
Abstract
Ubiquitin-like with PHD and RING finger domains 1 (UHRF1) is a multidomain protein that plays a critical role in maintaining DNA methylation patterns through concurrent recognition of hemimethylated DNA and histone marks by various domains, and recruitment of DNA methyltransferase 1 (DNMT1). UHRF1 is overexpressed in various cancers, including breast cancer. The tandem tudor domain (TTD) of UHRF1 specifically and tightly binds to histone H3 di- or trimethylated at lysine 9 (H3K9me2 or H3K9me3, respectively), and this binding is essential for UHRF1 function. We developed an H3K9me3 peptide displacement assay, which was used to screen a library of 44,000 compounds for small molecules that disrupt the UHRF1-H3K9me3 interaction. This screen resulted in the identification of NV01, which bound to UHRF1-TTD with a Kd value of 5 μM. The structure of UHRF1-TTD in complex with NV01 confirmed binding to the H3K9me3-binding pocket. Limited structure-based optimization of NV01 led to the discovery of NV03 (Kd of 2.4 μM). These well-characterized small-molecule antagonists of the UHRF1-H3K9me2/3 interaction could be valuable starting chemical matter for developing more potent and cell-active probes toward further characterizing UHRF1 function, with possible applications as anticancer therapeutics.
Introduction
Epigenetic signaling coordinated by DNA methylation and histone modification is an essential mechanism that dynamically regulates the chromatin structure for gene expression to determine cellular functions.
1
Abnormal alteration of genetic constitution and epigenetic regulation is the main cause of tumorigenesis.2,3 The 90 KDa
UHRF1 contains five functional domains: ubiquitin-like domain (UBL), tandem tudor domain (TTD), plant homeodomain (PHD), SET and RING associated domain (SRA), and really interesting new gene domain (RING). 8 Each domain could independently carry out epigenetic functions of UHRF1. In particular, the SRA domain is unique to UHRF1, which directly recognizes hemimethylated DNA and recruits DNA methyltransferase 1 (DNMT1) to chromatin for DNA methylation maintenance.9–11 Its TTD functions as a histone reader to bind the transcription repressive marker of di- and trimethylated histone H3 lysine 9 residues (H3K9me2 and H3K9me3). 12 Interestingly, the binding specificity of the tudor domain is determined by the specific recognition of the PHD to the wild-type arginine residue 2 and lysine 4 of histone H3 to cooperatively contribute to the pericentric heterochromatin organization of UHRF1.13–15 TTD recognition of the methylated H3K9 mark is reported to be essential for the recruitment of DNMT1 to daughter strands during DNA replication, suggesting the importance of the cooperative actions between SRA and TTD to establish and maintain DNA methylation. 16
Tumorigenesis is associated with abnormal DNA methylation patterns that affect the expression of tumor suppressor genes. Given the importance of UHRF1 in maintaining DNA methylation, its overexpression could lead to the disruption of the epigenomic homeostasis of DNA methylation, leading to the development and progression of cancer. 8 In breast cancer, UHRF1 overexpression induces the hypermethylation and inhibition of BRCA1 by forming an inhibitory transcriptional complex comprising HDAC1, DNMT1, and G9a. 17 Consistently, UHRF1 is highly expressed in hepatocellular carcinoma (HCC), leading to delocalization of DNMT1, induced DNA hypomethylation, and Tp53-mediated senescence. 18 Accordingly, HDAC inhibitor trichostatin A– and DNMT1 inhibitor 5-Aza-CdR–treated hepatic cancer cell line BEL-7402 showed reduced binding of UHRF1 to the 3OST2 tumor suppressor gene, leading to the inhibition of the cancer cell proliferation. 19 Therefore, antagonism of UHRF1 emerges as an attractive therapeutic strategy for the treatment of a variety of cancers associated with UHRF1 overexpression.
A few natural products have been reported to downregulate UHRF1 expression. However, these compounds most likely act through an indirect mechanism, such as p53-dependent apoptosis.20,21 A microRNA-9 was reported to inversely correlate with UHRF1 expression in colorectal cancer cell line. Upregulation of microRNA-9 downregulated UHRF1 function and triggered the growth inhibition and apoptosis of cancer cells. 22 Given the large size of the UHRF1 protein and multiple domain-orientated functions, it is very challenging to develop antagonists through binding to full-length UHRF1. To overcome this obstacle, targeting individual domains was an alternative option. The TTD is important in regulating UHRF1 functions through histone H3K9me2 or H3K9me3 recognition and therefore is an interesting target.
Accumulated progress has been made in developing antagonists targeting histone reader proteins.23–25 For example, a variety of antagonists have been discovered to modulate acetyl-lysine reader bromodomains, and many have been advanced to human clinical trials. 26 Potent and selective antagonists of L3MBTL3 have demonstrated target-dependent cellular activities. 27 To our knowledge, small-molecule antagonists for tudor domains remain elusive.
Here we report the discovery of single-digit micromolar antagonists of UHRF1-TTD. We screened a diverse library of 44,000 compounds and identified NV01 as a hit. We then determined the crystal structure of UHRF1-TTD in complex with NV01. The compound directly binds to the H3K9me3 pocket. Exploratory structure-based optimization was carried out and the potency was modestly improved. Site-directed mutagenesis studies further revealed the details of interaction of these compounds with UHRF1-TTD. Our studies provide a solid starting point for future development of chemical probes to study the cellular functions of the UHRF1 histone reader tudor domain and the therapeutic potential of its inhibition.
Materials and Methods
Expression and Purification
Wild-type (residues 139–298) and mutant (UHRF1_D145A, UHRF1_F152A, UHRF1_Y188A, UHRF1_Y191A, and UHRF1_N147A) human UHRF1-TTD, and a PHD and TTD (UHRF1-TTD-PHD; residues 147–380), as well as the construct, which we call full-length UHRF1 (14–806) in this article (FL-UHRF1), were cloned, expressed, and purified as described in the Supplementary Material and Methods.
Binding Assays
Fluorescence Polarization–Based Peptide Displacement Assay
This assay is designed based on the concept that binding of a fluorescein-labeled peptide to a larger molecule, such as a protein, results in a higher fluorescence polarization (FP) signal that will decrease upon displacement of the peptide by small molecules. To discriminate against the term “IC50 value,” which indicates the concentration of the compound that causes 50% reduction in activity of the target enzymes, we have previously introduced the term “Kdisp” to quantify the potency of the compounds, which refers to the same concept but for displacement-based binding assay.28–32 The Kdisp values were calculated using the four parameters logistic equation using GraphPad. The equation is as follows:
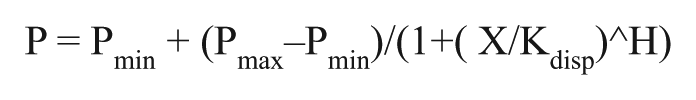
where P stands for polarization; Pmin, minimum polarization; Pmax, maximum polarization; and H, Hill slope.
The H3K9me3 (1–25) peptide (ARTKQTARK(me3)STGGKAPRKQLATKAK) was synthesized, N-terminal labeled with fluorescein isothiocyanate (FITC), and purified by Tufts University Core Services (Boston, MA). UHRF1-TTD titrations and compound binding assays were performed in a 10 µL volume at a constant labeled peptide concentration of 0.04 µM. For compound screening and titrations, a fixed protein concentration of 8 µM UHRF1-TTD (close to 90% of the maximum signal) was used. The assay buffer was 20 mM Tris (BioShop, Burlington, ON; cat. no. TRS001), pH 8.8, 50 mM NaCl (BioShop, cat. no. SOD001), and 0.01% Triton X-100 (Sigma, St. Louis, MO, cat. no. T9284). FP assays were performed in 384-well (Greiner, Monroe, NC, cat. no. 784209) plates using a Synergy 4 microplate reader (BioTek, Winooski, VT). An excitation wavelength of 485 nm and an emission wavelength of 528 nm were used. Similar assays were optimized for UHRF1-TTD-PHD and FL-UHRF1 to generate reliable signal (data not shown). The FP assay for UHRF1-TTD-PHD was performed in 20 mM HEPES (BioShop, cat. no. HEP001), pH 7.5, 0.1 mM ZnCl2 (BioShop, cat. no. ZNC222), 0.01% Triton X-100, 1.5 µM protein, and 0.04 µM labeled peptide. We observed about a threefold increase in signal for peptide binding to UHRF1-TTD-PHD at pH 7.5 compared with performing the experiment at pH 8.8. Change of assay pH did not have any significant effect on Kd values of H3K9me3 binding to UHRF1-TTD-PHD (1.7 and 1.8 µM at pH 7.5 and 8.8, respectively). The assay for FL-UHRF1 was performed in 20 mM Tris, pH 8.8, 50 mM NaCl, 0.01% Triton X-100, 30 µM protein, and 0.04 µM fluorescein-labeled peptide. Full-length UHRF1 behaved well at pH 8.8 but showed signs of aggregation at pH 7.5. DMSO (Sigma, cat. no. D4540) concentration in the initial screen of 44,000 compounds and follow-up assays was 2.5%. Note that high concentrations (
Differential Scanning Fluorimetry
Differential scanning fluorimetry (DSF) measurements were performed as previously reported. 33 The protein solutions with a final concentration of 0.1 mg/mL in a buffer consisting of 0.1 M HEPES, pH 7.5, 0.15 M NaCl in 20 µL volume were tested using a Light Cycler 480 II instrument from Roche Applied Science (Penzberg, Germany). Sypro Orange was purchased from Invitrogen (Waltham, MA; cat. no. S6650) as a 5000× stock solution and was diluted 1:1000 to yield a 5× working concentration. Invitrogen does not specify the concentration. Compounds were screened at 400 µM. DSF was carried out by increasing the temperature from 20 to 95 °C at a heating rate of 4 °C/min, and data points were collected at 1°C intervals. The temperature scan curves were fitted to a Boltzmann sigmoid function, and the Tm values were obtained from the midpoint of the transition as described previously.
Isothermal Titration Calorimetry
Purified UHRF1-TTD was dialyzed in isothermal titration calorimetry (ITC) buffer (0.02 M Tris, pH 7.5, and 0.05 M NaCl). Compound at 1 mM was injected into the sample cell containing approximately 300 µL of 40 μM UHRF1-TTD. ITC titrations were performed on a Nano ITC from TA Instruments (New Castle, DE) at 25 °C by using 2 μL injections with a total of 25 injections. Data were fitted with a one-binding-site model using Nano Analyze Software.
Crystal Structure Determination
Human UHRF1-TTD protein used for crystallization was produced as previously published. 12 Briefly, UHRF1-TTD, comprising residues 126–285, was cloned into pET28a vector with N-terminal 6His-tag and expressed by Escherichia coli strain BL21-CodonPlus(DE3)-RIL. The cells were sonicated, and supernatant containing UHRF1-TTD was purified by affinity chromatography using a Ni2+-NTA column (QIAGEN, Venlo, Netherlands) and subsequently cleaved by tobacco etch virus (TEV) protease at 4 °C overnight. The cleaved protein was passed through a Ni2+-NTA column (QIAGEN) to remove the His-tag and the TEV and was followed by a gel filtration column (Superdex 75, 10/60 from GE, Pittsburgh, PA). The purified UHRF1-TTD was concentrated to 8.5 mg/mL and stored in 20 mM Tris, pH 7.5, 100 mM NaCl, and 5 mM DTT. The crystal was grown using sitting drop vapor diffusion with a reservoir containing 2.7 M Na formate and 0.1 M Na acetate, pH 5.2. The crystal was soaked with 0.5 mM NV01 for 1 h and cryoprotected by 30% glycerol (v/v). Diffraction data were collected at the Shanghai Synchrotron Radiation Facility (SSRF) beamline BL17U1 and processed using HKL2000. 34
The published structure of UHRF1-TTD (Protein Data Bank [PDB]: 3DB3) was used to create a search model for molecular replacement using Molrep in the CCP4 suite.
35
NV01 was manually built in COOT,
36
and the structure was further refined by BUSTER.
37
The final structure was checked by PROCHECK.
38
The statistics of the structure refinement and the quality of the final model are summarized in
Chemistry (Figure 1)
Tricyclic intermediate
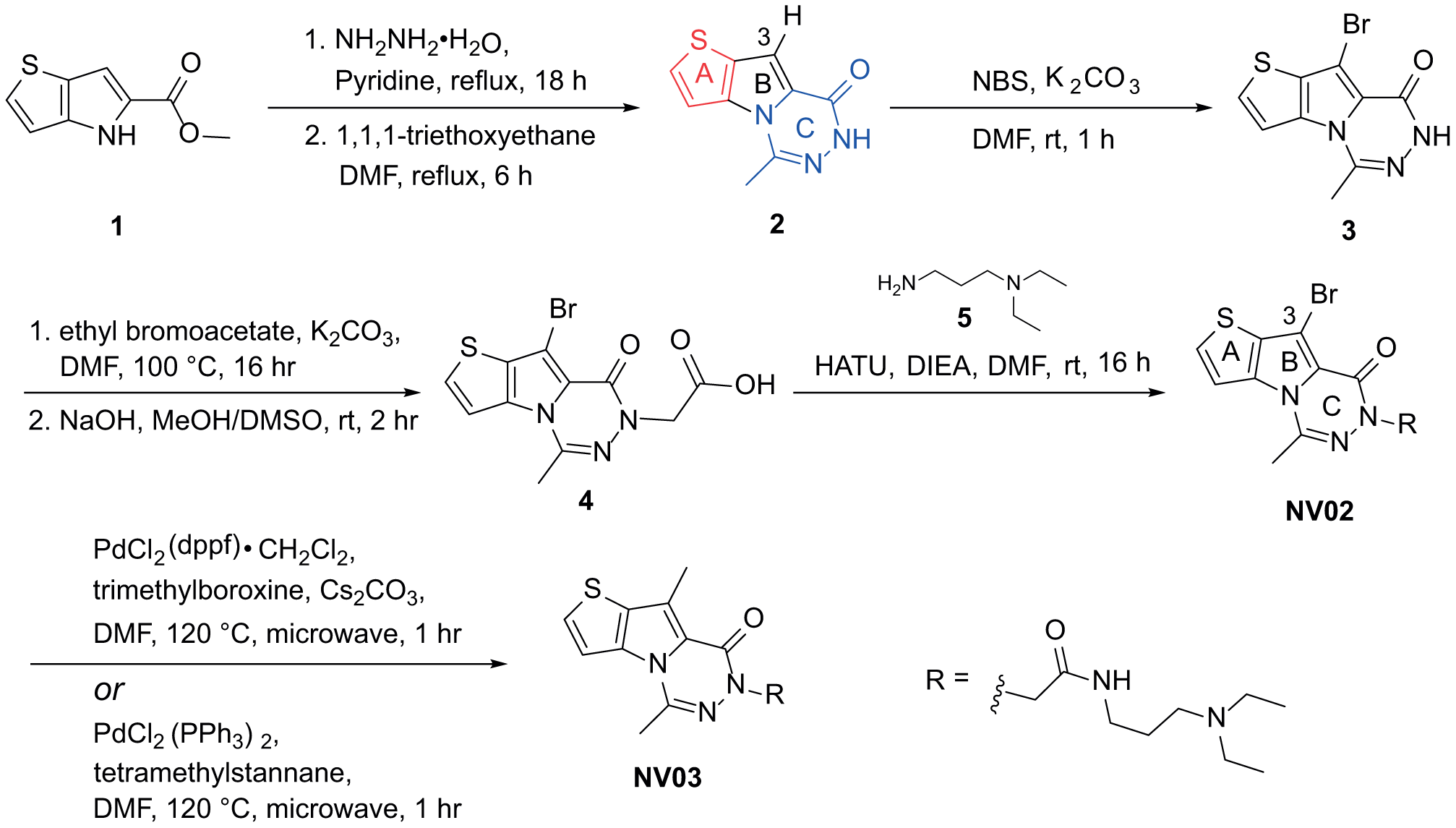
Representative synthetic route.
Following the same sequence, compounds
Results and Discussion
To identify compounds that disrupt the interaction between UHRF1-TTD and H3K9me3 peptide, we developed a peptide displacement assay. Binding of FITC-labeled H3K9me3- (1–25) peptide to UHRF1-TTD resulted in an increase in FP signal, which almost saturates at 10 µM UHRF1-TTD with an apparent Kd of 0.7 µM (
We employed this assay to screen a library of 44,000 diverse compounds. After eliminating compounds that interfere with the assay readout, we selected 121 hits showing more than a 50% decrease in FP signal (
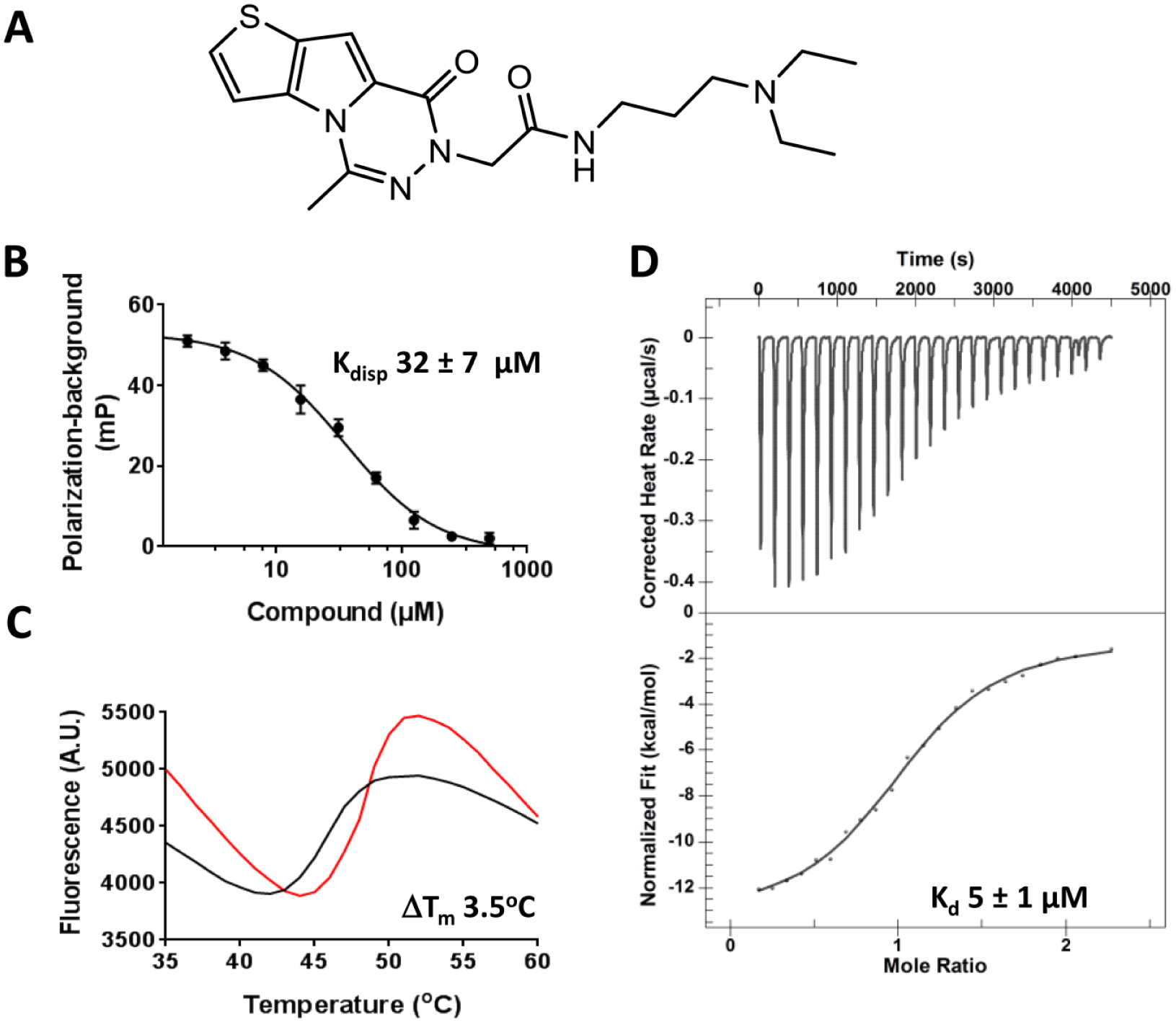
UHRF1-TTD binding confirmation of NV01. (
Interactions of Wild-Type UHRF1-TTD with NV01 to NV07 Were Assessed by FP, DSF, and ITC.
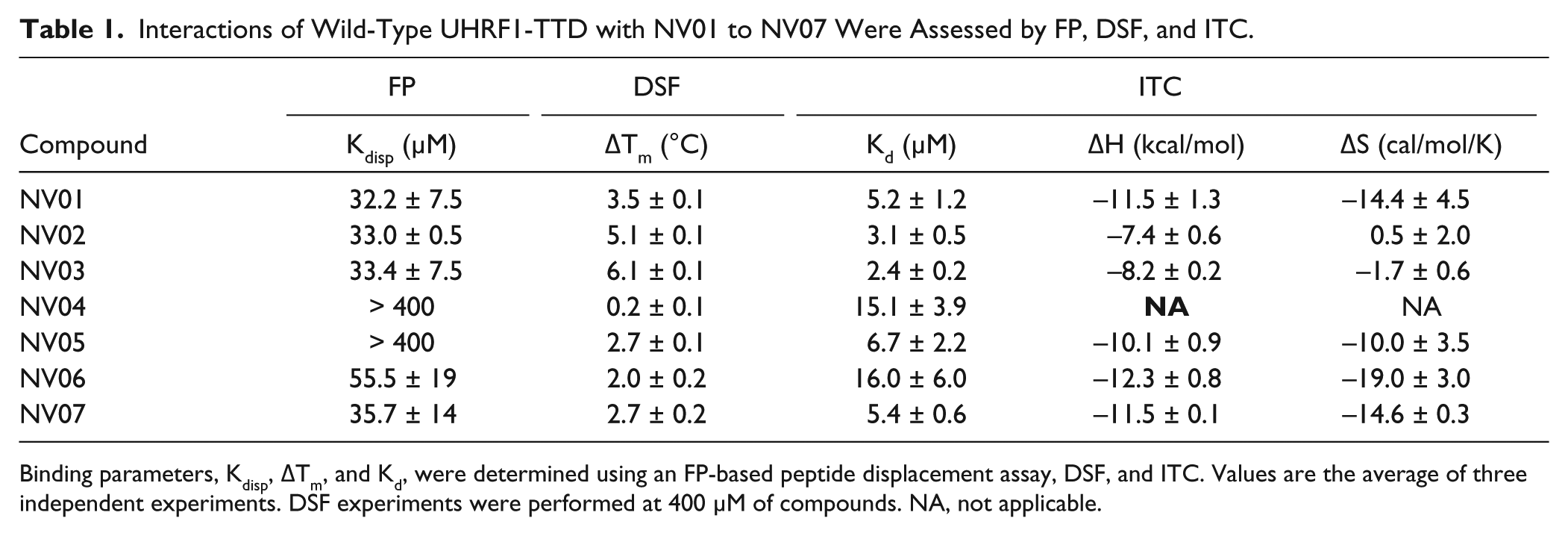
Binding parameters, Kdisp, ΔTm, and Kd, were determined using an FP-based peptide displacement assay, DSF, and ITC. Values are the average of three independent experiments. DSF experiments were performed at 400 µM of compounds. NA, not applicable.
Crystal Structure of UHRF1-TTD in Complex with NV01
To better understand the structural basis and binding mode of NV01, we solved the complex structure at 2.2 Å resolution (
Fig. 3
). The structure contains one molecule of UHRF1-TTD and one molecule of NV01 per asymmetric unit (
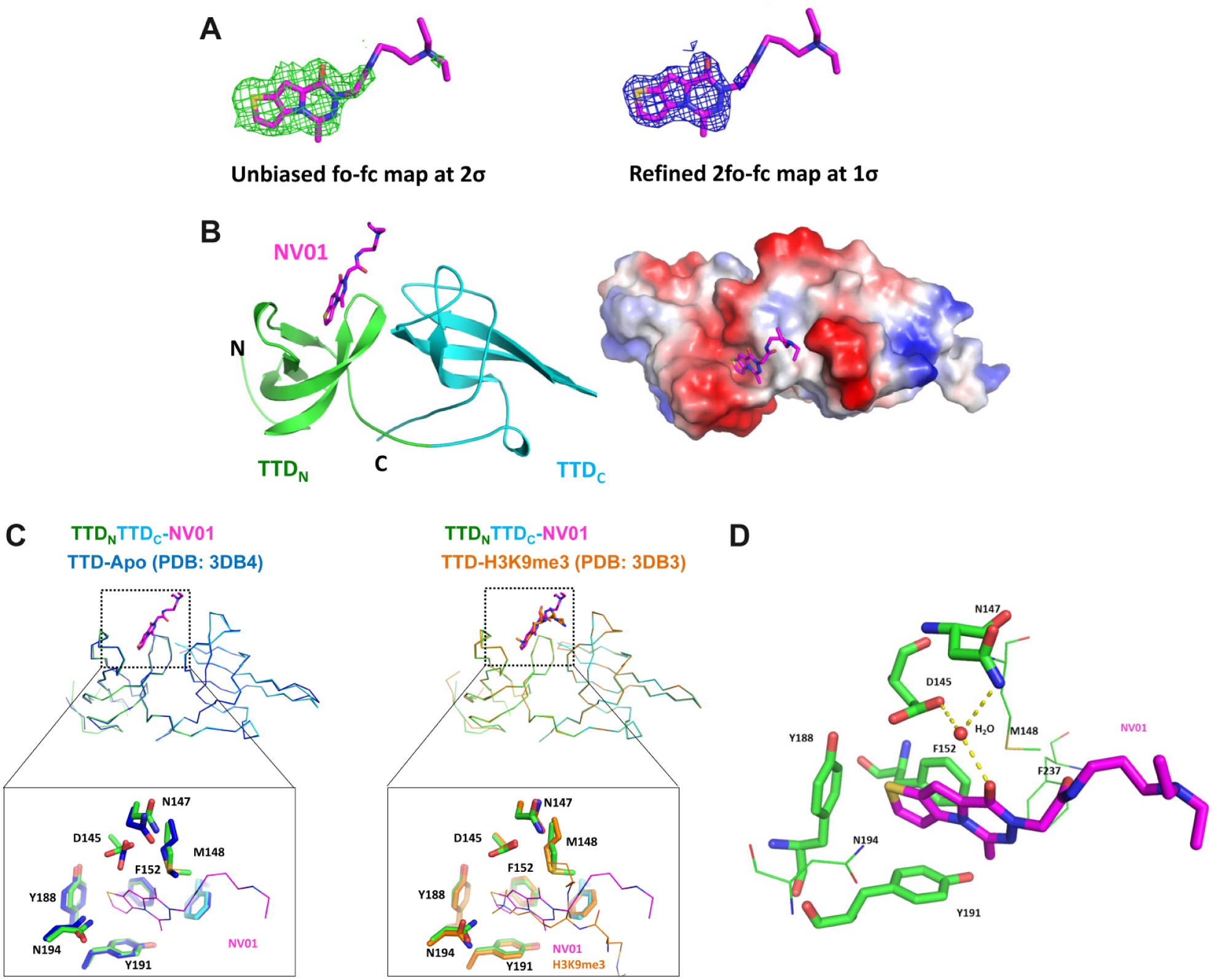
Crystal structure of UHRF1-TTD in complex with NV01. (
The overall structure of NV01-bound UHRF1-TTD is very similar to that of apo TTD (PDB: 3DB4; root mean square deviation [RMSD]: 0.275 Å for 119 Cα atoms), composed of TTDN and TTDC domains with a five-stranded β-barrel signature ( Fig. 3B , C ). The structure reveals that one NV01 molecule binds to the TTD at the H3K9me3-binding pocket, in which the oxothieno-pyrrolo-triazine moiety is deeply buried in the lysine channel, while the diethylamino-propyl-acetamide moiety extends to the solvent ( Fig. 3B ). The binding of NV01 at the H3K9me3-binding pocket does not induce global conformational change of TTD compared with the H3K9me3-bound TTD structure ( Fig. 3C ) (PDB: 3DB3; RMSD: 0.170 Å for 122 Cα atoms). The oxothieno-pyrrolo-triazine moiety of NV01 is packed by extensive π-stacking in a primary aromatic cage formed by F152, Y188, and Y191 from TTDN ( Fig. 3D ). It also forms van der Waal interactions with side chains of N194, M148, and F237. Comparison with the apo structure reveals that NV01 binding causes local adjustment of the side chains of Y188, N194, D145, and M148 to accommodate the ligand ( Fig. 3C , left). The conformation of these four residues induced by NV01 is very similar to that induced by H3K9me3 ( Fig. 3C , right). In addition, the keto group on the triazine contacts side chains of D145 and N147 through a water (H2O/36)-bridged H-bonding network ( Fig. 3D ). Lastly, we can trace the location of the diethylamino-propyl-acetamide moiety on the basis of its partial electron density map, but its detailed binding mode is missing, and the structure coordinates presented here best reflect the geometry.
Exploratory Structure-Based Optimization
Initial chemistry efforts were focused on the tricyclic core modification of NV01 since its amide tail was disordered in the x-ray structure. The bridging water (H2O/36) observed was part of the H-bond network between D145, N147, and the carbonyl of the NV01 C-ring. Subsequent water analysis using 3D RISM41,42 indicated that the water was relatively low energy (∆G = −0.57 kcal/mol), which could be replaced and compensated with a gain in entropy (
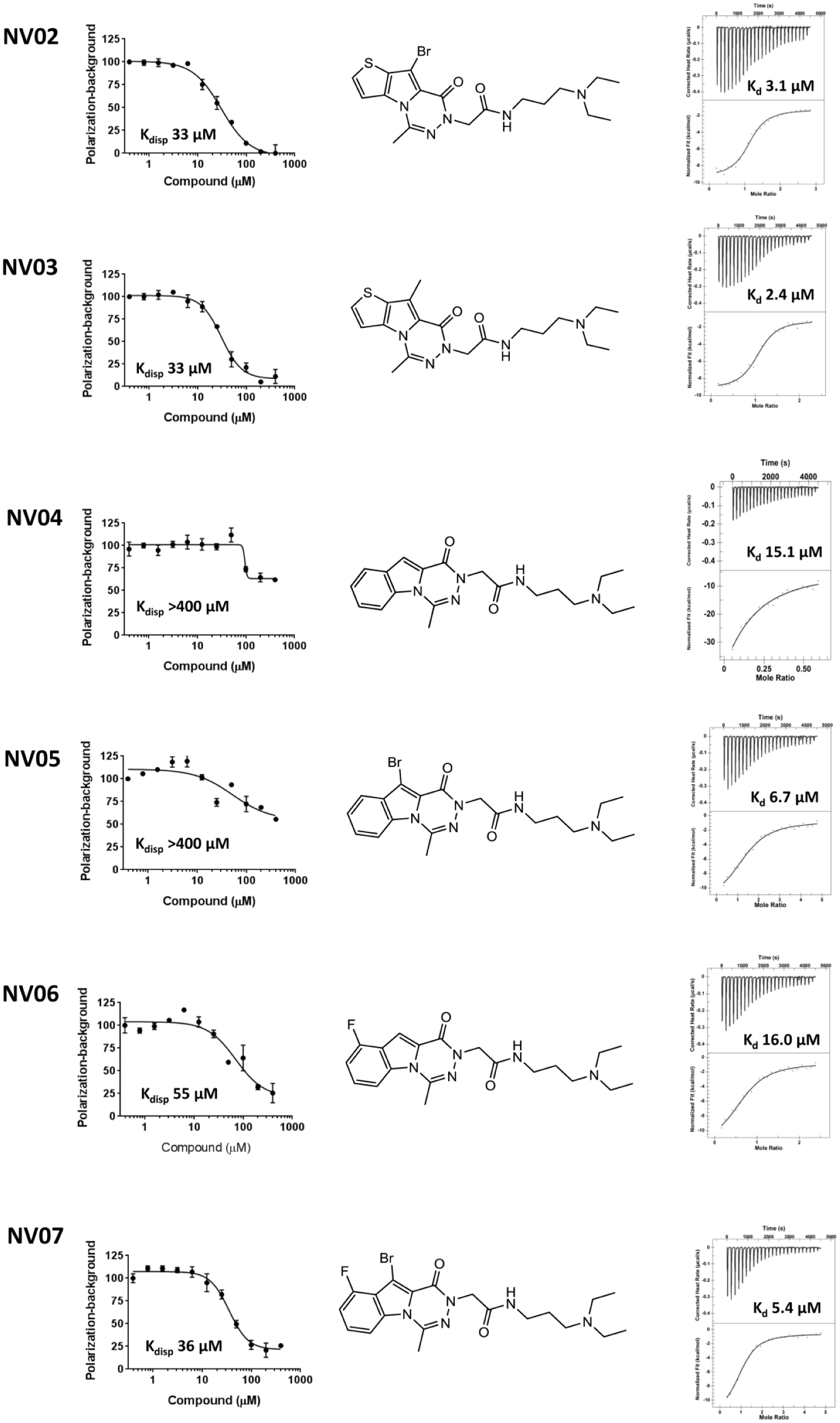
UHRF1-TTD binding confirmation of NV02-NV07. Binding affinities of NV02, NV03, NV04, NV05, NV06, and NV07 were measured by FP-based peptide displacement assay (left column; all y-axis values are normalized polarization [%]) and ITC (right column; corrected heat rate [µcal/s] and normalized fit [kcal/mol] for the upper and lower graphs, respectively). All experiments were performed in triplicate. The chemical structure of each compound is illustrated.
Ultimately, NV03 is the most potent antagonist for UHRF1-TTD interaction with H3K9me3. We next explored the selectivity of NV03 against a panel of 36 histone, DNA, and RNA methyltransferases, and an RNA demethylase using radioactivity-based enzyme assays (
Binding of NV03 to UHRF1-TTD-PHD and FL-UHRF1 Proteins
To investigate if the presence of other UHRF1 domains affects the binding of NV03 to UHRF1-TTD, we tested H3K9me3 displacement from FL-UHRF1 and UHRF1-TTD-PHD protein constructs. NV03 was much less effective in displacing the H3K9me3 peptide from the UHRF1-TTD-PHD protein (Kdisp ~110 µM) and had little effect on the interaction of the peptide with FL-UHRF1, a clear trend of decrease in ability of NV03 to displace the peptide ( Fig. 5 ). This was not unexpected, as UHRF1 undergoes conformational rearrangements in the presence of other domains that affect the accessibility of the TTD,44,45 and affinity of UHRF1-TTD for H3K9me3 peptide.14,46 However, the presence of hemimethylated DNA opens the closed conformation of UHRF1 and promotes H3K9me3 recognition by TTD-PHD. 11 In such conditions, the antagonists of H3K9me3 binding to UHRF1-TTD are expected to be more effective. The compounds presented here were not potent enough for cell-based assays and require further optimization before being tested in cells reliably.
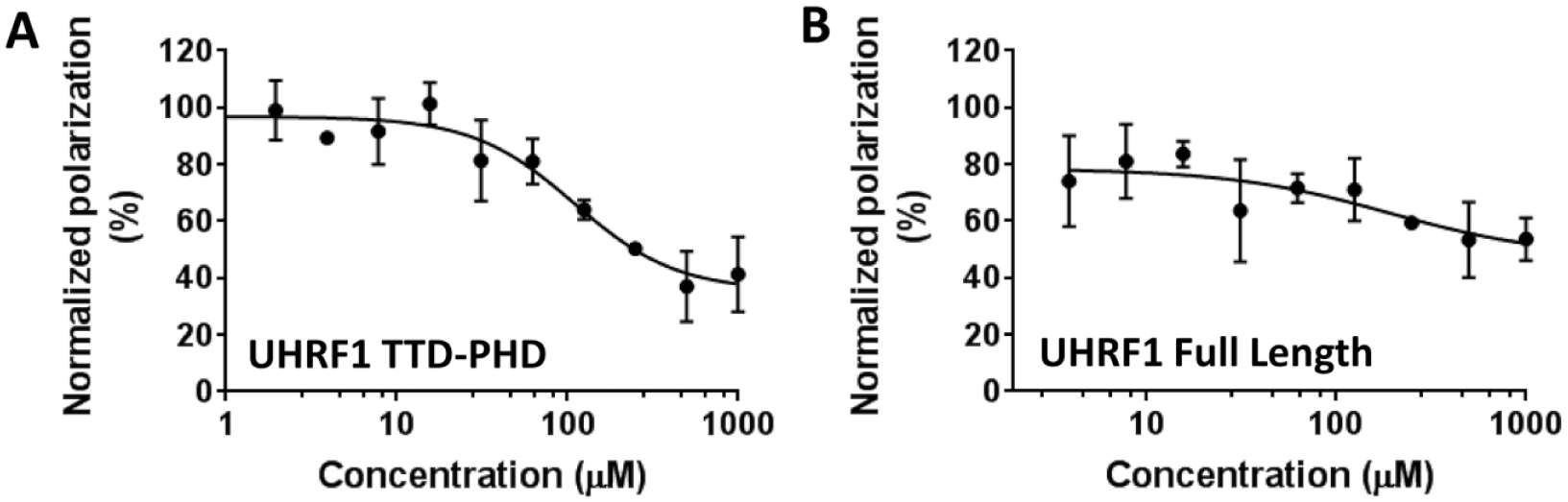
Binding of NV03 to UHRF1-TTD-PHD and FL-UHRF1. Binding of NV03 to (
Site-Directed Mutagenesis to Understand Binding Mode of NV01
To understand the narrow SAR and further investigate the interaction of NV01 with UHRF1-TTD, we tested three mutants of UHRF1-TTD that were expected to disrupt the aromatic cage (F152A, Y188A, and Y191A) and two mutants aimed at disrupting water-mediated H-bonds (D145A and N147A). The stabilities of these mutants were assessed by DSF (
The UHRF1-TTD structure in complex with NV01 revealed that an aromatic cage governs the shape of the pocket, a common feature also observed in other reader domain proteins, such as EED and L3MBTL3. The aromatic cage of EED, composed of Tyr365, Tyr148, and Phe97, forms the wall of the H3K27me3 pocket. 47 Likewise, aromatic cage residues F387, F405, W408, and Y412 in L3MBTL3 define the width of the H3K20me pocket. 27 These aromatic cages display shallow cavities in a pocket-free form, while adopting conformational change to accommodate the ligand interactions. NV01 binding to the UHRF1-TTD further confirmed that the conformational change at the aromatic cage upon compound binding may be a general phenomenon for histone reader proteins. The relatively flat SAR of the NV01 series suggests that other domains of the UHRF1 may be important to further improve the potency. Indeed, the PHD of UHRF1 plays an essential role in dramatically increasing the binding affinity and specificity of H3K9me3 to UHRF1 by providing an anchoring point for H3R2 interaction (PDB: 3ASK). 13 Future efforts in exploring the interaction of NV01 derivatives with TTD-PHD may contribute to developing more potent antagonists of UHRF1 that would be more suitable as probes for testing in cells.
In summary, we report on the discovery of small-molecule antagonists of the UHRF1-TTD interaction with the H3K9me3 histone peptide. The crystal structure of the UHRF1-TTD in complex with NV01 clearly confirms binding to the UHRF1 H3K9me3 recognition pocket. Limited structure-guided optimization led to a modest improvement of its inhibition potency. Although these compounds are not potent enough for cell-based assays, our chemical, biochemical, and structural data provide a good starting point for future development of more potent chemical probes for investigating the roles UHRF1 plays in health and disease and potential therapeutic applications.
Supplemental Material
DS_DISC766278 – Supplemental material for Discovery of Small-Molecule Antagonists of the H3K9me3 Binding to UHRF1 Tandem Tudor Domain
Supplemental material, DS_DISC766278 for Discovery of Small-Molecule Antagonists of the H3K9me3 Binding to UHRF1 Tandem Tudor Domain by Guillermo Senisterra, Hugh Y. Zhu, Xiao Luo, Hailong Zhang, Guoliang Xun, Chunliang Lu, Wen Xiao, Taraneh Hajian, Peter Loppnau, Irene Chau, Fengling Li, Abdellah Allali-Hassani, Peter Atadja, Counde Oyang, En Li, Peter J. Brown, Cheryl H. Arrowsmith, Kehao Zhao, Zhengtian Yu, and Masoud Vedadi in SLAS Discovery
Footnotes
Supplementary material is available online with this article.
Declaration of Conflicting Interests
The authors declared no potential conflicts of interest with respect to the research, authorship, and/or publication of this article.
Funding
The authors disclosed receipt of the following financial support for the research, authorship, and/or publication of this article: The SGC is a registered charity (number 1097737) that receives funds from AbbVie, Bayer Pharma AG, Boehringer Ingelheim, the Canada Foundation for Innovation, the Eshelman Institute for Innovation, Genome Canada, the Innovative Medicines Initiative (EU/EFPIA) (ULTRA-DD grant no. 115766), Janssen, Merck & Co., Novartis Pharma AG, the Ontario Ministry of Economic Development and Innovation, Pfizer, the São Paulo Research Foundation-FAPESP, Takeda, and the Wellcome Trust.
References
Supplementary Material
Please find the following supplemental material available below.
For Open Access articles published under a Creative Commons License, all supplemental material carries the same license as the article it is associated with.
For non-Open Access articles published, all supplemental material carries a non-exclusive license, and permission requests for re-use of supplemental material or any part of supplemental material shall be sent directly to the copyright owner as specified in the copyright notice associated with the article.