
Abstract
We have developed and validated label-free, liquid chromatography–mass spectrometry (LC-MS)-based equilibrium direct and competition binding assays to quantitate small-molecule antagonist binding to recombinant human and mouse BLT1 receptors expressed in HEK 293 cell membranes. Procedurally, these binding assays involve (1) equilibration of the BLT1 receptor and probe ligand, with or without a competitor; (2) vacuum filtration through cationic glass fiber filters to separate receptor-bound from free probe ligand; and (3) LC-MS analysis in selected reaction monitoring mode for bound probe ligand quantitation. Two novel, optimized probe ligands, compounds
Keywords
Introduction
BLT1 (LTB4R) is the high-affinity receptor for leukotriene B4 (LTB4). 1 It is a G-protein-coupled receptor primarily expressed on white blood cells, including granulocytes (neutrophils, basophils, and eosinophils), monocytes, macrophages, and naïve lymphocytes. 2 LTB4 is a potent pro-inflammatory mediator. 3 It is synthesized in myeloid cells from membrane phospholipid-derived arachidonic acid by the sequential action of 5-lipoxygenase and LTA4 hydrolase. 2 LTB4 mediates numerous inflammatory processes.3–5 It stimulates leukocyte activation, 6 prolongs neutrophil survival, 7 and as part of host immune response, contributes to the accumulation of effector leukocytes at sites of pathogen invasion. 8 Elevated LTB4 levels are present in patients with cystic fibrosis, 9 chronic obstructive pulmonary disease, 10 asthma, 11 and many other inflammatory diseases.12–14 In addition, LTB4 is secreted by select cancer cells and promotes tumor cell proliferation both in vitro and in vivo.15,16
In view of the well-documented pro-inflammatory effects of the LTB4/BLT1 pathway, extensive effort has been focused on the discovery of BLT1 antagonists as potential anti-inflammatory drugs. 3 Several of these compounds have exhibited efficacy in animal models of inflammatory disease. Preclinical studies have also demonstrated that BLT1 antagonist LY293111 inhibits the proliferation of, and induces apoptosis in, pancreatic and other cancer cells. 15 Unfortunately, none of these molecules have demonstrated registerable efficacy in humans.
More recently, BLT1 antagonism has been reported to have beneficial metabolic effects in rodents. 17 Knockdown of BLT1 receptor function by both genetic and pharmacological methods induced the expected anti-inflammatory effects and led to improved glucose homeostasis in insulin-resistant mouse models.17,18 Insulin resistance is a major defect in individuals with metabolic syndrome/prediabetes and a major contributor to hyperglycemia in type 2 diabetics. These results are of high interest, as there remains a compelling need for safe and effective drugs that improve insulin sensitivity. 19
As part of an effort to identify highly selective, novel BLT1 antagonists suitable for in vivo studies in insulin-resistant preclinical models, we needed robust assays to assess the binding affinity and kinetics of such ligands. In addition to various functional assays, 20 numerous direct binding assays for BLT1 have been described, most of which involve the use of radioactive LTB4 and synthetic antagonists or, more rarely, fluorescent ligands.20–23 Such assays are limited by the need to prepare a suitably labeled ligand for each compound of interest. 24 We now describe the development and application of label-free, liquid chromatography–mass spectrometry (LC-MS)-based direct binding assays to quantitate both the affinity and kinetics of antagonist binding to recombinant human and mouse BLT1 receptors. In addition, we report the validation and utilization of label-free, LC-MS-based competition binding assays to quantitate the affinity, and rapidly establish the structure–activity relationships (SARs), of mammalian BLT1 antagonists.
Materials and Methods
Materials
Tween 20, gliclazide, 3-isobutyl-1-methylxanthine (IBMX), forskolin, and LTB4 were purchased from Sigma-Aldrich (St. Louis, MO); 1 M HEPES (pH 7.4) was from Boston Bioproducts (Ashland, MA); complete protease inhibitor tablets were from Roche (Indianapolis, IN); and 40% weight/volume glucose solution was from Teknova (Hollister, CA). All cell culture media and reagents were purchased from GIBCO/Thermo Fisher Scientific (Waltham, MA), and HEK 293 cells were from ATCC (Manassas, VA). GF/B UniFilter 96-well plates and the MultiScreen high-throughput screening (HTS) vacuum manifold were purchased from EMD Millipore (Billerica, MA), and the 96-well plate evaporator was from Biotage (Charlotte, NC). All (potential) BLT1 antagonists tested were synthesized, purified, and authenticated by Merck Research Laboratories (Kenilworth, NJ).
Recombinant Cell Lines
The cDNA sequence for human BLT1 (NM_181657.3) was cloned into pDONR221 using standard techniques. The resulting plasmid was transfected into HEK 293 cells using Lipofectamine 2000, and stable clones selected using G418 (Geneticin). Clones were further characterized and selected by flow cytometry using a BLT1 antibody from Novus Biologicals (Littleton, CO; cat. NBP2-27422). HEK 293 cells stably expressing human BLT1 were grown in Dulbecco’s modified Eagle media (DMEM) containing 10% heat-inactivated fetal bovine serum (FBS) and 800 µg/mL G418.
The cDNA sequence for mouse BLT1 (NM_008519.2) was cloned into the pJTI-R4-DEST-CMV-pA destination vector using standard techniques. This vector, along with the pJTI-R4-Int integrase vector, was cotransfected into Jump-In HEK 293 cells using Lipofectamine 2000. Stable clones were selected using blasticidin and characterized using a functional calcium release FLIPR response to LTB4. Jump-In HEK 293 cells stably expressing mouse BLT1 were grown in DMEM containing 10% dialyzed FBS, 200 µg/mL hygromycin B, and 10 µg/mL blasticidin.
All cells were grown at 37 °C with 5% CO2. To passage the cells, 0.05% trypsin-EDTA was used.
BLT1-Expressing Membrane Preparations
Cells from a single 10-stack flask (Corning, Tewksbury, MA) were detached using an enzyme-free, phosphate-buffered saline (PBS)–based cell dissociation buffer. The cells were collected by centrifugation (450g) at 4 °C for 10 min, the supernatant discarded, and the resulting cell pellet resuspended in 90 mL of cell lysis buffer consisting of 20 mM HEPES (pH 7.4), 5 mM MgCl2, 180 mM sucrose, and complete protease inhibitor. The sample was maintained at 4 °C and dounced seven times at 2100 rpm using the Schuett Homgenplus homogenizer (Belenos, Vienna, Austria), the homogenate was centrifuged at 1150g for 12 min, and the supernatant was carefully removed and saved. This process was repeated on the resulting pellet twice using 60 mL of cell lysis buffer each time. The pooled supernatants from the cell lysis protocol were then centrifuged at 125,100g for 20 min at 4 °C in a Beckman Optima XL-100K ultracentrifuge using 70 mL Beckman polycarbonate ultracentrifuge bottles. The resulting membrane pellets were resuspended in 3.8 mL of membrane resuspension buffer consisting of 20 mM HEPES (pH 7.4) and 100 mM NaCl, using a 10 mL syringe fitted with a 20 G needle to disperse the membrane pellets. The protein concentration of the resulting membrane preparation was determined using the bicinchoninic acid (BCA) protein assay, with bovine serum albumin as standard (Pierce/Thermo Fisher Scientific).
Methods
Initial Screening to Identify Optimal Probe Ligands
For the initial compound screen, 20 structurally distinct, high-affinity BLT1 antagonists were individually incubated at two concentrations (10 and 100 nM) with 50 µg/mL HEK 293 membranes expressing recombinant human BLT1 in a total volume of 200 µL of buffer A (50 mM HEPES, 5 mM MgCl2, 1 mM CaCl2, 150 mM NaCl, 3 mM KCl, and 2 mM D-glucose [pH 7.4]). All incubations were performed in triplicate and maintained for 2 h at 22 °C with gentle shaking. Since we did not know initially whether all these potential ligands bound to the same versus distinct sites on the receptor (and therefore whether an excess of a single competitor ligand could be employed), nonspecific binding (NSB) was assessed for each compound by means of parallel incubations using mock HEK 293 membranes lacking BLT1. Binding was terminated, free ligand was separated from bound, and samples were prepared for LC-MS analysis, as described below.
Direct Saturation Binding Assays
Direct binding assays were performed using HEK 293 membranes expressing recombinant human or mouse BLT1 receptor in buffer A containing the indicated concentration of probe ligand
Competition Binding Assays
For competition binding assays, the indicated concentration of test ligand was added to incubations containing BLT1-expressing membranes in buffer A and initiated with the probe ligand at a fixed concentration close to its Kd value. The amount of probe ligand bound in the absence of competitors was defined as 0% inhibition, while that bound in the presence of a saturating amount of competitor was defined as 100% inhibition. For the human BLT1 competition assay using probe ligand
Dissociation Experiments
Human BLT1-expressing membranes were incubated in buffer A with probe compound
LC-MS Analysis
LC-MS analysis was carried out on a Thermo TSQ Vantage triple quadrupole mass spectrometer with an electrospray ionization source (Thermo Fisher Scientific). A Thermo Accucore C18 column (30 × 3.0 mm, 2.6 µm particle size) and a binary gradient were employed for chromatographic separation under the following conditions: column temperature 25 °C, mobile phase A: water/0.1% formic acid, mobile phase B: acetonitrile/0.1% formic acid, flow rate 0.5 mL/min, and injection volume 10 µL. The mobile phase gradient started with 85% A and was ramped to 95% B in 4 min, where it was held for 1 min, and then stepped down to 85% A and held for 0.5 min prior to the next injection. Data were acquired in either positive or negative ion mode, as indicated in
Table 1
. The operating parameters of the MS detector were as follows: spray voltage 3000 V, sheath gas pressure 60 arbitrary units, auxiliary gas pressure 20 arbitrary units, vaporizer temperature 400 °C, capillary temperature 270 °C, and collision pressure 1.4 mTorr. The S-lens and collision energy were set as 164 and 33 V for probe ligand
Rapid Evaluation of Potential BLT1 Probe Ligands Using an LC-MS-Based Approach.
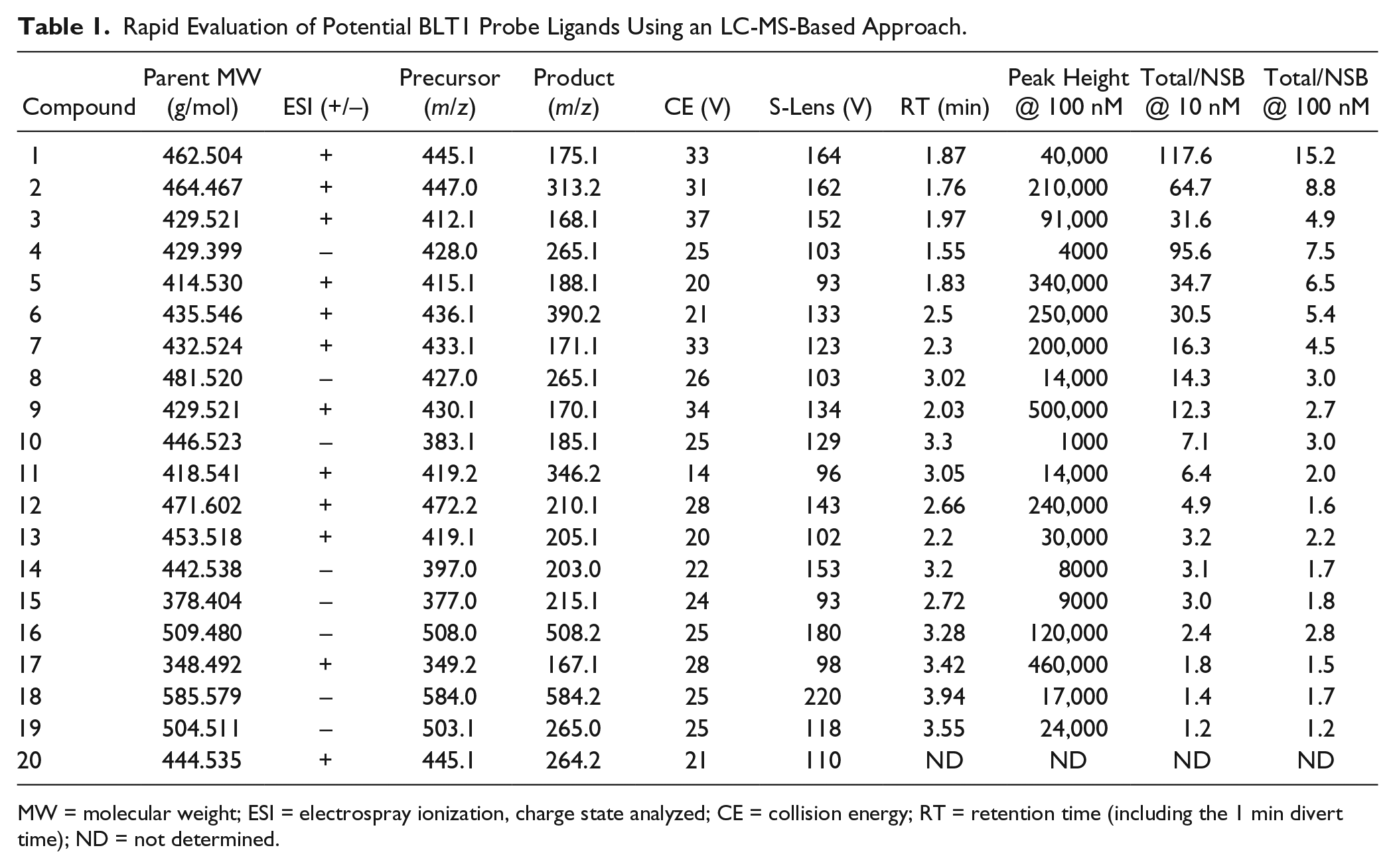
MW = molecular weight; ESI = electrospray ionization, charge state analyzed; CE = collision energy; RT = retention time (including the 1 min divert time); ND = not determined.
Human BLT1-Mediated cAMP Functional Assay
HEK 293 cells expressing human BLT1 were plated in 384-well plates at a density of 1250 or 5000 cells/well in Hank’s balanced salt solution (HBSS) containing 20 mM HEPES and 500 µM IBMX (5 µL). The BLT1 antagonist of interest (10 nL) was added to each well by use of an ECHO liquid dispenser (Labcyte, Sunnyvale, CA). The plate was incubated at 37 °C for 20 min, and then HBSS/HEPES/IBMX containing 2× (4 µM) forskolin and 2× EC80 (300 pM) LTB4 (5 µL) was added to each well. The plate was incubated for 30 min at 37 °C, at which point the cAMP dynamic homogeneous time-resolved fluorescence (HTRF) assay (Cisbio, Bedford, MA) was performed according to the manufacturer’s instructions by adding the d2/cryptate antibodies (10 µL), followed by incubation for 45 min at 22 °C. The assay plate was then read using an EnVision Multilabel Plate Reader (PerkinElmer, Waltham, MA). The ratiometric HTRF signal was calculated by dividing the 665 nm acceptor emission signal by the 620 nm donor emission signal and multiplying by 10,000. The ratiometric signals were used to calculate the cAMP produced (nM) using the cAMP dynamic HTRF assay standard. EC50 values were calculated using a four-parameter logistic fit based on the Levenberg–Marquardt algorithm.
Results and Discussion
Versatile LC-MS-Based Method for Quantitating Ligand Binding to Membrane-Associated Receptors
Ligand–receptor binding assays play a key role throughout the drug discovery and development process, providing validation of screening hits and defining the ligand affinity and binding kinetics SARs for the target receptor during lead identification and optimization efforts. 24 Receptor-bound ligand quantitation has traditionally required synthesizing the ligand of interest with radioisotope or highly fluorescent labels. In practice, the direct binding of only a limited number of ligands can be investigated by this approach due to resource-intensive synthetic requirements and/or because the modified ligands either cannot be prepared or are unsuitable for their intended purpose due to insufficient specific activity, high NSB, and so forth. Accordingly, a label-free method that can quantitate bound ligand would be advantageous.
MS offers exceptional specificity and sensitivity for compound identification and quantitation. As a tool for initial hit identification, extremely high-throughput LC-MS assays have been described that are capable of screening large libraries for the purpose of identifying ligands that bind to soluble or membrane-bound proteins.25,26 These methods have also been used to successfully rank-order ligand affinities; 27 however, detailed quantitation of binding affinities, and especially characterization of binding kinetics, is generally outside the scope of these techniques, with certain exceptions. 28 In addition, the application of the coupled LC-MS methodology to study low-molecular-weight ligand binding to membrane-associated receptors in a label-free manner was pioneered and extensively developed in the Wanner laboratory.29–31 The relative simplicity of this label-free approach to studying a wide array of biochemical binding processes held great appeal to us. We now report studies employing a modified, highly flexible, LC-MS-based method to quantitate small-molecule antagonist binding to membrane-associated BLT1 receptors in both equilibrium direct saturation binding and competition binding formats.
A successful, generic LC-MS-based protocol for the analysis of small-molecule ligand–receptor binding interactions requires two key components: (1) an LC-MS method that can easily quantitate a broad range of structurally diverse small-molecule ligands and (2) a versatile, moderate-throughput method for separating receptor-bound from free ligand. Whereas full-scan analysis using high-resolution mass spectrometers is ideally suited to rapid detection of ligands from large screening libraries with little to no requirement for up-front method development, we chose to employ selected reaction monitoring on a triple quadrupole mass spectrometer in these investigations because the number of ligands we wished to explore had already been highly curated (minimizing the amount of method development required) and because this approach generally affords high precision and lower limits of detection, important for robustly quantifying binding affinity and kinetics. For analytical separations, we employed an Accucore C18 reverse phase LC column with 2.6 µm particle size packing and volatile water/acetonitrile gradients containing 0.1% formic acid. This allowed the use of reasonable flow rates at standard high-performance liquid chromatography (HPLC) pressures (to minimize run times/maximize throughput) and provided good retention of a broad range of analytes. The LC effluent was directly coupled, using electrospray ionization, to a Thermo TSQ Vantage triple quadrupole mass spectrometer that facilitated ligand quantitation with high specificity by a combination of LC retention time and selected reaction monitoring of optimized parent-to-fragment ion transitions. Gliclazide was chosen as a “universal” internal standard for all assays to simplify the analysis and avoid the resource-intensive additional step of preparing a stable isotope standard for each compound of interest. All results were analyzed as the peak area response ratio of the analyte (ligand)/internal standard.
For separation of membrane receptor-bound ligand from excess free ligand, we employed the highly versatile method of rapid vacuum filtration through cation-derivatized glass fiber filters in a 96-well plate format. This approach captures anionic membranes containing the ligand-bound receptor and allows free ligands to be removed by washing the membranes with cold buffers of appropriate composition to reduce NSB. Rapid filtration, in combination with the use of cold buffers, minimizes receptor-bound ligand dissociation. Ligands retained on the filter after washing were eluted with 1:1 methanol–water, which was evaporated; the resulting residue was reconstituted in the presence of 100 nM gliclazide as the internal standard; and the sample was analyzed by LC-MS. A detailed protocol is described in the Materials and Methods section.
Identification of “Superior” BLT1 Antagonists to Use as Probe Ligands for LC-MS Binding
A theoretical advantage of the label-free LC-MS-based binding assay is the ability to screen a variety of molecules to identify an optimal probe ligand for further study. We successfully employed such a screen to identify an optimized BLT1 antagonist to use as an LC-MS probe ligand. To this end, 20 structurally distinct, proprietary BLT1 antagonists (established using functional assays) from our internal chemistry effort were analyzed using the Thermo TSQ Vantage mass spectrometer. For each compound, MS/MS parent-to-fragment ion transitions were optimized, the C18 LC retention time was defined, and the peak height/100 nM compound was determined as a representative measure of total signal intensity. We also applied a rapid screening assay in which the total binding of each antagonist at 10 and 100 nM to human BLT1 membranes (50 µg/mL) was measured by LC-MS after a 2 h incubation. The NSB of these 20 potential ligands was also assessed in parallel, otherwise identical incubations using mock HEK 293 membranes lacking BLT1. The data from these studies are summarized in
Table 1
. The ratios of total binding/NSB for each ligand at both concentrations were calculated to provide a measure of the binding window. This varied widely, with only 9 of the 20 BLT1 ligands evaluated demonstrating a total/NSB ratio greater than 10 at 10 nM. Based on their superior properties, compounds
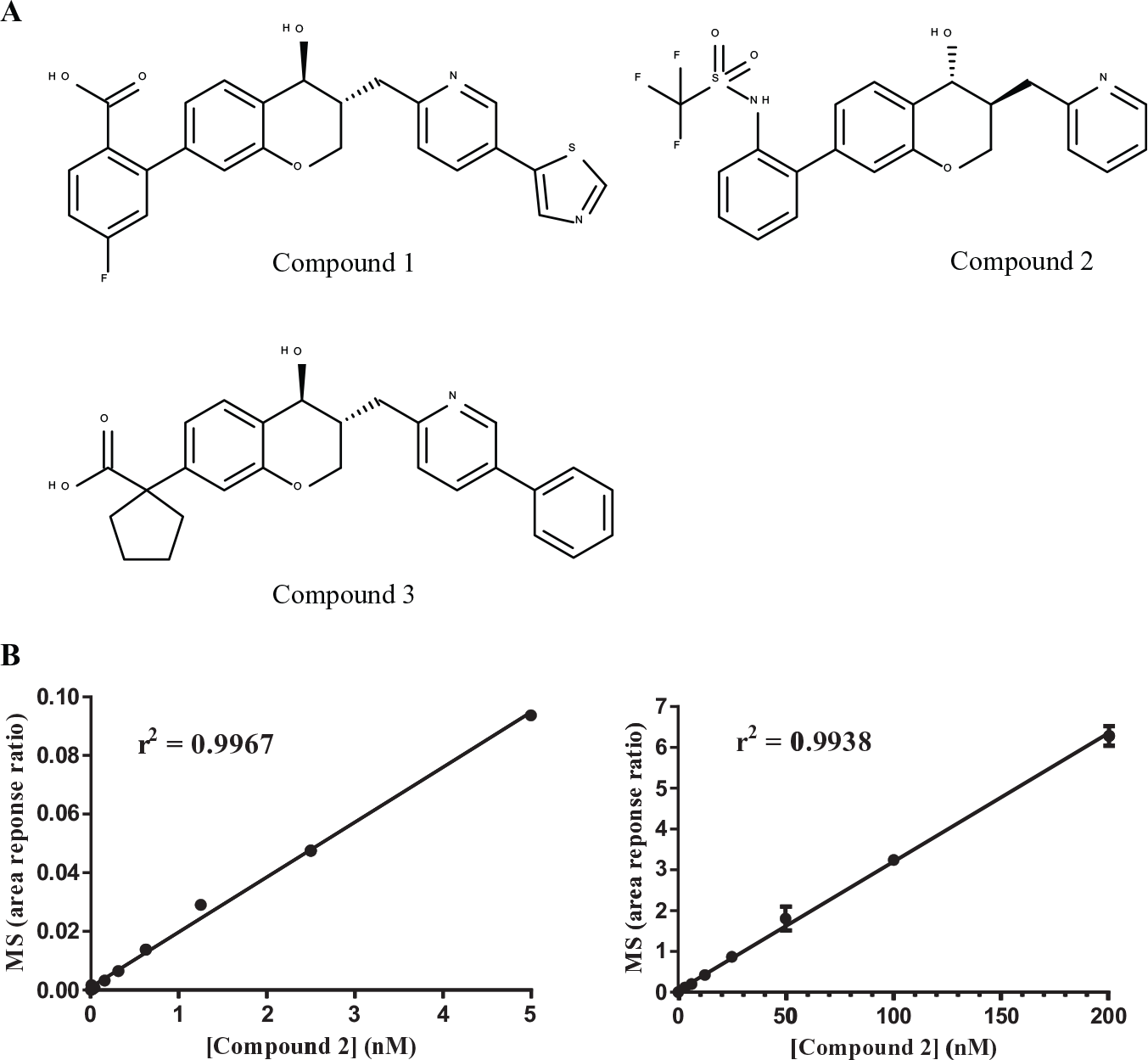
(
To define the dose–response linearity, a lower limit of detection (LLOD), and processing losses during the filtration/concentration steps for compounds
Direct Saturation Binding Data for BLT1 Antagonists 1 and 2
Equilibrium saturation binding studies are the method of choice for determining both ligand affinity (as a dissociation constant, Kd) and the number of ligand binding sites (Bmax) present in a biological source. We first applied this method to investigate the binding of compound
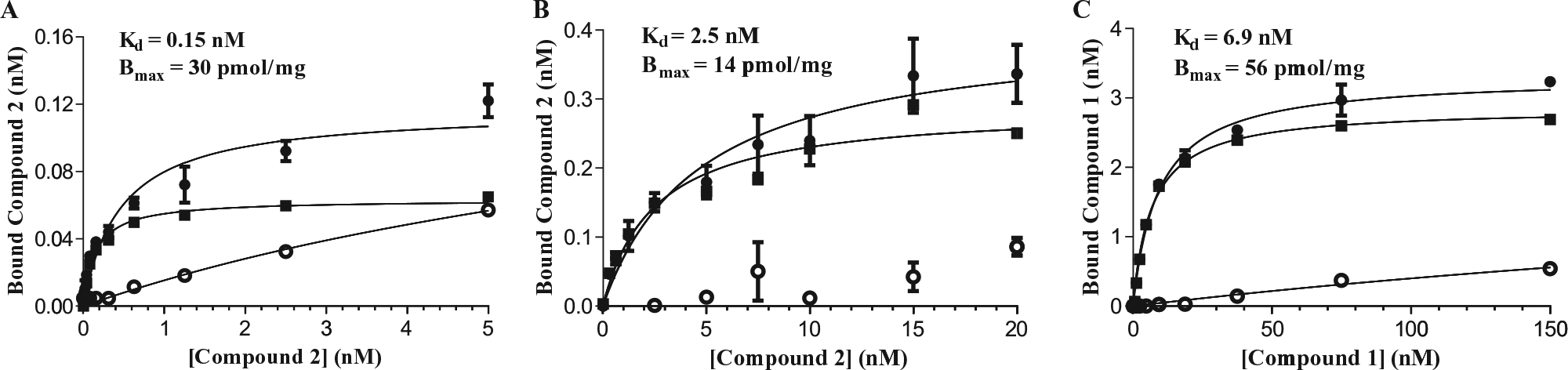
Representative data for equilibrium saturation direct binding of compounds
Several preclinical insulin resistance and diabetes models are of murine origin. As a prelude to using these models to evaluate the efficacy of our proprietary antagonists, we needed to establish murine BLT1 antagonist binding assays. We therefore examined the equilibrium saturation binding of compound
Compound
BLT1–Antagonist Complex Dissociation Studies
The kinetics of ligand binding, namely, the receptor–ligand association (kon) and dissociation (koff) rates, have profound effects on receptor-mediated pharmacology and are therefore important parameters to establish for key molecules in any drug development program.
24
To determine koff for the human BLT1–compound
The human BLT1–compound
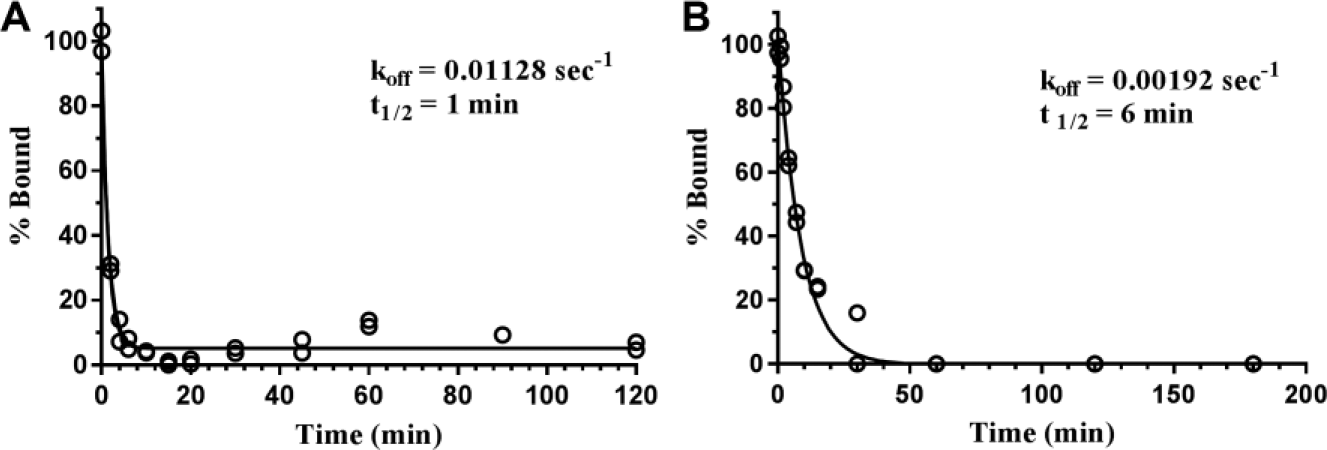
Dissociation time course of probe ligands from human BLT1. (
Using a similar protocol, we also measured koff for the human BLT1–compound
BLT1–Antagonist Competition Binding Assays
Competition binding assays offer a method to assess whether small molecules bind at either an allosteric or an orthosteric site on a receptor that modulates probe ligand binding. These assays offer a rapid method for establishing the affinity of numerous such competitors (as inhibition constants, Ki) and thereby facilitating the establishment of SARs within a compound series. To this end, we developed competitive binding assays for human BLT1 using probe ligands
To validate the human BLT1 competition assay using compound
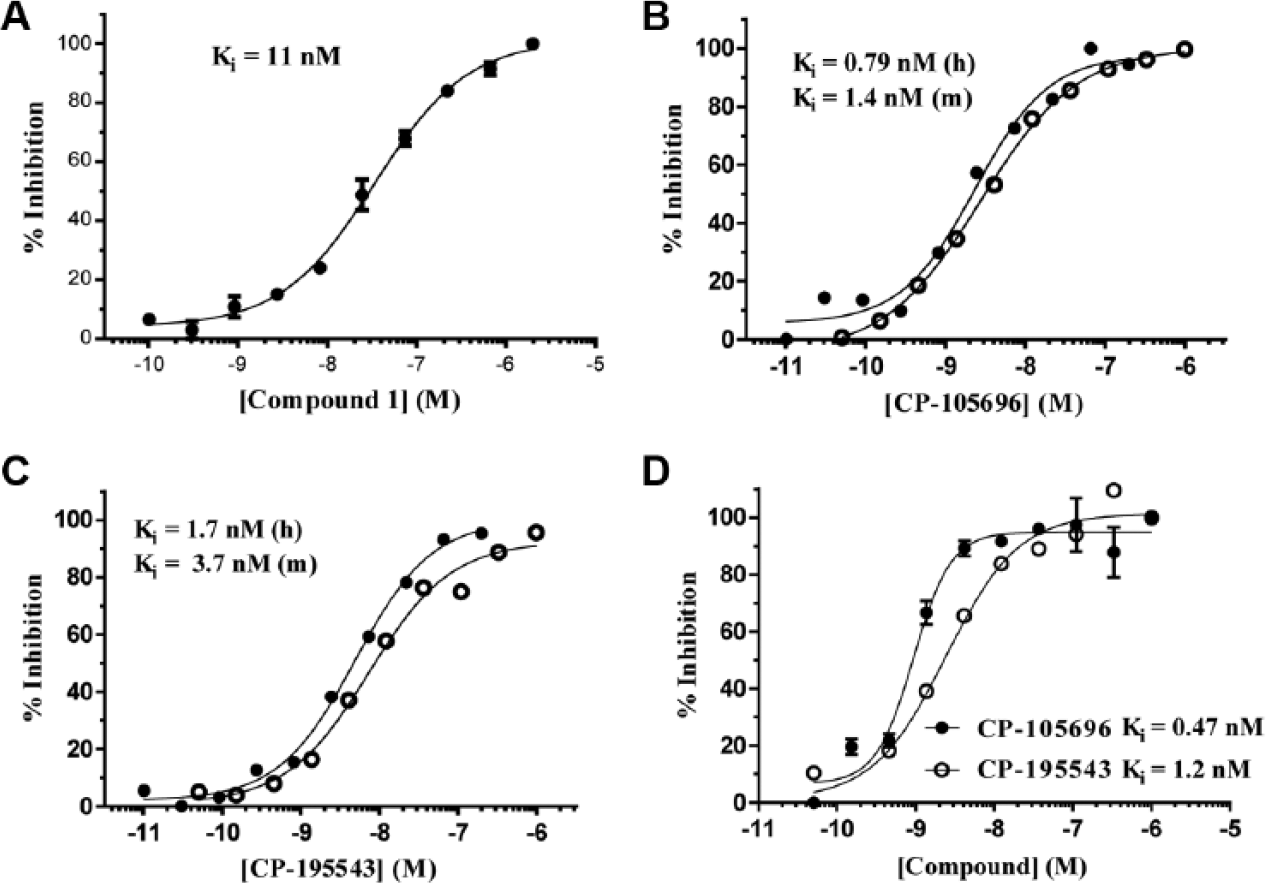
Validation of the competition binding assays using probe ligands
We also evaluated two previously described BLT1 antagonists from Pfizer, CP-105696 and CP-195543 in the human BTL1–compound
We also established a human BLT1 competition assay using probe ligand
Having successfully validated the competition binding assays, we measured the Ki values of numerous proprietary synthetic BLT1 modulators and ultra-high-throughput screening (uHTS) hits against human and mouse BLT1. We focused on assays employing probe ligand
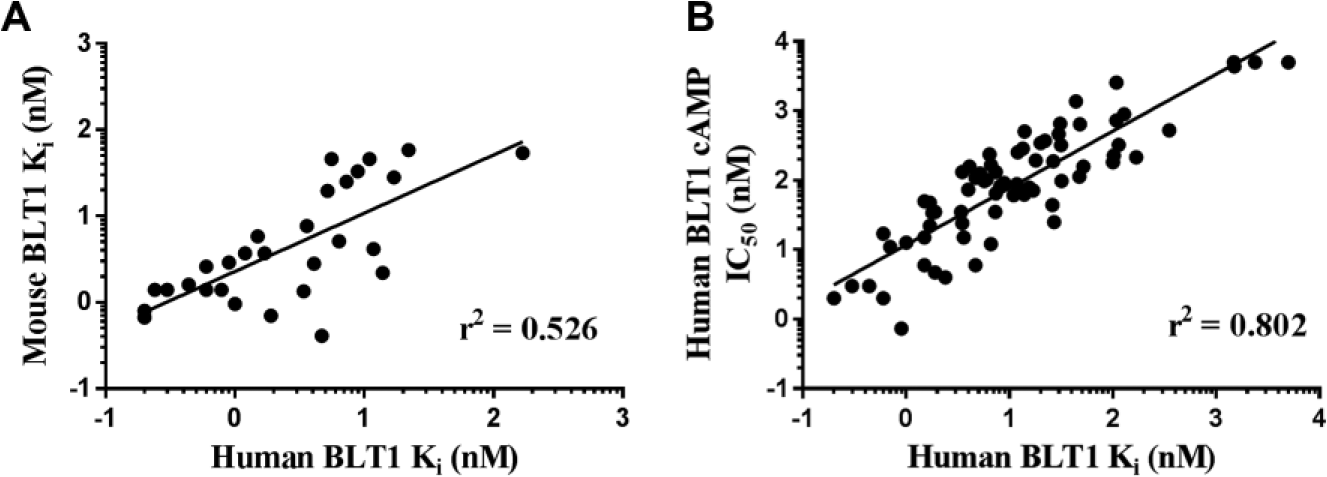
Correlation plots from BLT1 antagonist studies. (
In summary, we have developed and validated label-free, LC-MS-based equilibrium saturation direct binding and competition binding assays that enable the evaluation of small-molecule antagonist binding to recombinant human and mouse BLT1 receptors expressed in HEK 293 cell membranes. Using this approach, we rapidly scanned 20 antagonists to identify two “optimal” probe ligands and used these compounds to establish the expression level of binding-competent recombinant BLT1 receptors in membrane preparations. The binding affinity and kinetics of these two key BLT1 antagonists were characterized and exploited to develop moderate-throughput competition binding assays to assess the Ki values of numerous synthetic antagonists and uHTS hits. These efforts significantly expedited the selection of lead BLT1 antagonists for in vivo mouse studies. Building on the pioneering studies by Wanner and colleagues,29–31 our laboratory now routinely applies LC-MS-based, label-free binding assays to study receptor–ligand interactions in a wide array of drug discovery programs. We highly recommend that this methodology be more widely adopted. Furthermore, we challenge the field to explore new methods 34 that would increase the (moderate) throughput of the procedures described herein, which would be a welcomed complement to future studies.
Footnotes
Declaration of Conflicting Interests
The authors declared the following potential conflicts of interest with respect to the research, authorship, and/or publication of this article: All authors are, or were, employees of Merck & Co., Inc. and may own shares of Merck & Co., Inc. stock.
Funding
The authors received no financial support for the research, authorship, and/or publication of this article.