
Abstract
PRRs are sentinels of the innate immune system, with TLRs being the most important. Assays for TLR ligand interactions have been used to gain insights into their function and signaling pathways. As significant differences exist between species with regard to ligand recognition, it is necessary to adapt these tools for TLRs of other species. In the present work, we describe a species-specific cell-based assay adapted for the analysis of single PRRs. Human embryonic kidney 293T cells were stably transfected with the NF-κB-inducible reporter gene secreted embryonic alkaline phosphatase (SEAP) together with bovine TLR2. We compared the SEAP response with an existing luciferase NF-κB reporter assay for correlation with IL-8 production. A dose-dependent response was detected upon stimulation using both methods with good correlation to IL-8 secretion. Lower stimulant concentrations were detected by SEAP assay than IL-8 secretion. The luciferase assay produced high non-specific background for all ligand concentrations. Of all assays tested, we found the bovine-specific SEAP reporter assay to be the most convenient and delivered results in the shortest time. The developed reporter cell line would lend well to rapid, high-throughput TLR ligand screening for cattle.
Introduction
The innate immune system is the first line of defense against pathogenic microbes. It can be classified into two parts: the afferent (sensing) and the efferent (effector) system. PRRs like TLRs, dectins, CD14 or NLRs belong to the afferent system, together with the humoral components, for example LPS-binding protein, collectins and C3b. 1 PRRs are involved in early detection of specific pathogens because they recognize highly conserved and class-specific motifs called microbe associated molecular patterns. The best characterized class of the PRRs is the evolutionarily conserved TLR family, first discovered in Drosophila. 2 In most mammalian species 10 TLRs have been described to date and they differ from each other in expression patterns, ligand specificity and in the target genes they induce. 3 TLRs control the activation of innate immunity and induce activities against invading pathogens by the production of inflammatory cytokines. They link innate and adaptive immunity through the induction of antigen-presentation and co-stimulatory molecules.
TLRs are expressed on a variety of immune and non-immune-related cells. 4 Targeting TLRs with specific ligands shows great potential for application as vaccine adjuvants.5,6 Whereas ligand specificities for most TLRs are fairly well described, novel interactions are continuously being discovered; 7 however, no known ligand has been described for TLR10 in any species. Despite the fact that TLRs are highly conserved between mammals, most available information on TLR function was generated in murine or human systems and cannot be readily extrapolated to other, economically important farm or companion animal species. 8 The identification of new vaccine targets/adjuvant and immunomodulatory targets is becoming an increasing aim of research, given the increasing abuse and subsequent ban of antimicrobial compounds for use in farm animals. 9 For these reasons, several methods have been developed to screen potential TLR ligands in a fast and reproducible manner. The majority of these assays rely on the ability of a known ligand to stimulate transcription factors such as NF-κB, activating protein-1 or interferon regulatory factor family. 10 To do so, cellular assays involving either primary immune cell subsets or cell lines expressing specific TLRs are normally employed. Testing TLR activation in vitro with established cell lines is easier to standardise and effectively reduces the use of experimental animals. Although such lines may not be as biologically relevant as primary cells, ligand specificity of the receptor in question can be tested and compared regardless of which downstream events are activated. The decision of cell lines to be used has to take endogenous TLR expression, need for potential co-receptors and unresponsiveness to standard ligands, such as LPS into account. Transfection of such cell lines with specific TLRs allows for end products of activation (transcription factors, cytokines) to be detected and quantified.
Indeed, several colorimetric reporter gene assays have been developed allowing for the detection of transcription factor activation. Luciferase-based assays, such as dual luciferase assays are among the most popular reporter assays used for PRR activation. 11 Here, TLR-specific activation of NF-κB is measured using a reporter gene coding for the firefly (Photinus pyralis) luciferase, which is measured against the activity of the sea pansy (Renilla reniformis) luciferase using a luminometer. Another popular assay utilizes secreted embryonic alkaline phosphatase (SEAP) as a reporter gene. The application of SEAP as a reporter for TLR activation was introduced by Burger-Kentischer et al., 12 with the aim of substituting the Limulus amoebocyte lysate test for endotoxin and pyrogen detection. The advantage of this assay is that it does not rely on a luminometer to detect activation of NF-κB, but rather on a colorimetric change based on the activity of alkaline phosphatase (AP), which can be either detected by eye or quantified using a conventional ELISA plate reader. The SEAP assay has been further developed and stable reporter cell lines expressing human or murine TLRs are commercially available, along with substrate containing cell culture media enabling real-time quantification of SEAP activity; however, the assay has to be carefully controlled to avoid detection of endogenous AP activity from cells or culture media.
Lastly, activation of transcription factors can also be assessed by analysing production of downstream molecules, such as secreted cytokines/chemokines. This method has the advantage that it can be easier correlated with the response in primary cells but requires a direct correlation between transcription factor activation and cytokine/chemokine protein production.
In the present study, we compared the three different test systems mentioned above using luciferase, SEAP and IL-8 production to assess bovine TLR2 activation, for which we already had identified species-specific ligand differences. 13
Materials and methods
Generation of SEAP reporter cell line
All cell lines used in these experiments were generated using human embryonic kidney 293T cells, kindly provided by Dr. Bradley Cobb (The Royal Veterinary College, University of London), and were negative for endogenous TLR1, 2 and 6 expression, as established by RT-PCR and flow cytometry (data not shown). The cells were maintained in complete 293T medium; DMEM supplemented with GlutaMAX (Gibco, Paisley, UK), 10% FBS (Sigma, St. Louis, MO, USA), 100 IU/ml–100 µg/ml penicillin–streptomycin (Gibco) and 1 mM sodium-pyruvate (Sigma). The vector pNifty2-SEAP (Invivogen, San Diego, CA, USA) was transfected into 293T cells using TurboFect reagent (Thermo Scientific, Waltham, MA, USA) according to the manufacturer’s protocol for six-well plates. Transfectants (293T/SEAP) were clonally selected according to the method described by Burger-Kentischer et al., 12 using 100 µg/ml Zeocin (Invivogen) and maintained in complete selective medium afterwards. Endogenous SEAP activity was found to be minimal in response to a panel of TLR ligands for 293T and 293T/SEAP cells (data not shown).
Generation of bovine TLR2-expressing 293T and 293T/SEAP reporter cell lines
The bovine TLR2 gene was cloned into the mammalian expression vector pTracer-CMV/Bsd (Thermo Scientific) and transfected into 293T and 293T/SEAP reporter cells, followed by clonal selection using 10 µg/ml blasticidin (Sigma-Aldrich, St. Louis, MO, USA).
13
TLR2 expression was confirmed by flow cytometry (Figure 1). Both cell lines were maintained in complete selective medium (containing zeocin and blasticidin for 293T/SEAP/TLR2 and blasticidin only for 293T/TLR2 cell lines).
Bovine TLR2 expression of reporter cell lines. Surface expression of TLR2 was detected by staining 293T, 293T/TLR2, 293T/SEAP and 293T/SEAP/TLR2 cells using human anti-bovine CD282 Abs (shaded grey) conjugated with Alexa Fluor 647 (BioRad, Hercules, CA, USA). HuCAL Fab-dHLX-MH Ab (BioRad) was used as negative control, as recommended by the manufacturer (dotted outline). Then, 10,000 events were recorded and single cells (> 80% of all events) were gated at analysis using FlowJo version 10.1 (TreeStar, Ashland, OR, USA). Data were acquired with CellQuest Pro Version 6.0 (BD Biosciences, San Jose, CA, USA) using a FACSCalibur E3160 cytometer. One representative experiment of three is shown.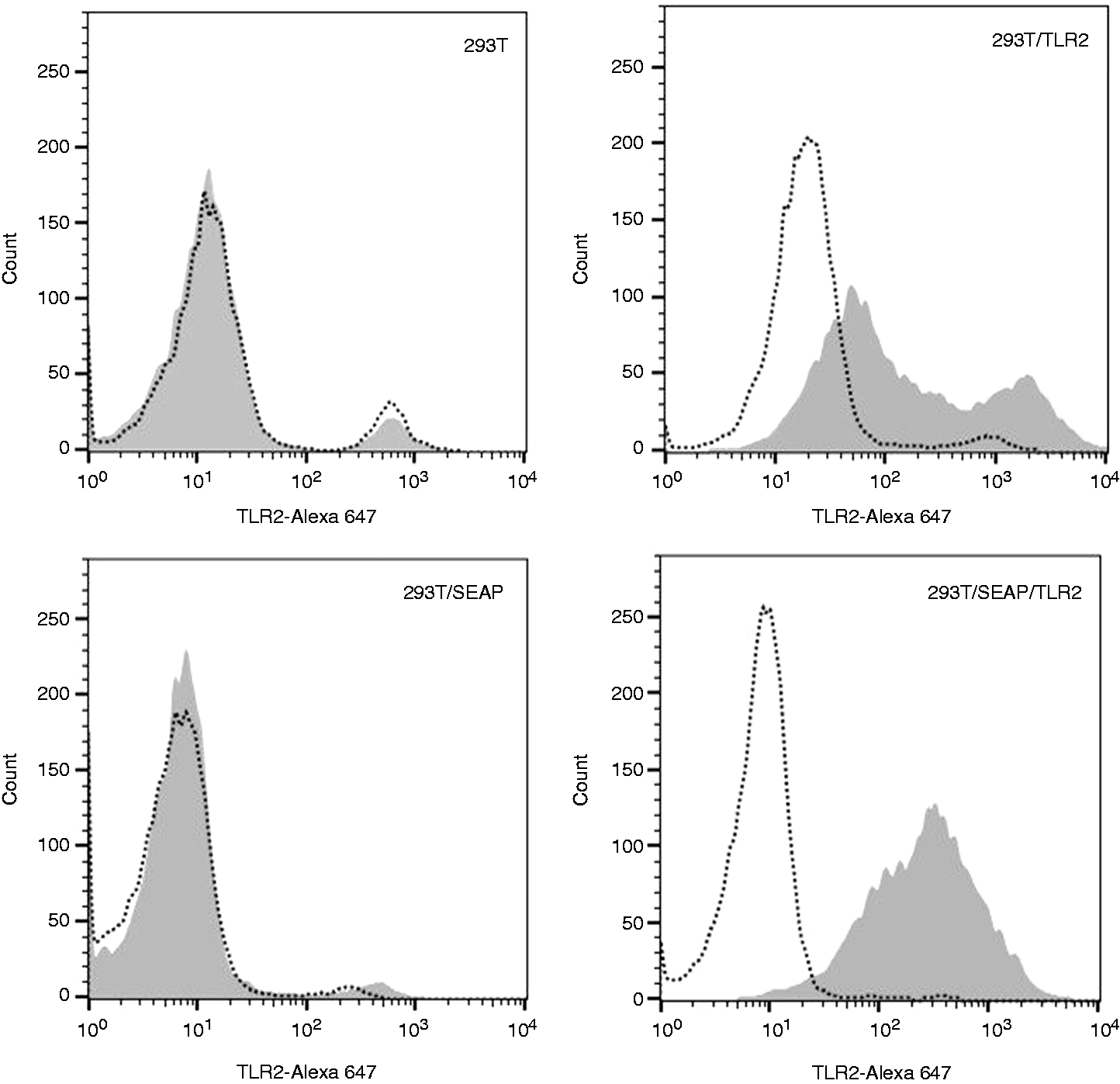
Stimulation assay of 293T/SEAP/TLR2 reporter cells
293T/SEAP/TLR2 cells were seeded into a 96-well plate at 2 × 104, 3 × 104 and 4 × 104 cell/well densities in 200 µl/well of complete selective media (using 12 wells of each concentration), and incubated in a 37℃ 5% CO2 humidified incubator. After 24 h, culture medium was exchanged for stimulation media (DMEM-Glutamax, supplemented with 2% FBS and 1 mM sodium-pyruvate) and 1, 10, 100, 500 or 1000 ng/ml FSL-1 (Invivogen), 200 ng/ml phorbol 12-myristate 13-acetate (PMA; Invivogen) or no stimulant in duplicate wells. FSL-1 is a synthetic lipoprotein derived from Mycoplasma salivarium, used as a specific TLR2 agonist, and PMA is a non-specific NF-κB stimulator, added as positive control with optimal stimulation levels previously determined (data not shown). Following an additional 24 h incubation, supernatants were collected and stored at −20℃ until measurements of SEAP activity and IL-8 production. SEAP activity was measured in a 96-well clear flat-bottom cell culture plate; 10 µl of each sample supernatant was added to 200 µl prewarmed QuantiBlue reagent (Invivogen) and incubated at 37℃. After 24 h, ODs at 635 nm were measured using a Tecan Infinite M200 Pro plate reader. IL-8 production was measured with Quantikine Human CXCL8/IL-8 ELISA kit (R&D Systems, Minneapolis, MN, USA) according to the protocol suggested by the manufacturer. Fifty µl of each cell supernatant was used as sample and absorbance was measured at 450 nm.
Stimulation assay of 293T/TLR2 cells
293T and 293T/TLR2 cells were seeded at 105 cell/ml density in 4 ml complete media/well, into three wells each on a six-well plate. After 24 h incubation, one well of each cell line was transfected with 500 ng luciferase reporter plasmid NF-κB -Luc, 14 4 µg pNifty2-SEAP or mock (4 µg irrelevant plasmid vector). DNA amounts were previously optimized (data not shown). After an additional 24 h, the supernatants were discarded, the cells lifted and re-seeded into 12 wells on a 96-well plate at 4 × 104/well density. Stimulation media was prepared with 125, 250, 500 or 1000 ng/ml FSL-1, 200 ng/ml PMA or no stimulant. After 24 h of stimulation, luciferase activity from mock transfected cells and cells transfected with luciferase reporter gene was measured as described by Patterson et al., 14 and supernatants were frozen until further processing. Human IL-8 levels and SEAP activity were assayed as described above.
Statistical analyses
For each assay, data collected from three biological repeats were combined, except for the IL-8 ELISA from stable reporter cell supernatants, where measurements from wells seeded with 4 × 104 cells were repeated. Calculations were performed using Prism 6 for Windows (GraphPad Software Inc). OD values imported from ELISA results were extrapolated using four-parameter logistic regression. Invalid results were excluded from further analyses. OD values from SEAP assay were used as results in further comparisons. Signals measured throughout 10 s of the luciferase assay plate were averaged separately for each well. Multiple unpaired t-tests with Bonferroni correction were used to compare responses within measurement groups between stimulant doses. Spearman’s correlation was computed and Deming’s regression was plotted to compare methods separately for biological repeats.
Results and discussion
Responses of stable reporter cells
In order to develop a fast screening assay to identify bovine-specific TLR ligands, we created a stable bovine TLR2-expressing 293T SEAP reporter cell line, by inserting the reporter gene first, followed by the introduction of bovine TLR2. The 293T/SEAP/TLR2 cells responded to the synthetic lipopeptide FSL-1 in a dose-dependent manner (Figure 2a, b). Within the combined data from three repeats, the lowest detectable, although not significant, specific ligand dose was 1 ng/ml for all seeding densities. Cells seeded at 4 × 104/well gave the most pronounced OD differences for each FSL-1 concentration analysed. In contrast, when cells were seeded at 2 × 104 or 3 × 104/well, we observed a stagnating signal detection above 500 ng/ml FSL-1 dose, probably because lower amounts of cells reached maximum binding capacity at lower ligand concentrations, resulting in a lower overall SEAP activity. Using the same supernatant, we detected the first significant increase in IL-8 concentration when stimulated with 500 ng/ml FSL-1.
Results of reporter cell line stimulation and comparison of SEAP and IL-8 assays. (a) Dose-dependent SEAP activity in 293T/SEAP/TLR2 cells at different seeding densities, the first significant increment compared with unstimulated cells indicated in each group. (b) Dose-dependent IL-8 production in 293T/SEAP/TLR2 cells with the first significant increment compared to unstimulated cells indicated. Combined data of three biological repeats are shown (mean ± SEM). (c) Correlation of ODs measured in SEAP and ELISA assays of the same samples, fitted with Deming’s regression line and equation are shown for one representative experiment. Levels of significance: *P < 0.05; **P < 0.01; ***P < 0.001.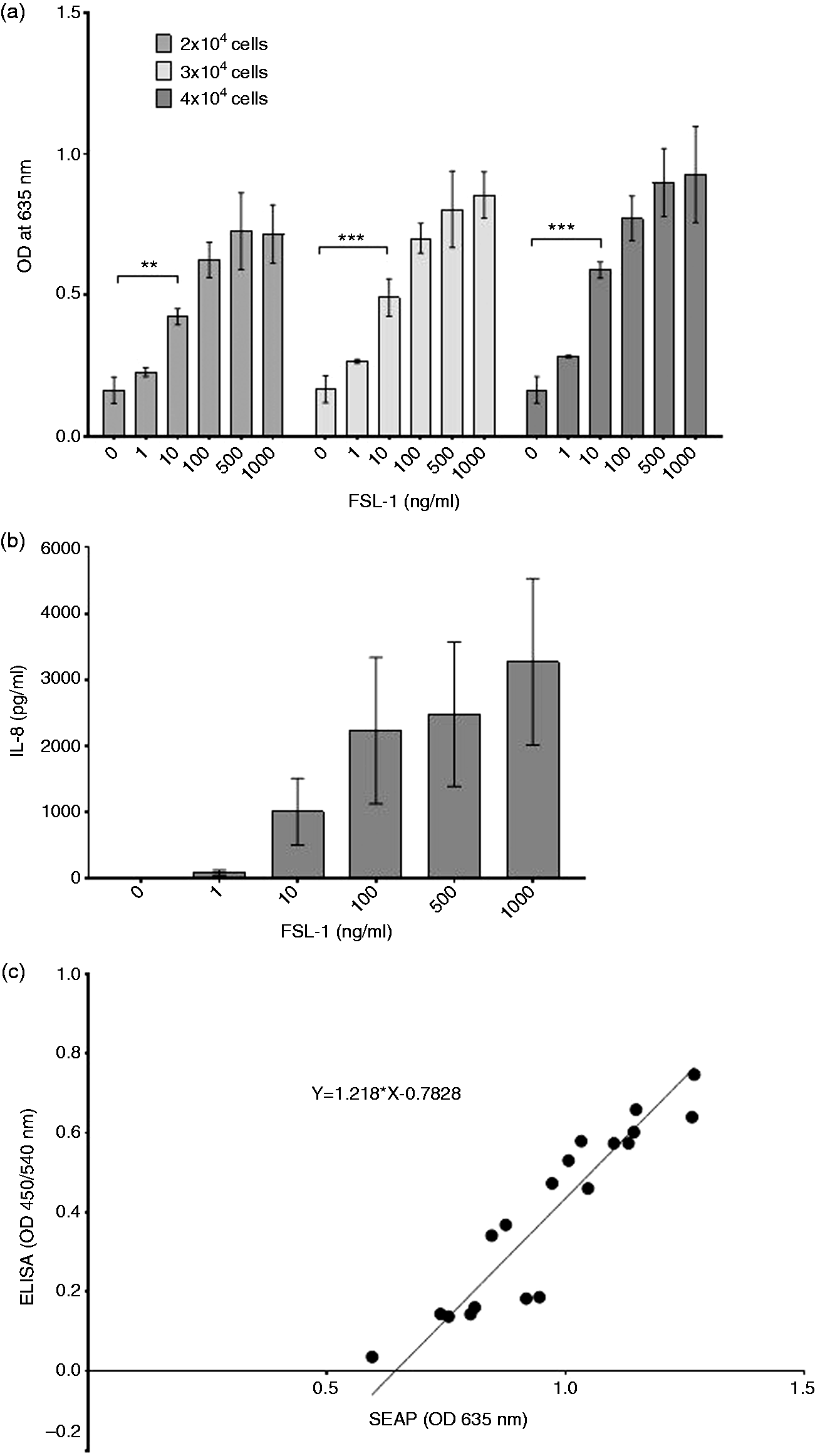
Owing to the fact that two different substances (SEAP vs. IL-8) were analysed, we could not apply Bland–Altman plot and Passing–Bablok regression, which are normally used to compare different measurement methods. Instead, a Spearman’s correlation between SEAP and IL-8 was used and shown to be significant (r = 0.9596, P < 0.0001). To demonstrate this correlation, a dot plot fitted with a Deming line was drawn (Figure 2c). The regression line shows a considerable shift along the x-axis (depicting SEAP values) towards the right (X = 0.6427 when Y = 0). Given that IL-8 values started to rise only at higher FSL-1 concentration, this shift was expected; however, at higher concentrations, the correlation remained strong between SEAP and IL-8 production. ODs measured in supernatants from cells stimulated non-specifically with PMA were at least 2.7-times higher (SEAP assay) or 17-times higher (ELISA) than those measured in non-stimulated cell media, validating the assays.
Stimulations of transient reporter cells
As the luciferase based reporter assay is based on a transient transfection system, we next compared the SEAP reporter system in a transient form with the luciferase system and IL-8 ELISA. To do so, we generated a bovine TLR2-expressing 293T cell clone (293T/TLR2), which was subsequently transiently transfected with luciferase or SEAP reporter genes, or mock transfected. IL-8 production was measured in all 293T/TLR2 cell clones. The results are presented in Figure 3. Similar to the data obtained using a stable transfection system, we measured a dose-dependent ligand response in the cells expressing TLR2 and transfected with the SEAP reporter gene, but not in cells lacking either the TLR, the reporter gene or both (Figure 3a). In contrast, transient transfection with the luciferase reporter gene resulted in a relatively high background in cells containing the reporter gene but lacking TLR2 expression (Figure 3b). This activity did not seem to be influenced by exposure to any of the FSL-1 concentrations used (Spearman’s r = 0.7; P = 0.2333). Lack of measurable IL-8 production in these cells (apart from the positive control) confirmed that this background reporter activity is unlikely to be NF-κB activation resulting from either endogenous TLR expression in undetectable levels or by non-specific means. As such, IL-8 levels were incomparable to those measured in cells where luciferase and also TLR2 were present (Figure 3c). ELISA results were valid in all 293T/TLR2 replicates and IL-8 production was induced in all TLR2 expressing cell clones (Figure 3d). Interestingly, the highest amount of IL-8 production was seen in mock-transfected 293T/TLR2 cells. One potential explanation for this observation is competition for NF-κB between binding sites. The high amount of vector DNA containing NF-κB-responsive elements introduced into transfected cells (500 ng/well in case of pGluc and 4 µg/well in case of pNifty-2) can engage a substantial amount of the transcription factor before translocation to the nucleus, while in mock transfected cells, all of these molecules can be employed in pro-inflammatory cytokine stimulation. In 293T/TLR2 cells, both reporter assays correlated well with IL-8 production (Spearman’s r = 0.9879 and P < 0.0001 in case of SEAP; and r = 0.7939 and P = 0.0088 with the luciferase assay within one biological repeat).
Results of FSL-1 stimulation in transient reporter assays and IL-8 ELISA. (a) Dose-dependent SEAP activity in TLR2-expressing cells transfected with SEAP reporter gene (293T/TLR2/pNifty), but not in cells lacking either (293T/pNifty; 293T/TLR2/mock) or both (293T/mock). (b) Luciferase activity measured in TLR2-expressing cells transfected with reporter gene (293T/TLR2/pGluc) or mock (293T/TLR2/mock), in 293T cells without TLR2 expression (293T/pGluc; 293T/mock). (c) Comparison of IL-8 production in cells showing luciferase activity (293T/pGluc; 293T/TLR2/pGluc). (d) IL-8 secretion detected in TLR2-expressing groups (293T/TLR2/pGluc; 293T/TLR2/pNifty; 293T/TLR2/mock). Combined data of three biological repeats, each run in duplicates are shown (mean ± SEM).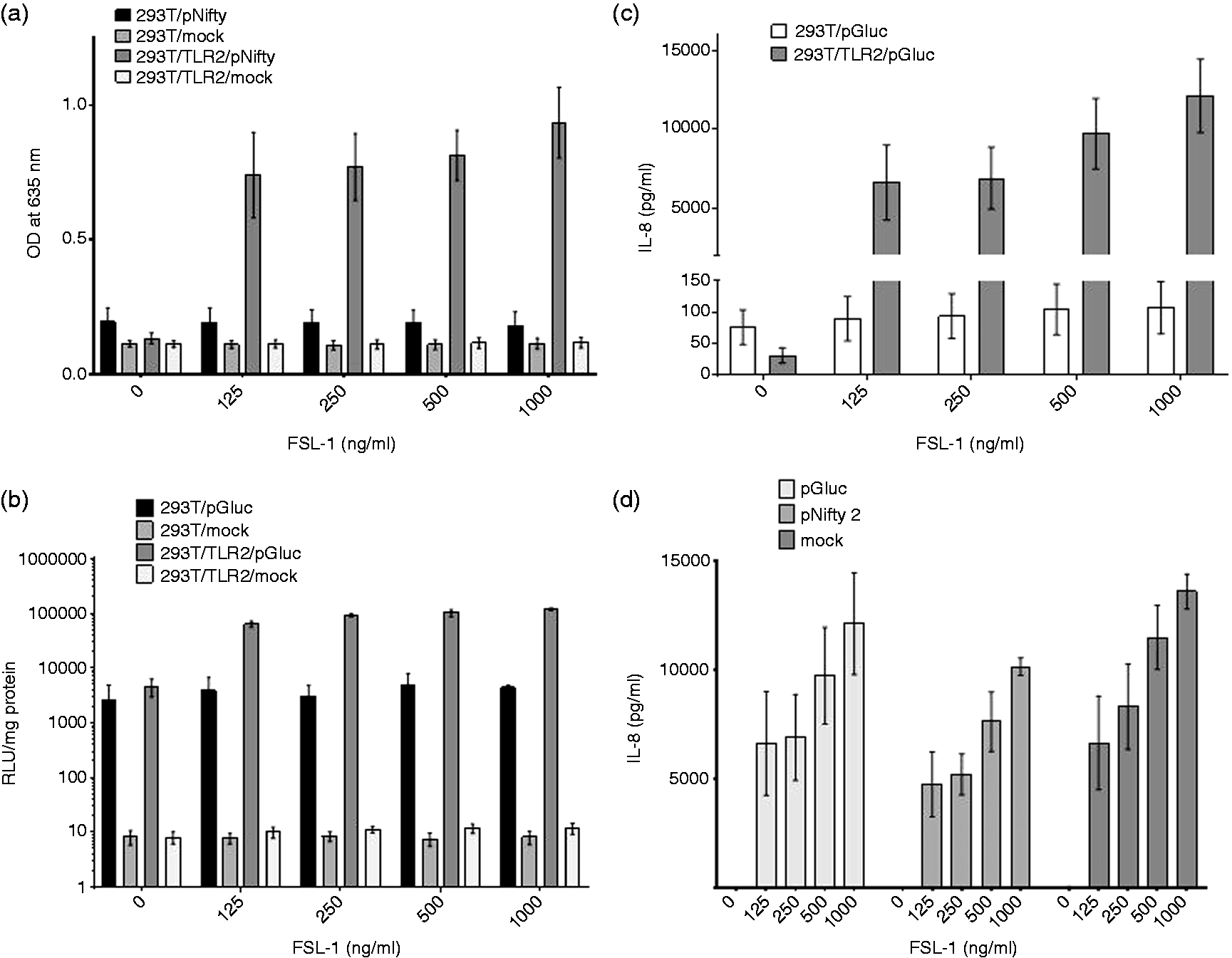
Conclusions
Our results show that the SEAP-based reporter system proved to be the most reliable and sensitive assay to detect bovine TLR dependent stimulation of NF-κB, and showed a good correlation with the resulting IL-8 production. Of all three assays tested we found the bovine-specific TLR2 NF-κB SEAP reporter assay the most convenient, with fewer steps and shortest duration. The developed reporter cell line would lend well to rapid, high-throughput bovine TLR2 ligand screening intended for supplementing vaccine adjuvant design strategies. Our findings extend recently published work by Tahoun et al. 15 Similarly to previous findings, 13 the TIR domain of the bovine receptor initiated adequate response in human cells in all three tested assays. In the present system, this response was already visible at 1 ng/ml, reaching significant differences at 10 ng/ml of specific stimulant doses in the SEAP assay when a stable reporter cell line was used. It was also observed that in stable reporter cell lines, a higher maximum signal was detected than in TLR2-expressing cells transiently transfected with SEAP gene (averaging at an OD of 1.27 for stable and at 0.76 for transient cells within the same biological repeat), indicating the benefits of creating stable reporter cell lines for farm animal TLRs, as already exist for human and murine systems. This way, there is no need for further transfections and the assay can be performed in a few simple steps, making it the assay of choice for small-scale laboratory experiments and also for high-throughput automated screening. In contrast, and as experienced before, a high background was detected in the luciferase assay, based on non-specific luciferase activity in 293T cells lacking TLR2, despite the fact that stimulation resulted in a marked increase of the signal. This phenomenon can be more problematic when detecting lower doses of the agonist or when applied for ligand screening, where effective stimulant concentrations are unknown. In most cases, assessment of IL-8 production provided a useful control for TLR-ligand screening methods; however, cytokine production was undetectable particularly at lower ligand concentrations. It also has to be kept in mind that cytokine production elicited by TLR-activation can also occur involving NF-κB-independent pathways.
Footnotes
Declaration of Conflicting Interests
The author(s) declared no potential conflicts of interest with respect to the research, authorship, and/or publication of this article.
Funding
The author(s) disclosed receipt of the following financial support for the research, authorship, and/or publication of this article: This work was supported by a grant of Zoetis Animal Health to D.W. The funder provided support in the form of salaries for authors K.T. and A.J.G., but did not have any additional role in the study design, data collection and analysis, decision to publish, or preparation of the manuscript. This manuscript represents number PPB_01454 of the RVC.