
Abstract
CC–chemokine receptor 7 (CCR7) is a G protein–coupled receptor expressed on a variety of immune cells. CCR7 plays a critical role in the migration of lymphocytes into secondary lymphoid tissues. CCR7 expression, however, has been linked to numerous disease states. Due to its therapeutic relevance and absence of available CCR7 inhibitors, we undertook a high-throughput screen (HTS) to identify small-molecule antagonists of the receptor. Here, we describe a robust HTS approach using a commercially available β-galactosidase enzyme fragment complementation system and confirmatory transwell chemotaxis assays. This work resulted in the identification of several compounds with activity against CCR7. The most potent of these was subsequently determined to be cosalane, a cholesterol derivative previously designed as a therapeutic for human immunodeficiency virus. Cosalane inhibited both human and murine CCR7 in response to both CCL19 and CCL21 agonists at physiologic concentrations. Furthermore, cosalane produced durable inhibition of the receptor following a cellular incubation period with subsequent washout. Overall, our work describes the development of an HTS-compatible assay, completion of a large HTS campaign, and demonstration for the first time that cosalane is a validated CCR7 antagonist. These efforts could pave the way for new approaches to address CCR7-associated disease processes.
Keywords
Introduction
CC–chemokine receptor 7 (CCR7) is a G protein–coupled receptor (GPCR) ordinarily expressed on several varieties of immune cells, including T cells, B cells, and dendritic cells. In vivo, CCR7 responds to two known ligands, CCL19 and CCL21, and plays a critical role in their chemotaxis and trafficking into secondary lymphoid tissue.1,2
The downstream signaling events following CCR7 ligation have been described previously. CCR7 is coupled to a Gαi/o subunit complexed to complementary Gβγ proteins. Upon receptor activation, these subunits dissociate so that Gαi/o is free to inhibit adenylate cyclase and the Gβγ proteins are available to stimulate phospholipase C. These respective events then result in a fall in cytoplasmic cAMP levels and a calcium flux from the endoplasmic reticulum. In addition, CCR7 ligation can result in the phosphorylation of its cytoplasmic domain by G protein receptor kinases (GRKs), which in turn results in the recruitment of cytosolic β-arrestin to the receptor tail and subsequent signaling through the ERK kinase pathway. Notably, the actions of CCL19 and CCL21 on the receptor are not entirely identical. While both appear to bind to CCR7 and to activate G protein signaling with similar potencies, CCL19 appears to have somewhat biased activity toward the β-arrestin pathway and in contrast to CCL21 is able to induce receptor desensitization.3,4
CCR7 is absolutely required for the normal movement of lymphoid cells into and through lymphatic tissue. While there are no known human conditions associated with the genetic loss of CCR7, CCR7 expression has been linked to graft-versus-host disease following hematopoietic stem cell transplantation and to the growth and lymphatic spread of a range of human malignancies.5–7 As a result, we deemed CCR7 to be an attractive therapeutic target.
Since there are no reported CCR7 small-molecule inhibitors, we undertook a high-throughput screen (HTS) to identity small-molecule antagonists of the receptor. Here we report for the first time that the antiviral compound cosalane, originally developed as an agent for the treatment of human immunodeficiency virus (HIV), 8 antagonizes human and mouse CCR7 signaling in response to CCL19 and CCL21 stimulation at nanomolar to low micromolar concentrations.
Materials and Methods
Cells and Cell Lines
PathHunter Chinese hamster ovary (CHO)–K1 CCR7 β-arrestin cells (cat. 93-0195C2 and 93-0528C2 for human and mouse CCR7, respectively) were obtained from DiscoveRx (Fremont, CA) and licensed for use. Human H9 cells (cat. HTB-176) were obtained from American Type Culture Collection (Manassas, VA). Primary mouse T cells were obtained from C57BL/6 mice purchased from The Jackson Laboratory (Bar Harbor, ME).
This study was carried out in strict accordance with the recommendations in the Guide for the Care and Use of Laboratory Animals of the National Institutes of Health. The protocol was approved by the Institutional Animal Care and Use Committee of the University of North Carolina at Chapel Hill (protocol number 15-208.0).
CHO-K1 CCR7 β-Arrestin Enzyme Complementation Assay
Human CCL19 (582102) and CCL21 (582202), as well as mouse CCL19 (587802) and CCL21 (586402), were purchased from BioLegend (San Diego, CA). PathHunter CHO-K1 CCR7 β-arrestin cells were plated in 20 µL Dulbecco’s modified Eagle’s medium (DMEM)–F12 (ThermoFisher, Waltham, MA) containing 2% bovine serum albumin (BSA) (Sigma-Aldrich, St. Louis, MO) at a concentration of 5,000 cells per well into Corning 384-well white-wall, clear-bottom plates (Corning, NY) and allowed to adhere overnight. After 24 h, 2.5 µL of 10× compounds diluted in Hank’s balanced salt solution (HBSS) containing 2 mM HEPES was added to cell assay plates at a final concentration of 10 µM using a MultiMek automated liquid dispenser (Nanoscreen, Charleston, SC) for 30 min. The final DMSO concentration in test wells was 0.5%, which was well within the DMSO tolerance of the cells. A Multidrop Combi Reagent Dispenser (ThermoFisher) was used to add 2.5 µL of 10× chemokine ligand diluted in 1% BSA/phosphate-buffered saline (PBS) at the EC80 concentration to the test wells and incubated for 90 min at room temperature. Last, 12 µL of PathHunter detection reagents (DiscoveRx) was added and the plates incubated at room temperature for 60 min before reading chemiluminescence on an EnVision Multilabel plate reader (PerkinElmer, Waltham, MA).
EC80 concentrations for the CCR7 agonists were determined by analyzing dose curves of CCL19 or CCL21 from a four-parameter curve fit (GraphPad Prism; GraphPad Software, La Jolla, CA) for use in the screen. A CCL19 ligand titration was included on each HTS plate to ensure assay conditions were consistently optimal along with high and low controls for Z factor determination (Z′). 9 All plates were subjected to PASS/FAIL quality control, and only those achieving Z factors >0.5 were assigned PASS and uploaded to the database for analysis (ScreenAble Solutions, Chapel Hill, NC).
Compound libraries were initially screened at single concentrations of 10 µM (“single shot”) using a final cutoff of 50% inhibition to determine hits for follow-up. For hit confirmation, 10-point dose-response data for compounds diluted 1:3 from a top 30-µM concentration were fit with a four-parameter curve model to determine IC50 values. Purity assessments for confirmed hits were made by standard analytical liquid chromatography/mass spectroscopy (LC/MS) and/or proton nuclear magnetic resonance (NMR). Error bars represent standard deviations.
Counterscreen
We used a counterscreen to eliminate false positives that directly inhibit β-galactosidase activity and that quench chemiluminescence independent of CCR7 activity. The counterscreen was performed using the same methods as described above with the following change: compounds starting at 5 µM were added 90 min after the addition and incubation of CCL19 ligand.
Cytotoxicity Screen
Active compounds from all screening efforts at the Center for Integrative Chemical Biology and Drug Discovery (CICBDD) are routinely evaluated for cytotoxicity using HeLa cells (ATCC) by the CellTiter-GLO (CTG) Luminescent Cell Viability Assay (Promega, Madison, WI) after a 72-h incubation with compounds in 10-point dose curves. Cytotoxicity for dose curves of compounds starting at 50 µM was calculated as the percent inhibition of luminescence normalized to vehicle control wells containing cells (0%) versus wells containing media without cells (100%).
Cosalane/Steroids
Cosalane was obtained from the Southern Research Institute (Birmingham, AL). Subsequently, additional cosalane was synthesized at the University of North Carolina using published methods.8,10
The steroids used in several chemotaxis assays were purchased from Sigma.
Chemotaxis Assays
Human H9 cells were expanded in T175 flasks. Murine T cells were isolated 1 day prior to chemotaxis by collecting spleens from C57BL/6 mice and using column purification methods previously described. 11 T cells were then cultured overnight on Costar ultra-low-attachment 24-well plates (Corning) in the presence of interleukin 2 (Peprotech, Rocky Hill, NJ) at 100 IU/mL. Chemotaxis assays were performed using a Neuro Probe 96-well chamber (Gaithersburg, MD) with cells suspended in serum-free, phenol red-free DMEM media (ThermoFisher). 2.0 × 105 cells were loaded into wells of the upper portion of the chamber with antagonist or vehicle. Cells sat on a polyvinylpyrrolidone (PVP)–treated filter with pore size of 3 µM for murine T cells or 5 µm for H9 cells. Chemotaxis media containing the appropriate chemokines filled the bottom portion of the chamber of a 96-well Microfluor black microtiter plate (ThermoFisher) below the PVP filter. All chemokines were obtained from Peprotech: murine CCL19 (250-27B), murine CCL21 (250-13), human CCL19 (300-29B), and CCL21 (300-35). The MBA96 apparatus was then incubated for 3.5 h at 37 °C. Upon removal from the incubator, the upper chamber of each well was aspirated. Migrating cells were then collected in each well by centrifugation as the microplate with PVP filter was spun at 400 rcf for 15 min. Cells at the bottom of the wells were quantified using a CyQUANT cell proliferation assay kit (ThermoFisher) and detected on a Biotek (Winooski, VT) Synergy 2 microplate reader at emission/excitation wavelengths of 480 nm/530 nm.
Data Analyses
Unless otherwise specified, continuous variables were compared using a two-sided Student t test. A p value ≤0.05 was considered statistically significant. Bar graph error bars represent SEM.
Results and Discussion
Design and Validation of the HTS Assay
For our primary diversity library screening assay, we elected to use a DiscoveRx PathHunter β-arrestin platform. We purchased and were thus licensed to use Chinese hamster ovary (CHO) K1 cells expressing either human CCR7 (hCCR7) or murine CCR7 (mCCR7). This platform allows for CCR7 activity to be monitored by detecting the interaction of β-arrestin with the activated receptor using β-galactosidase (β-gal) enzyme fragment complementation. In this system, cytosolic β-arrestin is fused to an N-terminal deletion mutant of β-gal, and the CCR7 receptor is fused to a smaller, complementary β-gal fragment. When CCL19 or CCL21 is added to the culture medium, β-arrestin is recruited to the cytosolic domain of the CCR7 receptor bringing the two β-gal fragments together. When this occurs, intact β-gal is formed, which in turn produces a luminescent signal upon the addition of substrate. As a result, a small-molecule diversity library can be screened for CCR7 inhibitors by examining a compound’s ability to block signal production in the presence of agonist at its EC80 concentration.
Our overall HTS strategy is depicted in Figure 1A . When designing our HTS approach, we believed that it was important to identify compounds with antagonist properties against both human and murine CCR7 given our ultimate goal of testing lead agents for efficacy and toxicity in mouse disease model systems. In addition, we wished to identify agents capable of blocking CCR7 signaling in response to CCL19 and CCL21, since both of these ligands function as CCR7 agonists and may in some instances serve a redundant function in vivo. 2 To minimize costs, however, we planned to use CHO-K1 cells expressing human CCR7 for all initial library screens in response to a single ligand, CCL19. CCL19 was chosen over CCL21 since the former had previously been shown to be a more consistent recruiter of β-arrestin following CCR7 stimulation.3,4 Any hits (defined as those compounds found to inhibit greater than 50% of the CCL19 response) identified during the initial runs would be confirmed in full 10-point dose-response curves spanning 4 log units of concentration in the hCCR7/CCL19 primary assay and assessed in parallel for purity and identity using analytical LC/MS and/or proton NMR spectroscopy. To eliminate apparent inhibitors that interfere nonspecifically with the luminescent assay, all hits would be evaluated in a counterscreen in which compounds are added at the very end of the assay after the agonist incubation period had occurred. This would allow us to determine if they were nonspecifically quenching the signal via direct action on the β-gal enzyme. All confirmed hits from the primary screen would then be tested using a Promega CellTiter-GLO cytotoxicity assay in a sentinel cell line. Confirmed hits showing activity against hCCR7 in response to CCL19 without evidence of activity in the counterscreen or cytotoxicity would then be tested for action against hCCR7 using CCL21 as the agonist. Those compounds exhibiting activity in the hCCR7/CCL21 assay would be profiled in the PathHunter β-arrestin mouse orthologue assay against CCL19 and/or CCL21. Compounds that were active in all four permutations would be progressed to secondary chemotaxis assays.
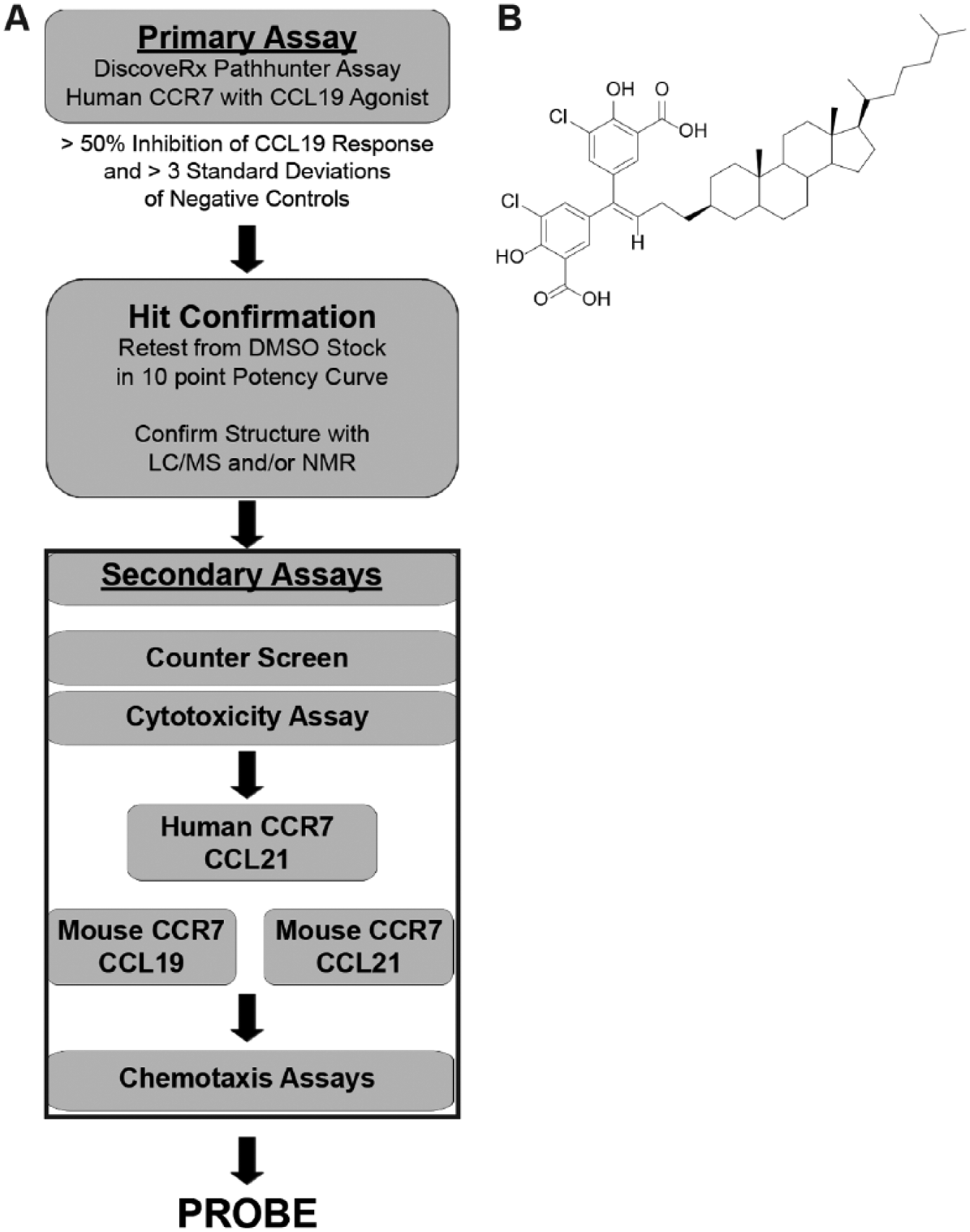
(
We began by confirming that our primary HTS assay behaved well in 384-well format and was sufficiently sensitive to identify potential inhibitory compounds. We fully optimized this assay by evaluating the following parameters: cell culture conditions, plate type and volume, time course, DMSO tolerance (up to 1%), and adaptation to automated equipment (data not shown). In addition, we formally validated the HTS in our facilities in accordance with National Institutes of Health (NIH) Chemical Genomics Center guidelines. 12 During our screening validation, our Z scores 9 were found to consistently be >0.85.
CHO K1 cells expressing hCCR7 produced a robust signal in a dose-dependent fashion in response to CCL19 and CCL21, with the former being the more potent agonist as expected ( Suppl. Fig. S1 ). Following up on these data, we also compared the ability of CCL19 and CCL21 to mobilize calcium using fluorescence imaging plate reader (FLIPR) detection in the DiscoveRx β-arrestin line. These results were subsequently compared with FLIPR results obtained with the Chem-1 CCR7 cell line (Millipore, Bedford, MA) containing a promiscuous G protein, Gα15 ( Suppl. Fig. S2 ). As reported by Zidar et al. 3 and Kohout et al., 4 our FLIPR calcium flux studies revealed that CCL19 and CCL21 have similar potencies and efficacies in activating G protein signaling in Chem-1 CCR7 cells where the promiscuous Gα15 is overexpressed. However, in the DiscoveRx β-arrestin line, which does not express Gα15, ligand-stimulated calcium flux mirrored the β-arrestin translocation responses, with CCL19 eliciting a more potent and robust ability to mobilize calcium than CCL21. Collectively, these data indicated that the DiscoveRx cells were generally more sensitive to CCL19 ligation versus CCL21 and supported our original plan to use CCL19 as agonist for all initial diversity library screens.
Cosalane Is Active against Human and Murine CCR7 in β-Galactosidase Enzyme Fragment Complementation Reporter Assays
Over several years, we screened numerous compound libraries (http://cicbdd.web.unc.edu/resources/) totaling 150,375 structures. For primary screening, a cutoff of 50% was chosen to provide a suitable number of compounds to progress to hit confirmation. A total of 2597 compounds were identified, yielding a primary hit rate of 1.7%. Of these 2597 structures, 614 were available in sufficient quantities to evaluate in full dose-response curves, and 97 of 614 were found to demonstrate IC50s ≤10 µM (confirm rate of 15.8%). Based on compound potency/structure and supply considerations, 25 confirmed hits were subsequently progressed.
In total, we identified 14 hits that were active in our primary screening assay against hCCR7/CCL19, nonreactive in the counterscreen and cytotoxicity assays, and active against CCL21 and the corresponding murine orthologue. In general, their structures were fairly diverse, and no consistent structure-activity relationships could be identified between the compounds. Potencies for the hit compounds were modest, yielding IC50s against human CCR7 in the low micromolar range in response to both CCL19 and CCL21 ( Suppl. Table S1 ).
Of these 14 compounds, UNC10150582 was found to be the most potent. Its structure, depicted in Figure 1B , shows that it is a lipophilic, cholesterol-like analogue consisting of cholestane with a dichlorinated disalicylmethane moiety at the C3 position. Upon review of the literature, our group determined that UNC10150582 was structurally identical to an HIV medication termed cosalane. 8 Henceforth, in this article, UNC10150582 will be referred to as cosalane.
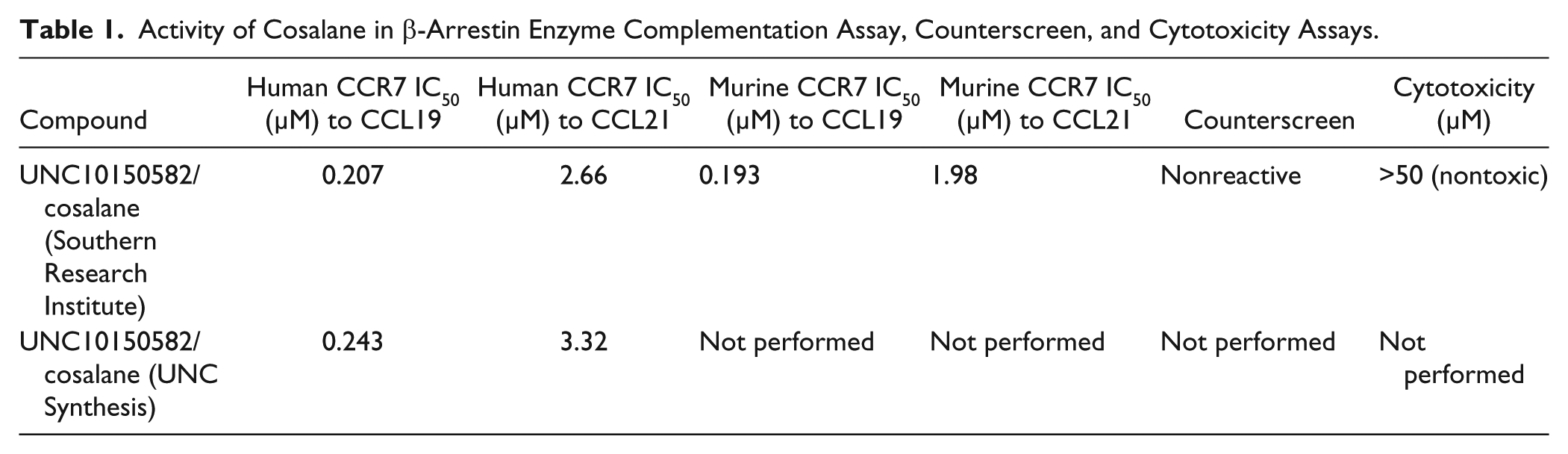
The primary assay data for cosalane (Southern Research Institute) as the structure was advanced through our progression strategy are depicted in Supplemental Figure S3 and summarized in Table 1 . Cosalane exhibited nearly identical activity against the human and murine orthologues of the receptor. Also, cosalane was consistently more active in blocking receptor activation by CCL19 than CCL21 by approximately one order of magnitude. To verify that similar results could be obtained with a separate batch of cosalane, we repeated chemiluminescence assays for hCCR7/CCL19 and hCCR7/CCL21 using cosalane synthesized at the University of North Carolina. Cosalane from both sources behaved similarly ( Table 1 ).
Activity of Cosalane in β-Arrestin Enzyme Complementation Assay, Counterscreen, and Cytotoxicity Assays.
Cosalane Impairs CCR7-Dependent Chemotaxis of Human and Murine T Cells
While useful from a screening perspective, our primary assays were not physiologic. As a result, it was important to confirm that promising hits could block an actual function mediated by native CCR7. For this, we used a well-described transwell chemotaxis assay system using human H9 cells, a human lymphoma known to express CCR7 and to migrate in response to its ligands, 13 and primary mouse T cells. As depicted in Figure 2A , human H9 cells in the upper chambers of the chemotaxis device migrated through the separating membrane when CCL19 was present in the lower chamber. Human H9 cells also migrated in response to CCL21, although this agonist was less potent as was the case in our primary HTS assay ( Fig. 2B ). When cosalane was added to the upper chambers, cell migration in response to CCL19 was inhibited versus DMSO ( Fig. 2A ). CCL21-driven chemotaxis was also reduced by cosalane, although this did not achieve statistical significance ( Fig. 2B ). Murine T-cell chemotaxis was also impaired by cosalane in response to both CCL19 and CCL21 ( Fig. 2C , D ).
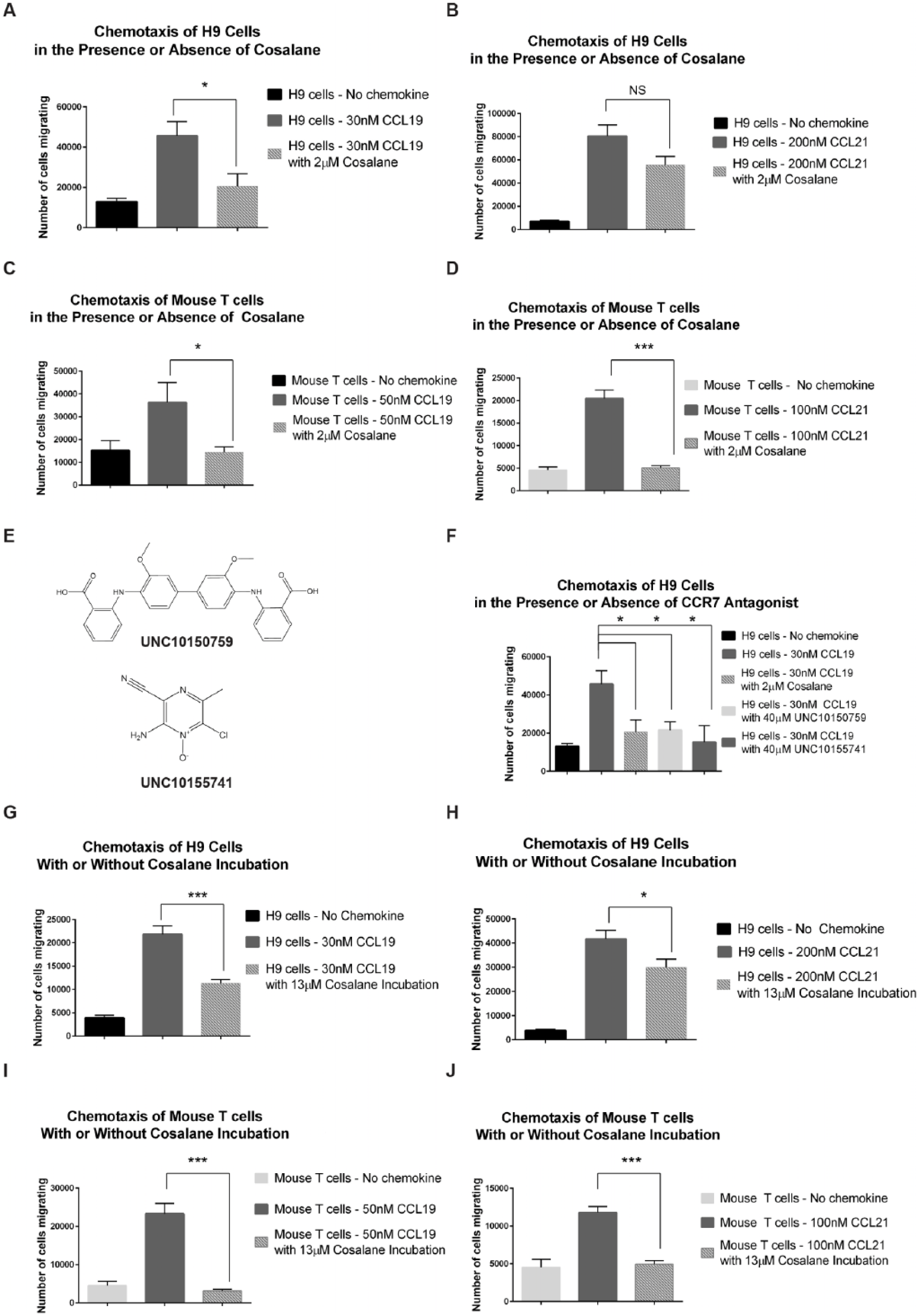
Cosalane impairs CCR7-driven human and murine T-cell chemotaxis. (
While these studies focused primarily on cosalane, we did evaluate two other hits identified during our HTS effort in this chemotaxis system. Both UNC10150759 and UNC10155741 (structures depicted in Fig. 2E ) were considerably less potent than cosalane in our primary assay system, demonstrating IC50s of 8.9 µM and 7.9 µM against hCCR7/CCL19, respectively. Nevertheless, they were also capable of blocking H9 chemotaxis in response to CCL19 when present at high concentrations ( Fig. 2F ). The fact that all three of these compounds significantly inhibited hCCR7/CCL19-driven chemotaxis at four or five times their respective IC50s in the primary reporter assay while maintaining their rank-order potencies further validated our overall compound progression strategy.
Cosalane was designed to imbed in the plasma membrane by way of its hydrophobic cholestane moiety. 8 Based on these data, we hypothesized that cosalane’s actions against CCR7 might prove to be durable and persist even after free cosalane was removed from the chemotaxis media. To evaluate this possibility, human H9 cells were incubated with 10 µg/mL (13 µM) of cosalane versus DMSO vehicle for 1 h, washed free of the compound, and then placed into the chemotaxis chamber. As shown in Figure 2G , H , chemotaxis in response to both CCL19 and CCL21 was significantly reduced by cosalane pretreatment alone. Similar experiments were performed with primary mouse T cells. As was the case with the human H9 cells, the chemotaxis of primary murine T cells in response to both CCL19 and CCL21 was significantly reduced by cosalane pretreatment versus DMSO vehicle ( Fig. 2I , J ).
Cosalane’s Ability to Impair CCR7-Dependent Chemotaxis Depends on the Entire Molecule and Not Just the Steroid Moiety
Previous reports in the literature have indicated that cholesterol itself plays a role in stabilizing chemokine receptors within the plasma membrane. 14 In addition, the extraction of cholesterol from the cell membranes of T cells or the addition of the oxysterol 22-hydroxycholesterol has been shown to reduce the binding of stromal cell–derived factor 1 (SDF-1) to CXC–chemokine receptor 4 (CXCR4) and the binding of CCL4 to CC–chemokine receptor 5 (CCR5).15–17 As a result, we hypothesized that cosalane’s anti-CCR7 properties may have been attributable to the steroid portion of the molecule. To test this hypothesis, we examined the ability of various nonoxidized and oxidized steroids to impair human H9 cell chemotaxis in vitro. We began by examining 5α-cholestane itself as it is most similar structurally to cosalane’s steroid moiety. As demonstrated in Figure 3A , 5α-cholestane afforded no significant inhibition of chemotaxis at three separate concentrations up to 20 µM (10 times that used for cosalane in Fig. 2 ). Similarly, no inhibition of chemotaxis was achieved with either cholesterol or cholestanol, its 5-dihydro derivative, over a range of concentrations ( Fig. 3B ). In addition, we examined a variety of oxysterols for their ability to impair CCR7-driven chemotaxis, including 25-hydroxycholesterol, androsterone, progesterone, testosterone, aldosterone, and hydrocortisone, as well as the LXR antagonist T0901317, which had previously been linked to chemokine receptor expression. 18 None of these compounds, however, was able to block CCR7-driven chemotaxis at 20-µM concentrations ( Fig. 3C , D ). Thus, we concluded that cosalane’s ability to impair CCR7-dependent chemotaxis does indeed require the complete molecule and is not a more generalized steroid effect.
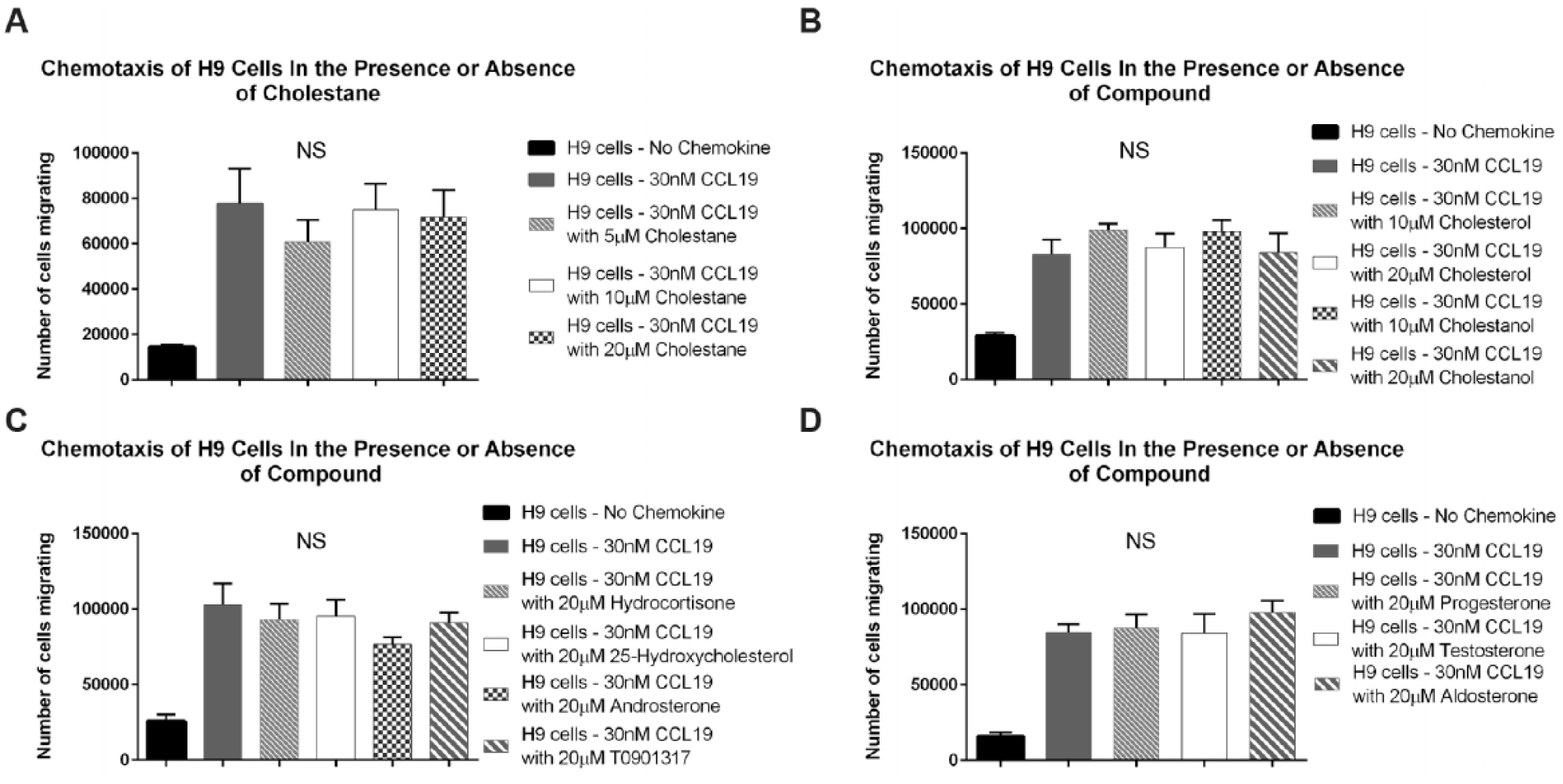
5α-Cholestane, cholesterol, cholestanol, and a variety of oxysterols fail to impair CCR7-driven human T-cell chemotaxis. (
Here we describe the design and implementation of a robust and efficient HTS strategy for the detection of small-molecule inhibitors of the CCR7 signaling axis. Our approach has allowed for the robotic screening of numerous diversity libraries totaling over 100,000 compounds and has resulted in the identification of multiple hits with limited in vitro toxicity that are active against both the murine and human orthologues. Importantly, several of these hits have been confirmed to impair CCR7 signaling in subsequent physiologic cellular chemotaxis studies and to perform with similar rank-order potencies in these secondary assays, as was observed in the primary screen.
To our knowledge, our work demonstrated for the first time that cosalane is an inhibitor of both human and murine CCR7. Cosalane and/or its derivatives, however, were previously shown to impair CCR1-dependent chemotaxis in response to its ligand, CCL5 (RANTES). In work by Howard et al., 19 cosalane’s inhibitory effects appeared to depend on both the chemokine agonist and its associated chemokine receptor, as cosalane was unable to block CCR1-driven chemotaxis in response to either of its other ligands, CCL3 (MIP-1a) or CCL4, and failed to inhibit CCL5-dependent chemotaxis using cells expressing its other binding partner, CCR5. Nevertheless, in that study, cosalane appeared to function primarily by binding to the chemokine agonist, as acetylation of CCL5 completely abrogated the ability of cosalane to inhibit CCR1-driven chemotaxis.
Our own results with CCR7 bear some similarities to these findings. In both our primary enzyme complementation assays and in several of our subsequent confirmatory chemotaxis studies, the nature of the CCR7 agonist affected the ability of the compound to block CCR7 signaling. Specifically, cosalane was approximately 10-fold more potent against CCL19-driven receptor activation versus CCL21 in our primary screening assay and was a more effective inhibitor of CCL19-driven human H9 chemotaxis. However, unlike the aforementioned findings for CCR1/CCL5, our data would suggest that cosalane inhibits CCR7 not via binding to chemokine ligand but rather via actions on the receptor itself. In our assays, cosalane was placed in the upper chambers of the chemotaxis device along with the migrating cells (as opposed to being mixed with chemokine and then added directly to the lower chambers). More important, however, cosalane was able to impair human and murine chemotaxis after an isolated 1-h preincubation step with the migrating cells, with no additional compound used during the subsequent assay. Consistent with previous reports, this would suggest that the compound was imbedding itself into the cell membrane where it then continued to inhibit cell surface CCR7 even after the cells were washed. Cosalane’s increased potency against hCCR7/CCL19 versus hCCR7/CCL21 in our primary reporter system was somewhat unexpected, as the assays were run with both ligands at their respective EC80 concentrations. While the mechanism for this difference is unclear, CCL19 and CCL21 are known to induce somewhat different CCR7 conformations. 20 We hypothesize that cosalane may bind the two receptor/ligand pairs with different affinity at a separate allosteric site. Future work will be directed toward better understanding cosalane’s mechanism of action in blocking CCR7 signaling.
In summary, we have developed an HTS approach that has allowed for the rapid screening of diversity libraries in search of small-molecule antagonists of CCR7 signaling. This effort has allowed us to demonstrate for the first time that the antiviral agent cosalane possesses an ability to impair CCR7 activity in response to both of its natural ligands. Additional efforts are ongoing to identify other inhibitors of the receptor.
Supplemental Material
Supplemental_Material_for_Hull-Ryde_et_al – Supplemental material for Identification of Cosalane as an Inhibitor of Human and Murine CC–Chemokine Receptor 7 Signaling via a High-Throughput Screen
Supplemental material, Supplemental_Material_for_Hull-Ryde_et_al for Identification of Cosalane as an Inhibitor of Human and Murine CC–Chemokine Receptor 7 Signaling via a High-Throughput Screen by Emily A. Hull-Ryde, Melissa A. Porter, Kenneth A. Fowler, Dmitri Kireev, Kelin Li, Catherine D. Simpson, Maria F. Sassano, Mark J. Suto, Kenneth H. Pearce, William Janzen and James M. Coghill in SLAS Discovery
Footnotes
Acknowledgements
We thank Dr. Stephen Frye for his advice and general support for the project. In addition, we acknowledge Dr. Jeffrey Aubé’s support for cosalane’s synthesis. FLIPR assays were generously provided by the National Institute of Mental Health’s Psychoactive Drug Screening Program.
Declaration of Conflicting Interests
The authors declared no potential conflicts of interest with respect to the research, authorship, and/or publication of this article.
Funding
The authors disclosed receipt of the following financial support for the research, authorship, and/or publication of this article: This work was supported by the National Institutes of Health (5K08HL111205-04) and the University of North Carolina’s University Cancer Research Fund and Translational and Clinical Sciences Institute.
References
Supplementary Material
Please find the following supplemental material available below.
For Open Access articles published under a Creative Commons License, all supplemental material carries the same license as the article it is associated with.
For non-Open Access articles published, all supplemental material carries a non-exclusive license, and permission requests for re-use of supplemental material or any part of supplemental material shall be sent directly to the copyright owner as specified in the copyright notice associated with the article.