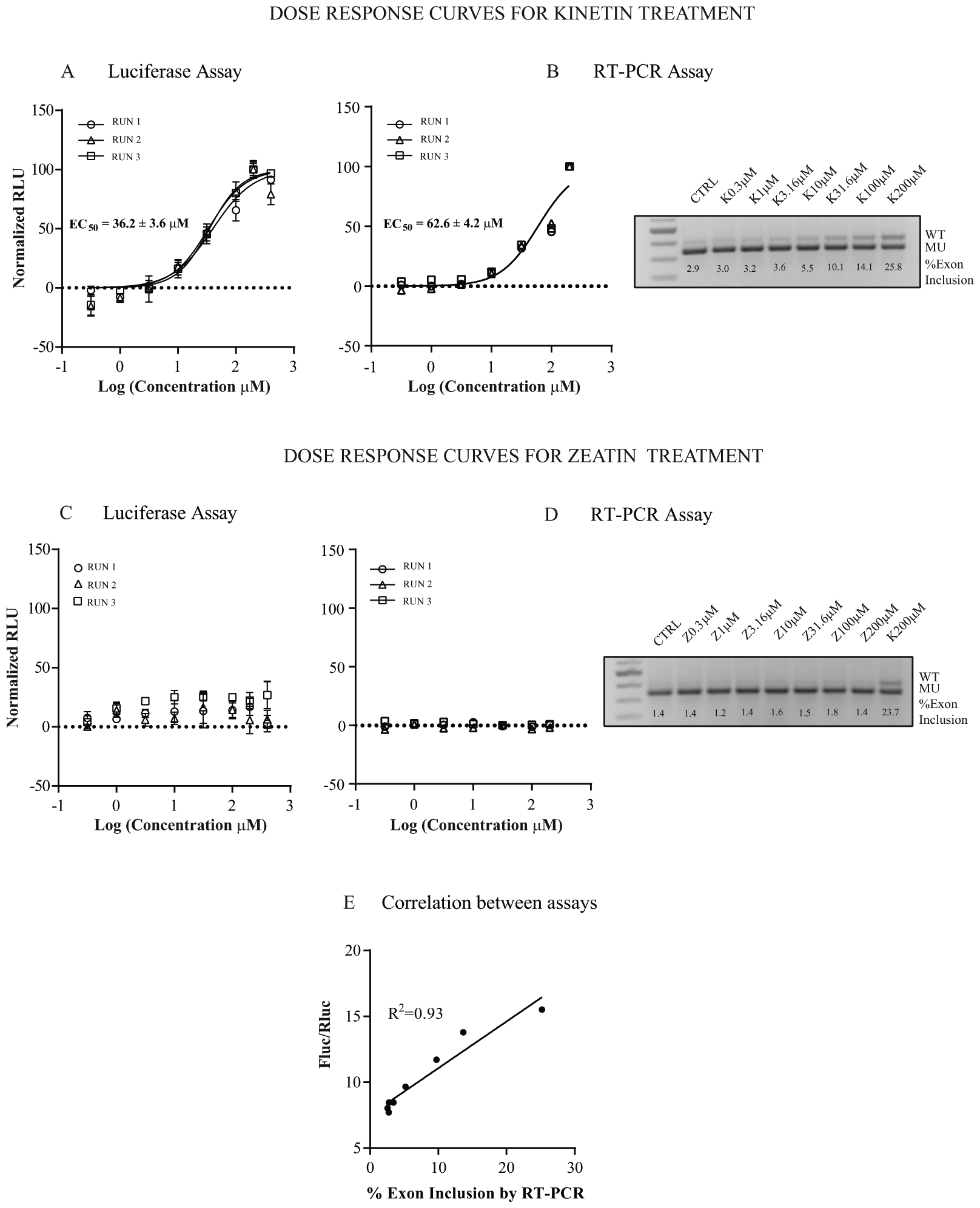
Dose–response curves for kinetin (
These data confirm the Fluc/Rluc ratio as an accurate measurement of splicing efficiency and a valid screening measure to identify SMCs that change exon 20 inclusion.
Luciferase Assay Optimization
To evaluate the performance of the luciferase assay, we tested positive and negative controls in 96-well plates and verified signal strength, reproducibility, and well-to-well percentage coefficient of variation (% CV). In addition to kinetin, we verified the performance of the assay by using a previously identified SMC, C-111, which displayed a 50-fold increase in efficacy ( Suppl. Fig. S1 ). Transfections were performed on three consecutive days in duplicate plates. In each plate, half of the wells were treated with C-111 and half with DMSO as a negative control. The results shown in Figure 3A confirmed that the assay is consistent across independent experiments. The calculated RLU is consistent across independent experiments, and the CVs for Fluc and Rluc were never greater than 20%. We calculated the Z′ value of Fluc activity and obtained an average value of 0.5, indicating a robust assay. We also performed a test–retest reliability assay by calculating EC50 values for 16 different compounds in two independent experiments ( Fig. 3B ). The test–retest reliability showed an R2 of 0.99, indicative of a high level of reproducibility.
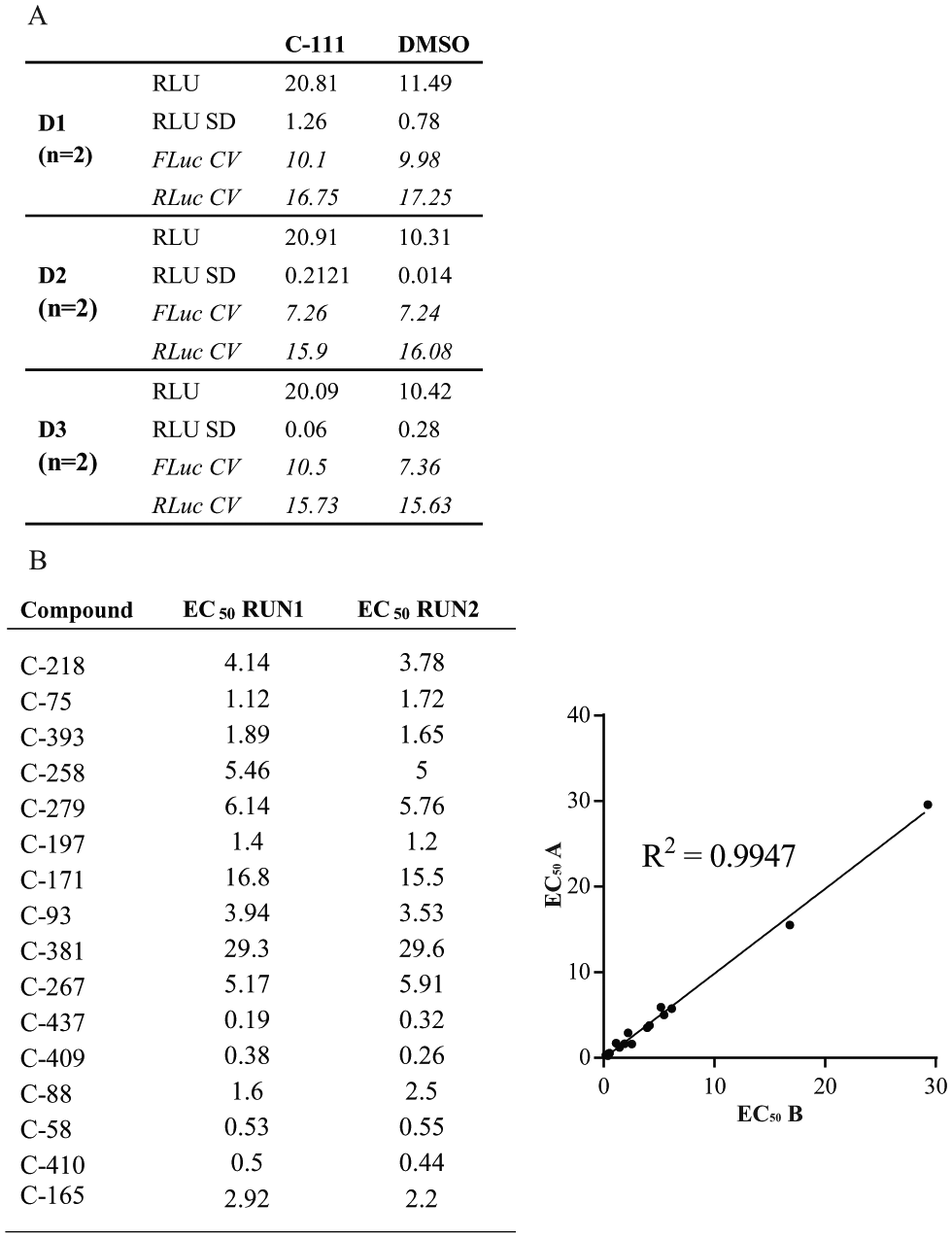
Luciferase assay optimization. (
Development of a Counterscreen to Identify Firefly Luciferase Inhibitors
While luciferase reporter assays are frequently used to assess gene activity, it is important to recognize that some molecules can directly increase luciferase reporter activity leading to nonspecific but highly reproducible dose-dependent activity.32,33 To avoid false positives, we developed an in vitro assay to measure changes in firefly enzymatic activity after incubation with SMCs. The concentrations of the enzyme and the substrate used in the assay were selected to be in the linear range of the assay ( Suppl. Fig. 2S ).
As shown in Figure 4 , compounds C-57 and C-438 had a statistically significant inhibitory effect on firefly activity at all concentrations ( Fig. 4A , D ), and they both showed reproducible activity in the luciferase assay ( Fig. 4B , E ). Nevertheless, this activity does not correspond to variation in exon 20 inclusion of the Rluc-FD-Fluc minigene as shown using RT-PCR analysis ( Fig. 4C , F ).
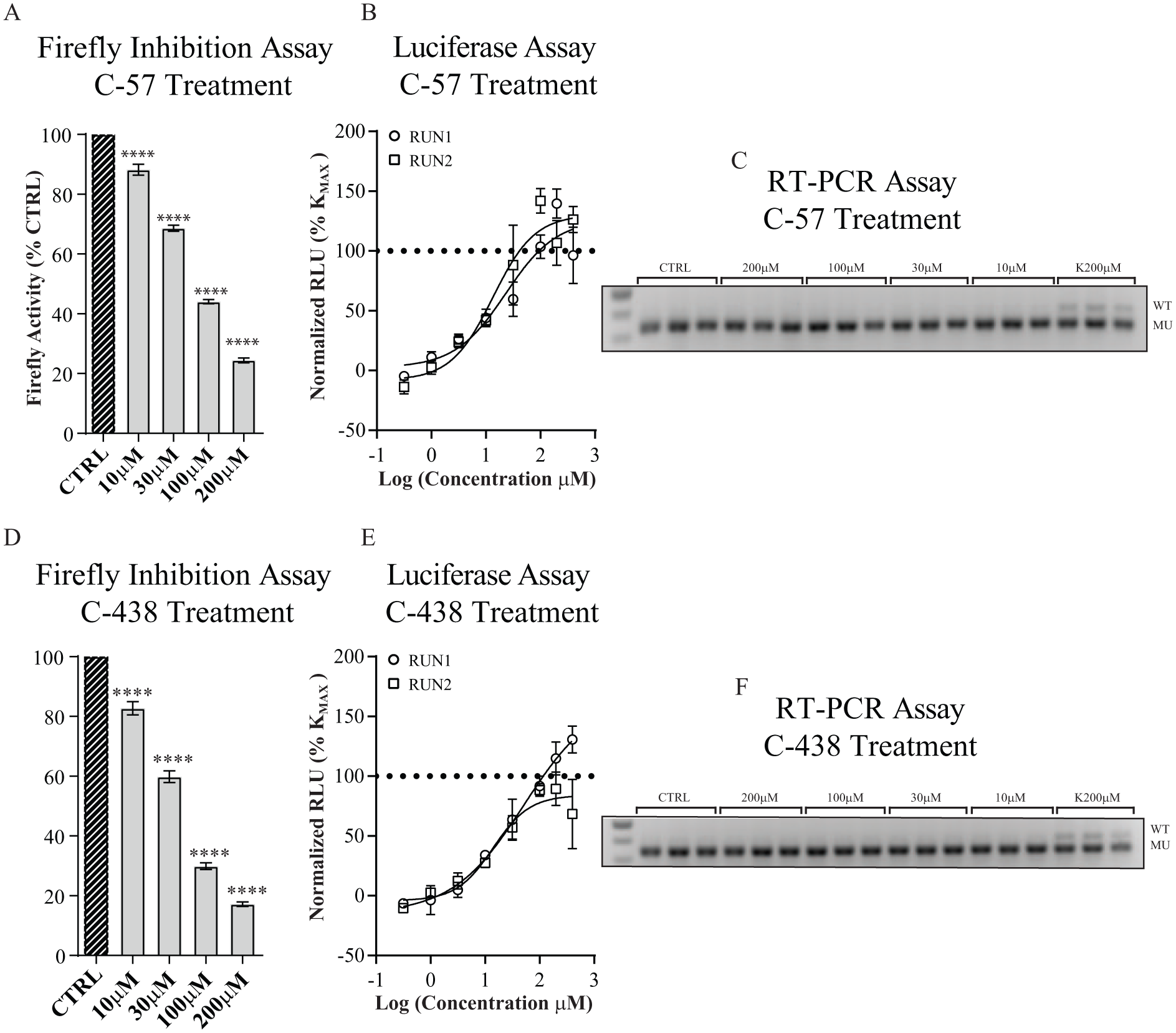
Effect of firefly inhibition on luciferase assay outcome. (
Therefore, the firefly inhibition assay was added as a counterscreen to minimize the occurrence of false positives. Compounds positive for firefly inhibition were assayed using RT-PCR rather than the luciferase assay.
Introduction of ECK to Rank-Positive Hits
To identify SMCs that are more potent and efficacious than kinetin, we coined the term ECK, which we define as the concentration at which the compound efficacy reached 100% of kinetin’s maximum activity (KMAX). This value proved more informative for ranking positive hits than EC50. As shown in Figure 5 , compound C-218 ( Fig. 5A ) and compound C-93 ( Fig. 5B ) have the same EC50; however, C-93 does not reach KMAX at any of the concentrations tested. Therefore, the ECK is not calculable. Compound C-218 is 37 times more potent than kinetin and has a calculated ECK of 5.4 ± 1.4 μM. Hence, we concluded that to classify the potency of our compounds, ECK is more appropriate than EC50. In total, 214 compounds were more active than kinetin in the primary assay.
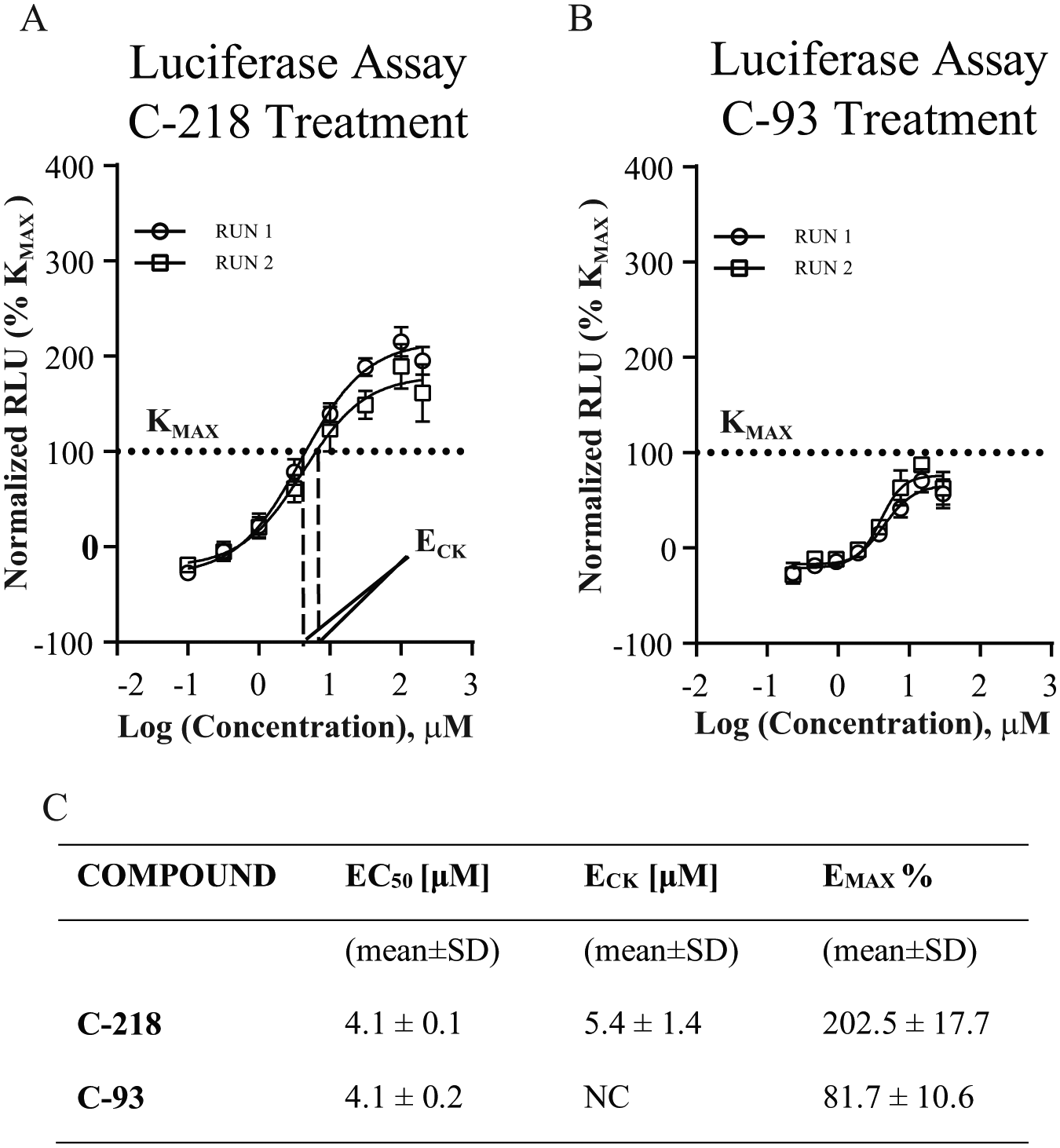
ECK. Rluc-FD-Fluc-transfected HEK-239T cells were treated for 24 h with SMCs generating a concentration–response curve over eight points. Measurements were performed in quadruplicate and plotted as the mean ± SD (n = 2). Curves were created by nonlinear regression using Prism4 (GraphPad Software). RLU were normalized and expressed as the percentage of kinetin maximum activity (KMAX) (
Secondary Assay: ELP1 Splicing Analysis in Patient Fibroblasts
The primary assay identifies SMCs that modify splicing of exon 20 in the minigene construct. To confirm their activity on the endogenous ELP1 pre-mRNA splicing, we tested a select group of SMCs in FD patient-derived fibroblasts using RT-PCR. We chose compounds with ECK values ranging from 0.2 to 6.8 μM (30–1000 more potent than kinetin). FD fibroblasts were treated for 24 h using two concentrations for each of the SMCs, and ELP1 splicing was analyzed using RT-PCR. Exon 20 inclusion was determined using band IDVs as previously described, and values were normalized to maximum kinetin activity (complete exon 20 inclusion is achieved at a concentration of 200 μM).
SMCs are ranked by potency in the secondary RT-PCR assay in Figure 6B , and the ECK determined using the primary luciferase assay is shown in Figure 6A , confirming that the primary assay accurately reflects compound potency in FD fibroblasts. For example, the most potent compound, C-437 (2-chloro-8-((3,3-difluorocyclobutyl)methoxy)-N-(thiazol-2-ylmethyl)-9H-purin-6-amine) is eight times more potent than C-165 (2-chloro-8-(2-methoxyethoxy)-N-(pyrimidin-4-ylmethyl)-9H-purin-6-amine) in the primary assay, and similar relative potency ranking was observed in patient fibroblasts.
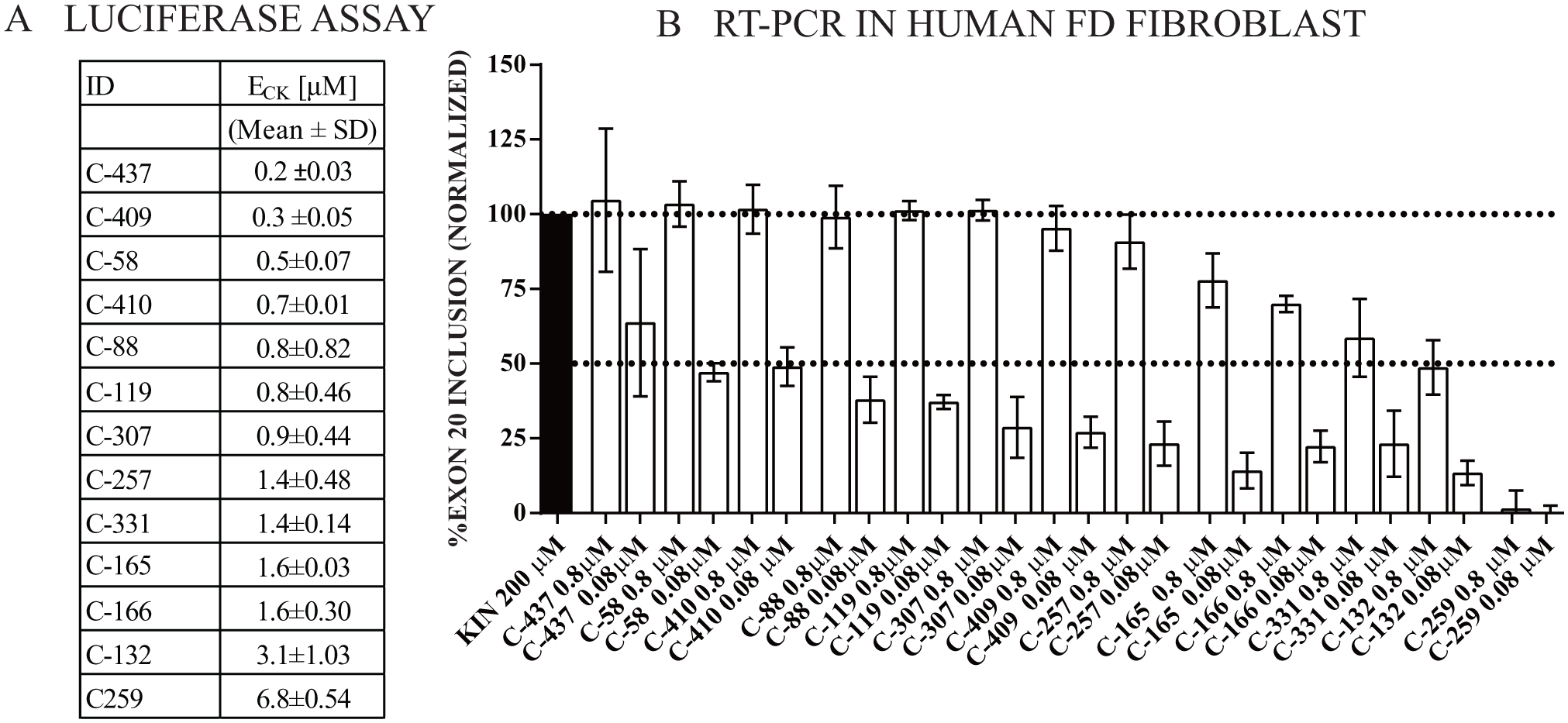
Secondary assay. Luciferase assay hit validation. (
In summary, we have generated a reliable and robust luciferase splicing assay that can be used to rapidly screen for ELP1 splicing modulators. We introduced ECK to compare splicing modulation efficiency of SMCs and to identify compounds that are more potent and active than kinetin. ECK can easily be used as a tool for prioritizing compounds for further analysis. More importantly, the results from our secondary assay using FD fibroblasts showed a striking similarity to those obtained in the primary reporter assay, supporting the validity of our newly developed drug screening approach.
Discussion
FD is a rare and fatal autosomal recessive disease that impairs the development of sensory and autonomic neurons. Because of the pervasive autonomic and sensory dysfunction, FD patients suffer severe morbidity and mortality. At present, FD is an incurable disease and available treatments are only supportive, often failing or generating unwanted side effects.34–36 Clearly, FD patients need new and better therapies. Development of a drug to precisely target the molecular defect underlying the disease would be a major advance and could potentially shift treatment paradigms for FD patients.
The recent progress in the development of therapies for spinal muscular atrophy (SMA), another genetic disorder caused by a splicing alteration, has validated the utility of splicing modification as a valuable therapeutic strategy for neurologic diseases.37,38
The advantage of targeting splicing defects pharmacologically is that, unlike loss-of-function mutations, the cells retain the capacity to make functional protein. Previously, we have shown that the plant cytokinin kinetin can restore normal levels of protein expression in patient cell lines,23,24 modify splicing in vivo in a mouse model in all tissues tested, and increase ELP1 protein production in brain. 25 However, the limited results obtained in a short-term safety trial of kinetin in FD patients highlighted the need for additional drug candidates that are more potent and efficacious than kinetin.26,27 Here we have described a rapid, reliable, and reproducible cell-based splicing assay to identify SMCs that are more potent and active than kinetin.
The study of factors that affect mammalian splicing is generally done by transfection of gene fragments (minigenes) into cells, followed by analysis of the mRNA transcripts generated. Direct analysis of the transcripts using RT-PCR is laborious and difficult to scale up for screening purposes. On the other hand, the use of a single-reporter system based on the successful expression of a protein after splicing is susceptible to variations between samples caused by differences in transfection efficiency, cell growth, transcription, or processing. To bypass variables that could potentially affect a single-reporter assay, we engineered a dual-reporter system using a previously validated minigene. Yoshida et al. used a dual-color splicing reporter system to identify a compound more potent than kinetin, RECTAS. 28 We decided to use a luciferase-based assay because it offers several advantages in the development of high- and medium-throughput screening due to broad linearity, robustness in complex biological samples, and high sensitivity. 39 The versatility of the luciferase assay allowed us to develop a systematic method to screen and categorize compounds based on rigorous parameter quantification. As expected, we identified RECTAS as one of the 214 compounds with improved potency over kinetin.
We have tested the luciferase assay in several ways to show that it reflects changes in the splicing efficiency. Our results showed that the Rluc-FD-Fluc minigene is responsive to kinetin but not to the splicing-inactive cytokinin zeatin, and that luciferase activity can be used as a quantitative measurement of exon 20 inclusion. These measurements showed very strong correlation with the exon inclusion values obtained using RT-PCR and band density analysis. Our cell-based assay has been optimized to obtain high reproducibility and sensitivity, and we established an efficient counterscreen method to avoid false positives due to possible interferences with the reporter system.
We had originally developed this assay to identify SMCs that are more potent than kinetin whose activity represents the required cutoff in the assay. To quantify the level of splicing optimization obtained, we introduced ECK as a convenient and more reliable measure of compound potency and efficacy relative to kinetin. The introduction of ECK allowed us to rank and classify our hits in the luciferase primary assay. Minigene constructs are useful tools for in vivo studies of splicing mechanism and regulation, but an important caveat to consider is that splicing of an exon in this artificial context might be quantitatively different from that in the context of the full-length pre-mRNA. 40 Hence, to confirm the hits obtained with our primary assay, we have established a secondary assay analyzing the effect of the SMCs in FD patient fibroblasts. The noteworthy concordance between splicing modulation activity measured with the primary and the secondary assay demonstrates the validity of our screening method. Furthermore, the secondary assay confirmed that ECK is a quantitative measure of the activity of SMCs not only in the minigene splicing assay but also in FD patient fibroblasts.
Using this assay, we screened more than 500 new chemical entities and identified several compounds that are significantly more active and potent than kinetin in both primary luciferase and secondary patient cell-based assay.
These results demonstrate that the Rluc-FD-Fluc minigene reporter can be used as a tool to systematically identify and characterize compounds that correct ELP1 splicing. Our work represents the first fundamental step in the drug development process, a major advancement in the pursuit of a better therapy for FD patients.
Supplemental Material
Supplemental_Material_for_Devel_Screen_Platf_to_ident_small_molecules_modyf_splicing_in_FD_by_Salani_et_al – Supplemental material for Development of a Screening Platform to Identify Small Molecules That Modify ELP1 Pre-mRNA Splicing in Familial Dysautonomia
Supplemental material, Supplemental_Material_for_Devel_Screen_Platf_to_ident_small_molecules_modyf_splicing_in_FD_by_Salani_et_al for Development of a Screening Platform to Identify Small Molecules That Modify ELP1 Pre-mRNA Splicing in Familial Dysautonomia by Monica Salani, Fabio Urbina, Anthony Brenner, Elisabetta Morini, Ranjit Shetty, C. Scott Gallagher, Emily A. Law, Sara Sunshine, Dylan J. Finneran, Graham Johnson, Lisa Minor and Susan A. Slaugenhaupt in SLAS Discovery
Footnotes
Acknowledgements
We thank the NIH Blueprint lead development team members, Dr. Charles Cywin, Dr. Bill Paquette, Dr. Ronald Franklin, Dr. Ronald White, Jamie Driscoll, Dr. Juan Marugan, Dr. Enrique Michelotti, Dr. Amir Tamiz, Dr. Bruce Molino, Dr. Douglas Kitchen, Keith Barnes, Dr. Katrina Gwinn, and Dr. Rebecca Farkas, for their long-standing collaboration and helpful discussions.
Declaration of Conflicting Interests
The authors disclosed the following potential conflicts of interest with respect to the research, authorship, and/or publication of this article: Susan A. Slaugenhaupt consults for PTC Therapeutics, Inc.
Funding
The authors disclosed receipt of the following financial support for the research, authorship, and/or publication of this article: This work was supported by National Institutes of Health grants (1U01NS078025-01 and 5R37NS095640-03 to S.A.S.).
Supplemental material is available online with this article.
References
Supplementary Material
Please find the following supplemental material available below.
For Open Access articles published under a Creative Commons License, all supplemental material carries the same license as the article it is associated with.
For non-Open Access articles published, all supplemental material carries a non-exclusive license, and permission requests for re-use of supplemental material or any part of supplemental material shall be sent directly to the copyright owner as specified in the copyright notice associated with the article.