
Abstract
The unfolded protein response (UPR) is an integrated, adaptive biochemical process that is inextricably linked with cell homeostasis and paramount to maintenance of normal physiological function. Prolonged accumulation of improperly folded proteins in the endoplasmic reticulum (ER) leads to stress. This is the driving stimulus behind the UPR. As such, prolonged ER stress can push the UPR past beneficial functions such as reduced protein production and increased folding and clearance to apoptotic signaling. The UPR is thus contributory to the commencement, maintenance, and exacerbation of a multitude of disease states, making it an attractive global target to tackle conditions sorely in need of novel therapeutic intervention. The accumulation of information of screening tools, readily available therapies, and potential pathways to drug development is the cornerstone of informed clinical research and clinical trial design. Here, we review the UPR’s involvement in health and disease and, beyond providing an in-depth description of the molecules found to target the three UPR arms, we compile all the tools available to screen for and develop novel therapeutic agents that modulate the UPR with the scope of future disease intervention.
Introduction
The endoplasmic reticulum (ER) is a large cellular organelle that can account for more than 50% of total membranes in eukaryotic cells. It was one of the last cellular organelles to be discovered in 1945 by Porter, Claude, and Thompson 1 and is one of the most structurally complex and evolutionarily diverse components of cellular function and maintenance. It is subcategorized into smooth ER, the specialized myocyte sarcoplasmic reticulum, and rough ER (RER). The RER is distinguished by ER membrane–bound ribosomes that play a significant role in secretory and transmembrane protein biogenesis. 2 The smooth ER is a site of sterol and lipid production, thereby majorly contributing phospholipids to mitochondrial and peroxisomal membranes as well as the Golgi apparatus, secretory vesicles, and, of course, the ER itself. 3 Moreover, it is the largest depository of intracellular calcium, paramount in a vast host of processes such as muscle fiber contraction, mitochondrial function, neurotransmitter release, and the regulation of the secretory pathway through signaling the release of stored proteins from secretory vesicles. 4 Collectively, the ER integrates all of these different properties to fulfill its main task that lies in the productive folding of secretory and transmembrane proteins. This highly active processing must be accompanied by a finely tuned regulation of ER homeostasis. As such, protein homeostasis (proteostasis) in the ER is ensured by the coordinated action of chaperoning, folding, quality control (QC), and degradation mechanisms. 5 Newly synthesized proteins enter the ER, where they face a folding prone environment comprising chaperones (e.g., glucose regulated proteins such as GRP78/BiP, GRP94, GRP170) and enzymatic complexes involved in posttranslational modification such as N-linked glycosylation (e.g., oligosaccharyl transferase complex) or disulphide bond formation (e.g., protein disulphide isomerases). 6 These newly synthesized proteins undergo a folding process, which, if successful, allows them to exit the ER and traffic toward their final destination through the secretory pathway. 7 If folding fails, improperly folded proteins are retained in the ER through QC mechanisms. 8 For instance, a specific QC machinery has been identified for N-linked glycoproteins that includes the lectins calnexin and calreticulin, which bind monoglucosylated glycans, and the UDP-glucose glucosyl transferases, whose role is to re-monoglucosylate improperly folded glycoproteins to provide them with another chance of acquiring a proper conformation.9–11 Terminally misfolded proteins, identified as such as a result of several failed folding attempts, are then retrotranslocated in the cytosol to be degraded via the proteasome. This process is called ER-associated degradation (ERAD). 12 Under homeostatic conditions, ER folding, export, and degradation are not saturated and cope with the protein folding demand. However, during certain physiological and pathological conditions, the protein misfolding burden overwhelms ER folding and export capacity, thereby leading to an accumulation of improperly folded proteins in this compartment and to a situation called ER stress. To cope with ER stress, the ER has evolved an adaptive pathway, called the unfolded protein response (UPR), whose main function is to restore protein homeostasis in this compartment. 13 If the stress is alleviated, the UPR is shut down and ER proteostasis is restored. 14 In contrast, if adaptation fails and homeostatic mechanisms are overcome, the UPR also triggers signals that will engage and destine cells to die through apoptosis. 15
The UPR: Canonical and Noncanonical Signaling Pathways
The canonical core of the UPR is composed of three major transmembrane stress sensors; PERK, IRE-1, and ATF6.16–18 In their inactive state, all three sensors are bound to the protein BiP, otherwise known as GRP78 or HSPA5.19–21 BiP is an ER chaperone that is indispensable to both basal and perturbed UPR as it displays antiapoptotic abilities, is heavily involved in calcium homeostasis, and promotes QC by either targeting misfolded proteins for ERAD-mediated degradation or promoting correct refolding and translocation.22,23 Upon stress, BiP dissociates from the three sensors of the UPR, allowing them to trigger prosurvival or, in the case of overwhelming stress, prodeath mechanisms. It is this function that has led to BiP to be considered a master regulator of the UPR. The three sensors and associated signaling pathways can be viewed in Figure 1 .
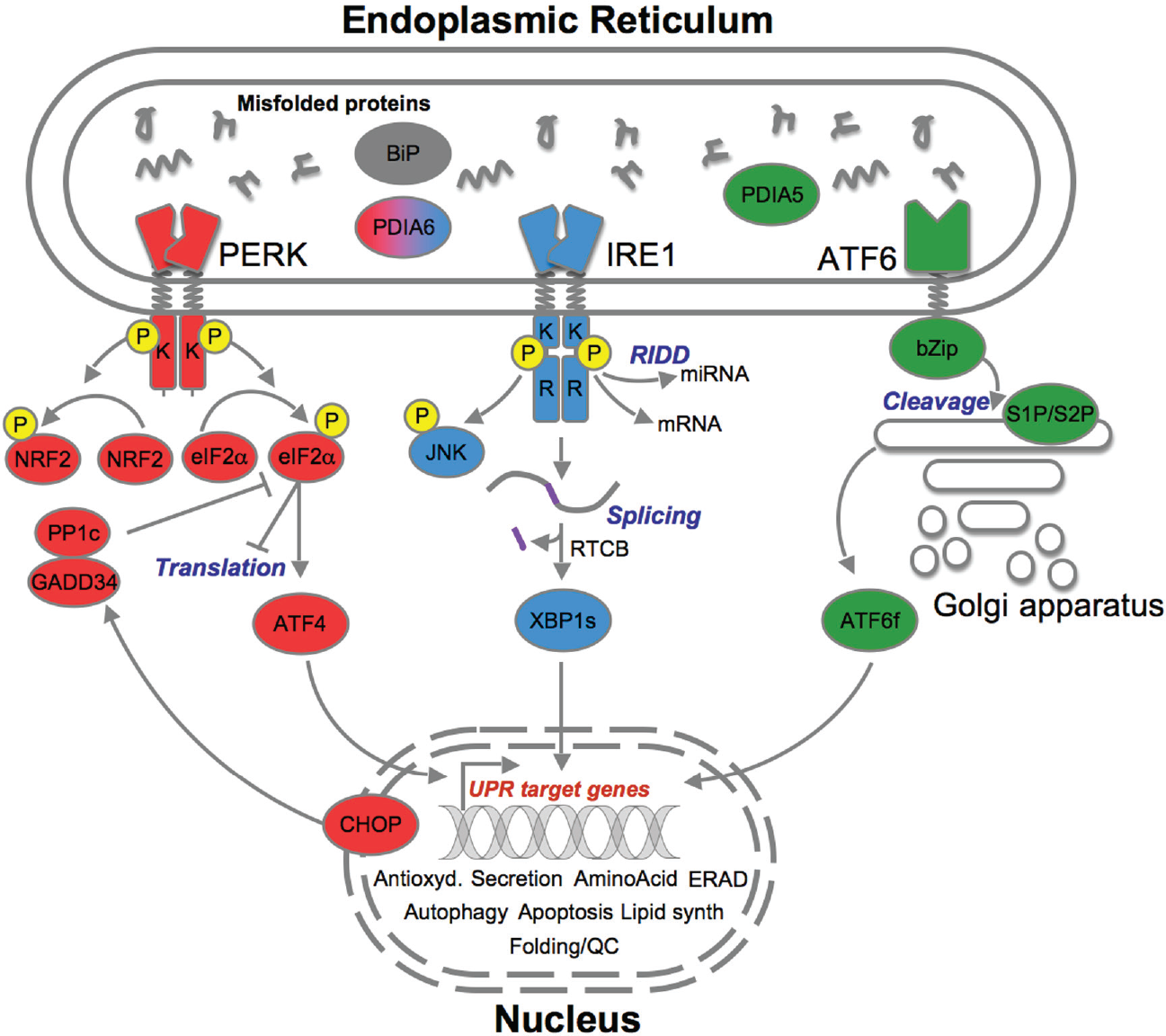
The three endoplasmic reticulum (ER) stress sensors (PERK [red], IRE1 [blue], ATF6 [green]) initially activate signaling events that increase protein-folding capacity and reduce protein load on the ER. These transcriptional and translational outputs tend to reestablish protein-folding homeostasis in the ER and promote cell survival.14,24
IRE1
IRE1 in humans exists in the form of two homologues: IRE1α and IRE1β. 25 IRE1α (referred to as IRE1 hereafter) is ubiquitously and abundantly expressed, whereas IRE1β is abundant only in intestinal and pulmonary epithelial and mucosal epithelial tissues.26,27 Interestingly, IRE1β displays higher structural similarity to murine IRE1 than human IPE1a. 28 IRE1 is a transmembrane type 1 protein. Its cytosolic domain carries out two enzymatic activities including serine/threonine kinase and endoribonuclease activities. 29 Recently, the activation profiles of IRE1 have revealed that the protein can form dimers and oligomers, which in turn confer selectivity in the output signals emitted from IRE1. 30 This includes not only the nonconventional splicing of XBP1 mRNA (which was recently found to be the result of the concerted action of IRE1 RNase and the RTCB tRNA ligase) 31 but also the regulated IRE1-dependent decay of RNA (RIDD, including mRNA 32 and microRNA 14 ). The coordinated action of these IRE1-dependent mechanisms yields select biological outputs. Interestingly, upon ER stress, the splicing of XBP1 mRNA has been shown to be transient and the subject of inactivation mechanisms that depend in part on the protein disulphide isomerase PDIA6. 33 In contrast, RIDD activity, which constitutively exists (tonic RIDD) appears to increase with stress, thereby leading to an uncontrolled and death-promoting mechanism (prodeath RIDD). 34 The precise mechanisms by which IRE1 operates XBP1 mRNA splicing and RIDD remain unclear.
PERK
PERK is similarly activated to IRE1, and it phosphorylates eukaryotic translation initiation factor 2α (eIF2α) that ultimately inhibits protein translation and thus synthesis as a potent prosurvival mechanism; this is based on the fact that the number of proteins entering the ER for processing is massively reduced. 35 In contrast to its induction of a generalized arrest in translation, eIF2α phosphorylation promotes the translation of the transcription factor ATF4. 36 ATF4 then acts as either a transcriptional repressor or a transcriptional activator and affects a multitude of biological processes such as bone resorption (RANKL), 37 amino acid metabolism (asparagine synthase), 38 neovascularization (VEGF), 39 as well as controlling expression of genes involved in ER chaperone and foldase production. 14 CHOP and GADD34 further contribute to PERK-mediated cell fate control by mediating apoptosis or PERK inhibition, respectively. 40 In a study by Novoa et al., 41 phosphorylated eIF2α was found to be greatly diminished in GADD34 overexpressing cells because of the formation of a complex between GADD34 and the catalytic domain of protein phosphatase 1 (PP1c) that resulted in the dephosphorylation of eIF2α and thus blocked attenuation of CHOP and interfered with ATF4 activation in a stress-dependent manner. As is the case with IRE1, PDIA6 controls the transition to the inactive form of the protein. 42 In addition to eIF2α, PERK has been shown to target Nrf2, thereby mediating the antioxidant responsive element response. 43
ATF6
ATF6 is a type 2 transmembrane protein that resides in the ER. It contains a transcription factor in its cytosolic domain. Upon ER stress, ATF6 is exported to the Golgi apparatus, where it is cleaved on both sides of the membrane by the proteases S1P and S2P.44,45 This prompts the release of the ATF6 cytosolic domain (ATF6f), which then translocates to the nucleus to act as a transcription factor for genes whose products are involved in ERAD and quality control (HERPUD1, SEL1L, OS9), folding (BiP, GRP94), and redox control (PDIA4, ERO1L). 46 Beyond the role of BiP in ATF6 activation, the protein disulphide isomerase PDIA5 has also been shown to be instrumental in this process. 47
In recent years, other events were described to transduce information about the ER status independently of the three UPR sensors, leading to their attribution to the noncanonical UPR. Those three different classes can be defined as (1) regulation of calcium fluxes, (2) transmembrane transcription factors, and (3) atypical transcription mechanisms. First, calcium release from the ER has been shown to represent an atypical signaling event upon ER stress. Indeed, inositol 3-phosphate–induced calcium release is increased during ER stress and is also involved in CHOP-mediated apoptotic signaling through ER oxidases 48 as well as being linked with the binding to members of the apoptotic BCL-2 family to modulate calcium release and modulate ER stress cell death. 49 Therefore, dysregulation of calcium fluxes can lead to the initiation or exacerbation of the maladaptive UPR and hence lead to apoptotic cell death. Moreover, apoptotic regulators such as the Golgi anti-apoptotic protein modulate intracellular calcium fluxes 50 and caspases 4 and 12 have been shown to be activated during ER stress in a calcium-dependent manner in Parkinsonism and prion neurotoxic disorders.51,52 Beyond ATF6, a large family of transmembrane bZIP proteins has been discovered and plays diverse functions in the ER homeostasis outside the scope of the canonical UPR. 53 This family comprises the transcription factors OASIS, BBF2H7, CREBH, AIbZIP, and Luman and has been implicated in diverse responses to cell differentiation, maturation, and homeostasis in different tissue distributions. 54 Each family member may play a distinct role in ER homeostasis, and it has been shown in an in vivo model of cerebral ischemia that BB2H7 is predominantly present in the peri-infarction region, pointing to an up-regulation during a specific phase of the UPR. 55 Another example is provided by CREBH, whose activation upon ER stress was shown to control iron regulation in the liver through the control of hepcidin expression. 56 More recently, the AAA+ ATPase CDC48(yeast)/valosin-containing protein (VCP)/p97(mammals), known to play a role in ERAD, 57 was shown to also regulate ER stress-induced gene expression. 58 This mechanism was shown to occur through the p97/VCP-mediated degradation of RUVBL-2, another AAA+ ATPase acting as a repressor of XBP1s-dependent transcription, thus indicating a dual role for p97/VCP in protein degradation and transcriptional control. 59
Control of ER Proteostasis in Diseases
The UPR has been extensively implicated in disease either as a cause or a consequence, playing roles in disease commencement or exacerbation. These effects are mainly mediated by prodeath and prosurvival properties of the UPR. 60 In certain clinical scenarios, prodeath signaling leads to degeneration and apoptosis, whereas in others, the same biochemical pathways lead to disease therapeutic resistance. Amyotrophic lateral sclerosis (ALS; reviewed in ref. 61) and glioblastoma multiforme (GBM; reviewed in ref. 62) are two examples of this diverse response in the central nervous system. P97/VCP, involved in the noncanonical control of the UPR, provides a genetic crossover between ALS and GBM. It has been shown to be able to induce ALS with or without frontotemporal lobar degeneration accompanied by Paget disease, mimicking osteopathies and inclusion body myositis by colocalizing in trademark ALS cytoplasmic protein inclusions with established common ALS proteins FUS and TDP-43.63,64 Conversely, VCP has been shown to regulate DNA-dependent protein kinase degradation and hence affect radiation sensitivity of GBM cells. 65 Despite this crossover, the hyperactivation of the perturbed UPR in these diseases has contrasting effects, leading to autophagy and apoptosis of motor neurons in ALS but to chemoresistance, neoangiogenesis, and migration regulation in cancer cells in GBM. We look into the prodeath and prosurvival effect of the UPR in different diseases in more detail below.
Prodeath Signaling of the UPR in Diseases: Neurodegeneration and Diabetes
Classical ALS is a predominantly adult-onset neurodegenerative genetic disorder characterized by rapidly progressive loss of both upper and lower motor neurons, with the most common cause of death being respiratory failure following diaphragmatic muscle failure. 66 Studies in in vivo models of ALS have shown that pharmacologically modulating the UPR can enhance ALS pathogenesis or indeed replicate it. 67 Superoxide dismutase (SOD1) mutations lead to severe oxidative stress by affecting the conversion of superoxide to water or hydrogen peroxide and the regulation of copper as a catalyst for SOD1. 68 Furthermore, the trademarks of TDP43 and VCP mutation causing ALS are cytoplasmic ubiquitinated inclusion protein aggregates and autophagy. Oxidative stress, autophagy, and protein aggregation are trademark triggers of ER stress and subsequently the UPR. 69 Furthermore, a number of UPR-related genes and proteins are directly altered and related to the course of ALS progression. GRP78/BiP, an ER chaperone, has been shown to be bound by SOD1 up-regulating its expression in ALS in vivo models, and the calcium-binding chaperone calreticulin leads to motor neuron apoptosis. 70 To further support this, administration of the eIF2α phosphorylation inducer salubrinal to SOD1 transgenic mice arrests disease progression, PERK haploinsufficiency exacerbates disease, and XBP1 down-regulation stalls disease onset and improves prognosis. 71 Furthermore, PDI and GADD34 have been shown in in vivo models upon disease onset to disseminate into ventral horn astrocytes and white matter microglia. 72 ALS is a complex spectrum disorder of unclear origin. As a predominantly sporadic (>80%) condition diagnosed late in its progression, it is extremely difficult to discern a specific pathognomonic pattern and therefore decide whether UPR involvement is causative or consequential. In some cases, it may well be true that it is both, encompassing a positive feedback loop of prodeath signals that are compounded by a multitude of exacerbating conditions that arise as part of the ALS comorbidity spectrum (frontotemporal dementia, osteopathies, and gastrointestinal disturbances due to bulbar involvement, among others). Therefore, targeting the UPR at this postdiagnostic stage would play a disease-limiting role. By blocking the UPR prodeath signals that dominate ALS progression, the patient’s prognosis could be drastically improved.
Diabetes mellitus is a disease that affects almost 430 million people worldwide. 73 The development of diabetes depends on loss of pancreatic β cells, which are the cells responsible for producing insulin to regulate glucose homeostasis in the islets of Langerhans. To respond to such high acute or chronic metabolic and proteostatic demand, β cells have an established ER that has been shown to be paramount in the homeostasis of β cells and a contributor in the development of diabetic symptoms when it fails. 74 The knockout of IRE1 and XBP1 in β cells produces a distinct decline in cell proliferation and also affects proinsulin and insulin synthesis and secretion/excretion. 75 Similarly, PERK inactivation displays diabetic symptoms of hyperglycaemia in in vivo models, and the UPR in general is triggered during the misfolding of preproinsulin, eventually leading to accumulated and overwhelmed ER stress and β cell apoptosis. 76 In the case of diabetes, targeting the UPR could play both a disease-limiting and a preventative role. Diabetes is a small- and medium-vessel disease, and the UPR has been strongly linked with VEGF regulation and neoangiogenesis. 77 By targeting these pathways, the quality of life of patients already suffering from diabetes could be vastly improved as harnessing and limiting such processes could have an effect on manifestations such as diabetic retinopathy and coronary artery disease.
Prosurvival UPR in Cancer: The Example of Glioblastoma
GBM is the most frequent central nervous system primary tumor. Its incidence is two to three new cases per 100,000 per year, and it has an extremely low prevalence; the 5-year survival levels are less than 3%, resulting from a dismal median prognosis of 15 months survival postdiagnosis. GBM is a solid tumor–occupying lesion and therefore carries trademark characteristics such as poor vascularization and high proliferation rates. 78 The resulting subsequent low pH and lack of nutrients offer major triggers of potentially proapoptotic UPR involvement. However, tumors manage to adapt to such environments by modulating the UPR. 79 Molecular chaperones have been heavily implicated in disease progression in GBM. GRP78/BiP (high proliferation, prolonged cell survival) has been shown to be up-regulated in a variety of cancers including GBM and shown to decrease in the presence of chemotherapeutic agents, resulting in an increase in neoplastic cell apoptosis. 80 Another chaperone, GRP94, displays similar properties to GRP78 and is linked to ROS UPR activation. 81 The current treatment of choice in GBM involves maximal tumor resection followed by the STUPP protocol that involves treatment with the alkylating agent temozolomide alongside radiotherapy. 78 This has variable success in patient cohorts as some high-grade gliomas are resistant to the effects of this particular course of chemotherapy and adjuvant radiotherapy. 79 Modulation of the UPR to sensitize resistant tumors to existing therapies could be an attractive therapeutic target in GBM. IRE1 is a key player involved in the apoptotic switch; however, it produces either adaptive or death signals via its RNase activity and was shown to play key roles in GBM development, with particular emphasis on tumor growth and neoangiogenesis. 82 It is currently not clear how IRE1 downstream signaling events integrate to yield those specific outcomes. Using a dominant negative approach in U87 cells, IRE1 was found to contribute to neoangiogenesis, and its RNase activity was involved in migration/invasion as well as in proinflammatory processes. 83 Silencing IRE1 RNase activity in glioblastoma cells pushed them toward a more motile phenotype, showing short-range infiltration at the immediate periphery of GBM cores and an extensive blood vessel cooption with formation of distal perivascular tumor microsatellites.82,83 It has been shown in orthotopic xenograft models of human glioma that invalidation of both the kinase and endoribonuclease domains of IRE1 produced avascular infiltrative tumors with vessel cooption, while invalidation of the endoribonuclease function alone did not affect angiogenesis, suggesting that the IRE1 domains play specific roles in the migration and vascularization of tumors. 83 Moreover, it has been shown that IRE1 plays an important role in the up-regulation of extracellular matrix proteins such as SPARC, further cementing IRE1’s involvement in tumor growth and migration.82,84,85 Finally, IRE1 has been implicated in the maintenance of the circadian clock gene PER1, directly involving it in the survival and growth of tumor cells. 86
The UPR as a Therapeutic Target: From the Tools to the Small Molecules
The first report of an ER stress–induced adaptive response in the yeast Saccharomyces cerevisiae was made in the late 1980s 87 using molecules perturbing protein folding in this compartment. As such, the alteration of protein N-glycosylation, for instance, by replacing glucose with 2-deoxyglucose or using tunicamycin (an antibiotic that prevents the generation of glycans), leads to the accumulation of improperly folded proteins in the ER. 88 Similarly, thapsigargin specifically inhibits the SERCA Ca2+ ATPases, thus preventing the refilling of ER calcium stores, leading to functional imbalance of this compartment. 89 Other compounds such as dithiothreitol, which is a reducing agent, or brefeldin A, which reversibly disables ER to Golgi transport, induce the UPR.88,90 None of these compounds were usable in a clinical context because of their lack of specificity and high toxicity. Attempts to bypass such effects are indeed under way, and it has been reported that mipsagargin, a prodrug analog of thapsigargin, did display acceptable tolerability and favorable pharmacokinetic profiles in patients with solid tumors. 91 Despite the wide-reaching application of ER stressors and the discovery of some currently used in clinics such as bortezomib in multiple myeloma, 92 it is evident that further characterization of the UPR and its potential modulation is needed for more effective therapies, justifying the approaches described below.
Screening Strategies
Taking into account the increasing number of reports incriminating the UPR in the development of multiple diseases, targeting this signaling pathway is becoming a central public health and economic issue for numerous laboratories and companies. The diversity of the in vitro and in vivo approaches employed to reach this goal can give a picture of the inherent complexity of this signaling pathway. In Table 1 , we provide a nonexhaustive review of the diversity of screening approaches that have led to the discovery of new UPR modulators. In some cases (e.g., cancer treatment), molecules that induce the UPR by initiating ER stress could represent a way of overloading cellular resistance to chemotherapy. 120 In that case, screening strategies employing cell protection from ER stress–induced cell death or GRP78-luciferase reporter as readout have led to the identification of new specific ER stressors. Consequently, drugs that inhibit a specific branch or protein of the UPR could represent a relevant targeted therapy, first, in cancers whose development appears to be dependent of specific branches of the UPR 121 or, second, in the case of treatment against viruses that use the secretory pathway to produce difficult-to-fold viral proteins. Identification of such drugs has been dependent on the development of in vitro methodologies or cellular reporters of the IRE1, PERK, or ATF6 pathways, using the specificity of their molecular activation (XBP1 splicing, ATF4 translation, and ATF6 nuclear translocation, respectively; Table 1 ). In contrast, other diseases, such as human amyloid diseases, are related to insufficient UPR. In such diseases, screening studies have mainly focused on the identification of UPR activators. In general, both in vitro and cellular strategies are employed in screening strategies aiming to identify UPR modulators. Although cellular strategies are usually coupled with efforts to determine the specificity of the hits, it is important to note that these approaches have enabled the identification of multitarget drugs, which are particularly relevant in bypassing neoplastic chemoresistance driven by mutation or compensatory mechanism processes. Finally, although in vivo models, such as Caenorhabditis elegans and Drosophila melanogaster, provide promising platforms for efficient drug discovery and drug target identification, their use in high-throughput screening (HTS) studies of the UPR are up to now underrepresented.
Screening Approaches That Have Led to the Discovery of New Unfolded Protein Response (UPR) Modulators.

Cellular Reporters of UPR Activation
Cell luminescent reporters have been widely used to identify UPR modulators. These reporters have been designed to target (1) transcriptional activation of the UPR, by fusing luciferase genes to regulatory sequences such as GRP78/BiP or CHOP promoters or ER stress response element sequences; (2) IRE1 activity, by cloning luciferase sequence downstream of the 26-nt ER stress–specific intron of human XBP1; and (3) PERK activity, by fusing the 5′ UTR of the human ATF4 containing the µORF to luciferase. Although these last methodologies were designed to screen for drugs that target one UPR branch, they usually require a second set of experiments to establish drug specificity. A study conducted by Walter and colleagues 122 can be cited as a representative experimental design of screening studies of the UPR. By screening 106,281 compounds using an ATF4-luciferase reporter system, they initially identified 460 hits. In a second phase, they discarded inhibitors that also affected the IRE1 branch of the UPR, by using a reporter of XBP1 splicing, restricting the hits number to 187. This number was then reduced to 77 in a microscopy-based screen that employed another ATF4 reporter. Subsequently, Western blot analysis was used in a quaternary screen. Finally, 28 compounds were selected for their ability to specifically target the PERK branch of the UPR and led to the identification of ISRIB, a potent inhibitor of PERK signaling. A similar approach was used by the same group to identify specific modulators of the ATF6 branch. 123 Other approaches to identify selective modulators of IRE1 have included the use of a chimeric CHOP-Gal4 transcription factor to monitor IRE1-mediated p38MAPK activation in a 384-well format and the monitoring of XBP1 activation through an UPR pathway element-Luc–based assay in a 1536-well format. 124 HTS also identified 2,9-diazaspiro[5.5]undecanes as cytotoxicity inducers in several cell lines through the depletion of intracellular calcium stores. 125 The UPR inducer borrelidin was also discovered through a cell HTS in which UPR-inducing natural extracts were purified by RP-18 high-performance liquid chromatography and iterative bioassay–guided C18 fractionation. 113
Reporters of ER Functions
Adding to the strategies employed to target the adaptation pathway of the UPR, we wanted to underline the efforts that have been made to identify drugs that target the molecular functions of the secretory pathway. In 2004, Fiebiger and colleagues 114 established a fluorescent reporter of (ER)-to-cytosol degradation pathway. They used a double construction consisting of a class I major histocompatibility complex (MHC) heavy chain–enhanced green fluorescent protein fusion protein (EGFP-HC) and the human cytomegalovirus protein US11. When both expressed in cells, US11 induces the dislocation of MHC class I heavy chains from the ER to the cytosol, leading to its degradation, producing a weak fluorescence signal. Using this system, the authors performed an HTS and identified two compounds, eeyarestatin I and II, which inhibit degradation of three dislocation substrates by retaining them in the ER. These were EGFP-HC, wild-type class I heavy chain and T-cell receptor. Bennet et al. 118 adapted a glycosylation-sensitive luciferase reporter by the addition of the EGFR N-terminal amino acid leader sequence to the luciferase gene and the incorporation of glycosylation consensus sites into the luciferase coding sequence. Through the screen of 2802 compounds, the authors identified aclacinomycin as a compound that reduces cell surface expression of glycosylated proteins. Fu and colleagues 116 developed two HTS functional screening systems that independently measure the free chaperone content and protein-folding capacity of the ER. The first system consists of the fusion of a human gene fragment that encodes a peptide derived from the ATF6 luminal domain to luciferase. This system allowed the measurement of the free chaperone content in the ER. In conditions where ATF6 is released from GRP78 (e.g., ER stress), the luciferase secretion is triggered, resulting in luminescence increase. The second system was constructed by associating the coding sequence of the membrane protein asialoglycoprotein receptor 1 (ASGR1) to luciferase gene. Considering that the ASGR1 expression is sensitive to ER function, this reporter monitors ER-folding capacities. Using these cellular reporters, the authors identified a small-molecule compound, azoramide, that regulates ER folding and secretion capacity without inducing ER stress and protects cells from the consequences of ER stress. This drug was proposed as a potential drug candidate for type 2 diabetes. These studies underline the multiplicity of processes that can be targeted in order to identify modulators of the ER functions (reviewed in ref. 121). The CHOP pathway has been targeted to identify compounds that promote proapoptotic CHOP-related cascades without triggering adaptive cascades. Through HTS, an optimized sulfonamidebenzamide compound was discovered that displayed antiproliferative effects spanning multiple cancer cell lines. 126
In Vitro Strategies
Fluorescence quenching–based, Alphascreen, or homogeneous time-resolved fluorescence, have been proposed as tools for the in vitro identification of modulators of central proteins of the UPR such as IRE1, PERK, or GRP94 ( Table 2 ). Note that these approaches are particularly relevant in the case of some highly regulated proteins such as chaperones, taking into account (1) that molecular processes could compensate for their inhibition and (2) the need of a high degree of specificity due to the existence of highly homologous paralogs. These in vitro strategies led to the identification of highly selective modulators. Furthermore, they were also often followed by optimization studies aiming at improving drug affinity, physicochemical properties, metabolism, and so forth. As a result, GSK2656157 was discovered as a decreased lipophilicity GSK2606414 derivative, ISRIB underwent EC50 improvement and optimization, IPA was engineered as a better IRE1 activator than APY29, and BnIm was shown to present better potency and selectivity for Grp94 than previously described purine-scaffold compounds ( Table 1 ). N-acridine-9-yl-N′,N′-dimethylpropane-1,3-diamine or DAPA and its derivatives such as N9-(3-(dimethylamino)propyl)-N3,N3,N6,N6-tetramethylacridine-3,6,9-triamine (3,6-DMAD) were also identified as IRE1 inhibitors through an HTS chemical library screen and were shown to be cytotoxic to multiple myeloma by inhibiting both IRE1α oligomerization and XBP1 splicing. 136 Moreover, a dissociation-enhanced lanthanide fluorescence immunoassay can be used to identify IRE1 autophosphorylation inhibitors by designing a 384-well HTS specific to this branch of the UPR to produce drugs targeting multiple myeloma and other secretory diseases. 137 This approach was used to screen a library of 2312 potential kinase inhibitors, identifying 30 compounds that were shown to bind to the IRE1 kinase domain, including some known inhibitors such as sunitinib and some unknown such as CCT249525. 137
Molecules Identified to Target Directly the ATF6, IRE1, or PERK Pathways.

Molecules Targeting IRE1 and Their Use in Disease Models
The detection of the accumulated unfolded proteins is an essential initial step for the activation of the three arms of the UPR. This detection is mediated by GRP78 or BiP, which, under normal physiological conditions, is bound to the luminal domain of the sensors, thereby keeping them deactivated. ER stress causes the dissociation of BiP from the sensors, priming them for activation.21,138,139 The understanding of the physiology of these targets forms the basis of their potential pharmacological modulation. Modulators have been synthesized to both activate and inhibit IRE1. Activators of IRE1 predominantly act through the kinase domain by stabilizing a conformation that mimics the phosphorylated from of IRE1. 139 Compound 3 is one such molecule shown to inhibit IRE1 kinase activity while promoting at the same time XBP1 splicing. 140 APY-29 and sunitinib displayed similar functions, filling the adenine binding site of the kinase domain with high affinity, leading to partial molecular activation of IRE1. 30 Compounds have also been explored acting upstream of IRE1, targeting BiP. A series of molecules (thiazole benzensulfonamides) have been shown to have antineoplastic properties in melanoma cells by inducing autophagic and apoptotic cell death. 108 MKC-3946 (a noncompetitive inhibitor) of the IRE1 RNAse site has been shown to inhibit growth of MM cell lines, avoiding toxicity in mononuclear cells. 141 Moreover, bortezomib-induced cytotoxicity was shown to be increased in the presence of MKC, and ER stress induction by bortezomib, deduced by the presence of XBP1s, was blocked by MKC-3946. 142 Apoptosis induced by these agents was enhanced by MKC-3946, associated with an increase in CHOP expression. Furthermore, MKC-3946 was shown to inhibit XBP1 splicing in vivo, while inhibiting the growth of multiple myeloma cells. 143 UPRM8 is a covalently bound RNAse inhibitor. 144 The development of kinase inhibitors involving the manipulation of an analogous DFG+2 nucleophile (C715) located allosterically to the ATP binding site has been postulated to enable the identification of IRE1 inhibitors that could overcome traditional specificity concerns. Chronic lymphocytic leukemia and non–small-cell lung cancer medications ibrutinib and afatinib have set some precedence in the clinical use of cysteine-directed covalent inhibitors, and therefore, UPRM8 shows promise in its use as an IRE1 modulator in GBM.144,145 The kinase-inhibiting RNase attenuators KIRA3 and 6 act on the RNAse domain of IRE1 indirectly and were discovered during the screening of type II ATP competitive ligands. KIRA3 (an optimized pyrazolopyrimidine structure) specifically directly antagonizes the effect of IRE1 activation by APY29. KIRA3 favors the prevalence of the monomeric inactive form of IRE1, and although effective, KIRA6 (a KIRA3 derivative; Table 2 ) was developed as a more thermodynamically stable alternative for use in in vivo models. KIRAs offer an alternative to modulation with salicylaldehyde inhibitors such as MKC as they inhibit not only the RNAse but also kinase domains of IRE1, offering a more complete blocking alternative. 146 Recent attempts to obtain novel tricyclic chromenone-based inhibitors have been presented as useful tools to study XBP1 suppression in whole cells. Inhibitors of the peptidyl-propyl isomerases and HSP90 inhibitors such as geldanamycin have been developed primarily targeting immunosuppression and anticancer treatment. This has followed the association of such inhibitors with the HSP90 ER paralog GRP94 and evidence pointing toward the HSP90 dependent destabilization of UPR signaling. 147 In a different study, 11 overlapping fragments of the IRE1 cytosolic domain (F1-F11) were generated as 6xHis fusion proteins to test their effect on IRE1 signaling. One of those, F6, its peptide derivative P4, and its modified version TAT-P4 (FIRE), have been shown to promote ER stress resistance by reducing RIDD and JNK activation and enhancing XBP1s production. However, the exploration of peptidomimetic design targeting pharmacological chaperones and protein folding control remains to be fully addressed. 104
Modulators have also targeted the other two arms of the UPR. ISRIB has been shown to be a potent inhibitor of the integrated stress response by modulating eIF2α phosphorylation and its translational effects. 112 Salubrinal and guanabenz inhibit the GADD34/PP1c complex, thereby preventing eIF2α dephosphorylation, although the direct or indirect mechanism by which this is achieved is not yet clear and is currently the subject of ongoing investigation. The GADD34/PP1c axis is also targeted by sephin1/IFB-088. In addition, GSK2656157 inhibits PERK directly.95,128,134 Modulators targeting the ATF6 pathway include the newly identified ceapins that trap ATF6α in ER-resident foci, thereby inhibiting its translocation to the Golgi and compound 147 that preferentially targets the ATF6 transcriptional repertoire ( Table 2 ).123,127
Conclusions
In this review, we have illustrated the critical functions played by the regulation of ER homeostasis in health and diseases, with particular attention to three examples of pathologies comprising ALS, diabetes, and cancer (glioblastoma). We also discuss the relevance of targeting the main ER homeostasis control pathway, namely, the UPR, in diseases either to improve an altered ER homeostasis and rescue cells from death (e.g., for ALS) or to weaken cells’ adaptive capacity to enhance death (e.g., cancer/GBM). Screening strategies have identified selective modulators of the UPR sensors for which a clinical future can be envisioned. On the other hand, clinically relevant compounds with applications in various cancers, such as the renal, hepatocellular, and thyroid carcinoma (sorafenib) or melanoma (vemurafenib), have been shown to mediate their toxic effects in part through the perturbation of ER homeostasis and activation of the UPR. As a consequence, the value of the UPR as a therapeutic target in diseases with dismal prognoses continues to be proven. Further investigations can only show how wide ranging the reach of UPR targeting therapeutics can be.
Footnotes
Declaration of Conflicting Interests
The authors declared no potential conflicts of interest with respect to the research, authorship, and/or publication of this article.
Funding
The authors disclosed receipt of the following financial support for the research, authorship, and/or publication of this article: This work was funded by grants from the Institut National du Cancer (INCa) and EU H2020 MSCA ITN-675448 (TRAINERS).